
Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations

 ,
,  ,
,  ,
,  , and
, and 
Abstract
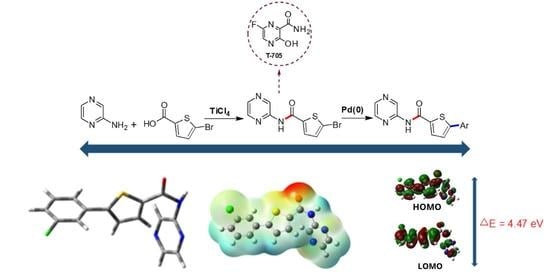
:
1. Introduction
2. Results and Discussion
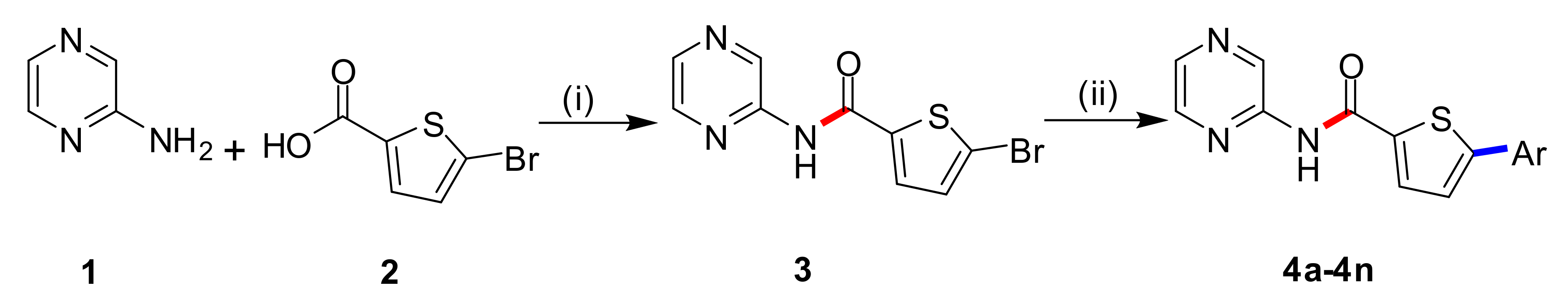
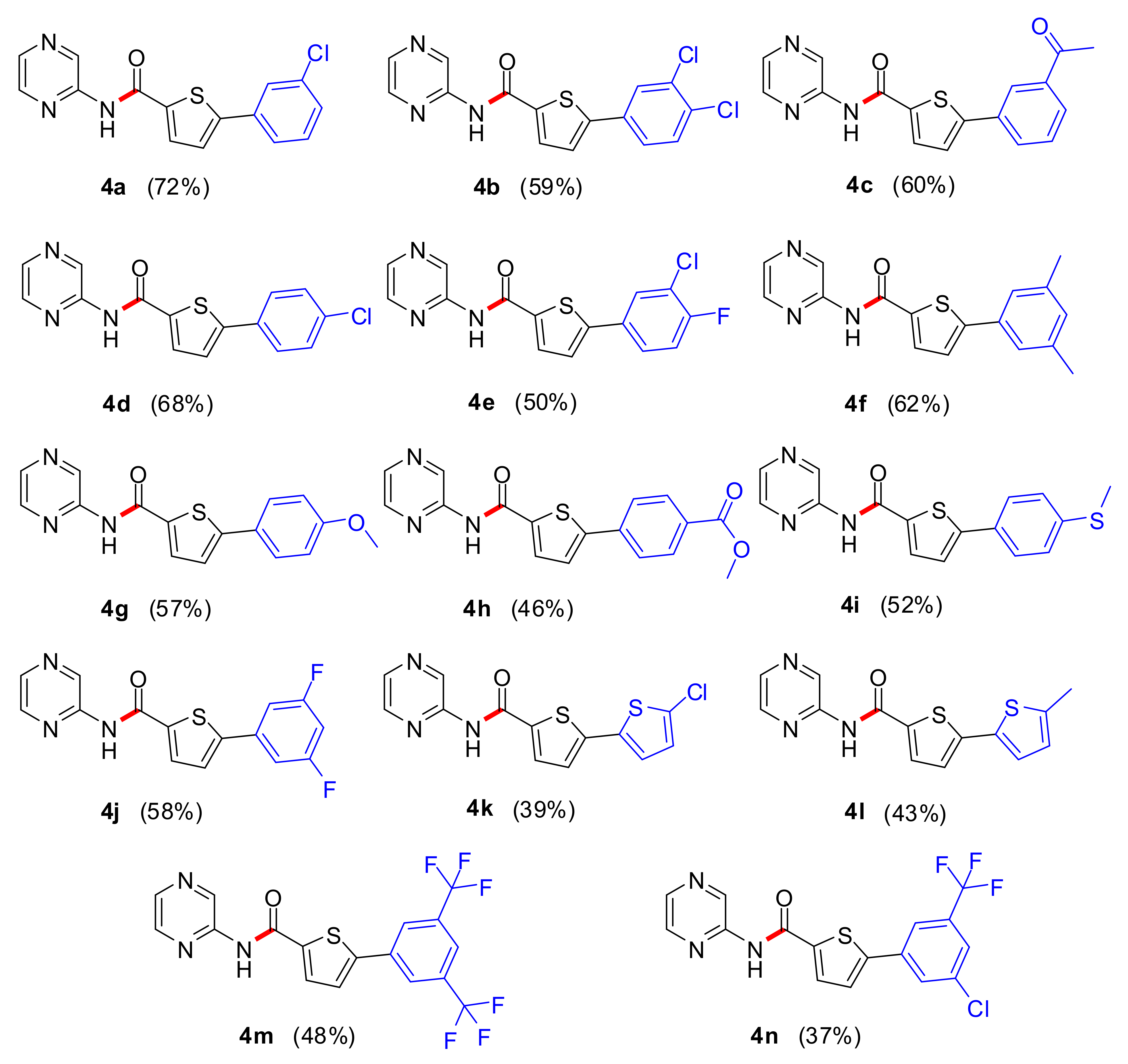
2.1. Chemistry
2.2. Computational Details
2.2.1. Optimized Geometries
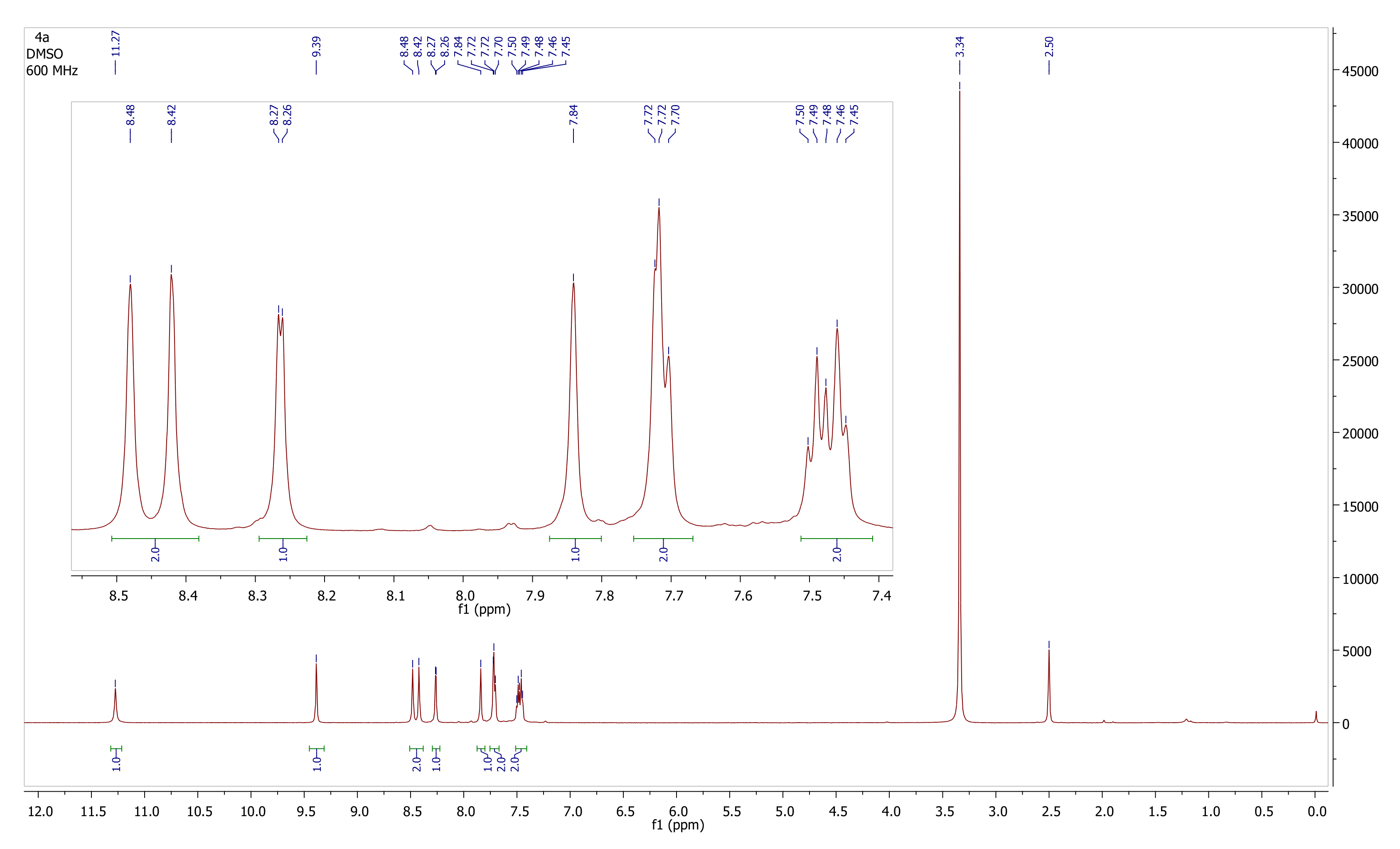
2.2.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
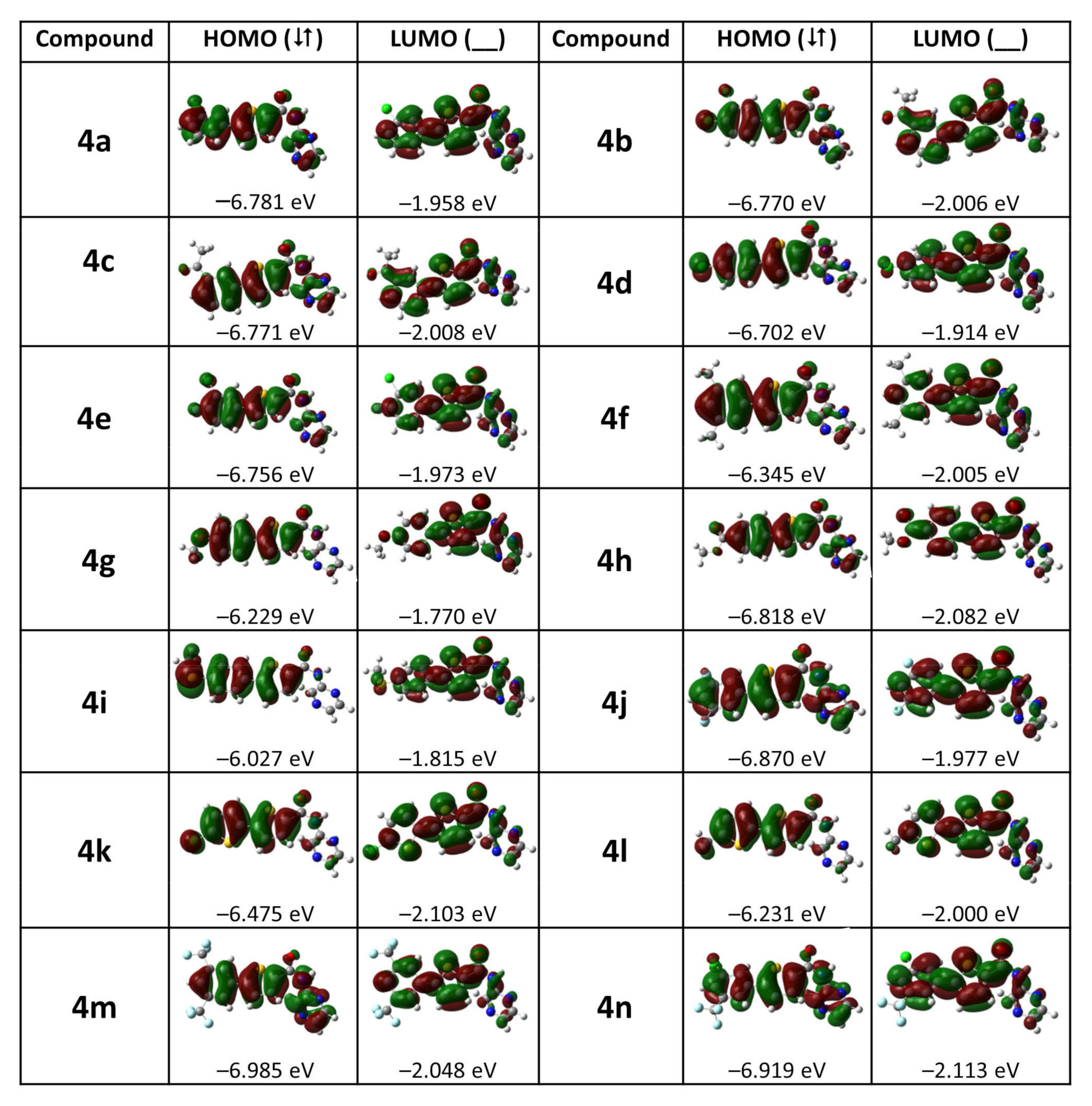
2.2.3. FMO and NLO Analysis
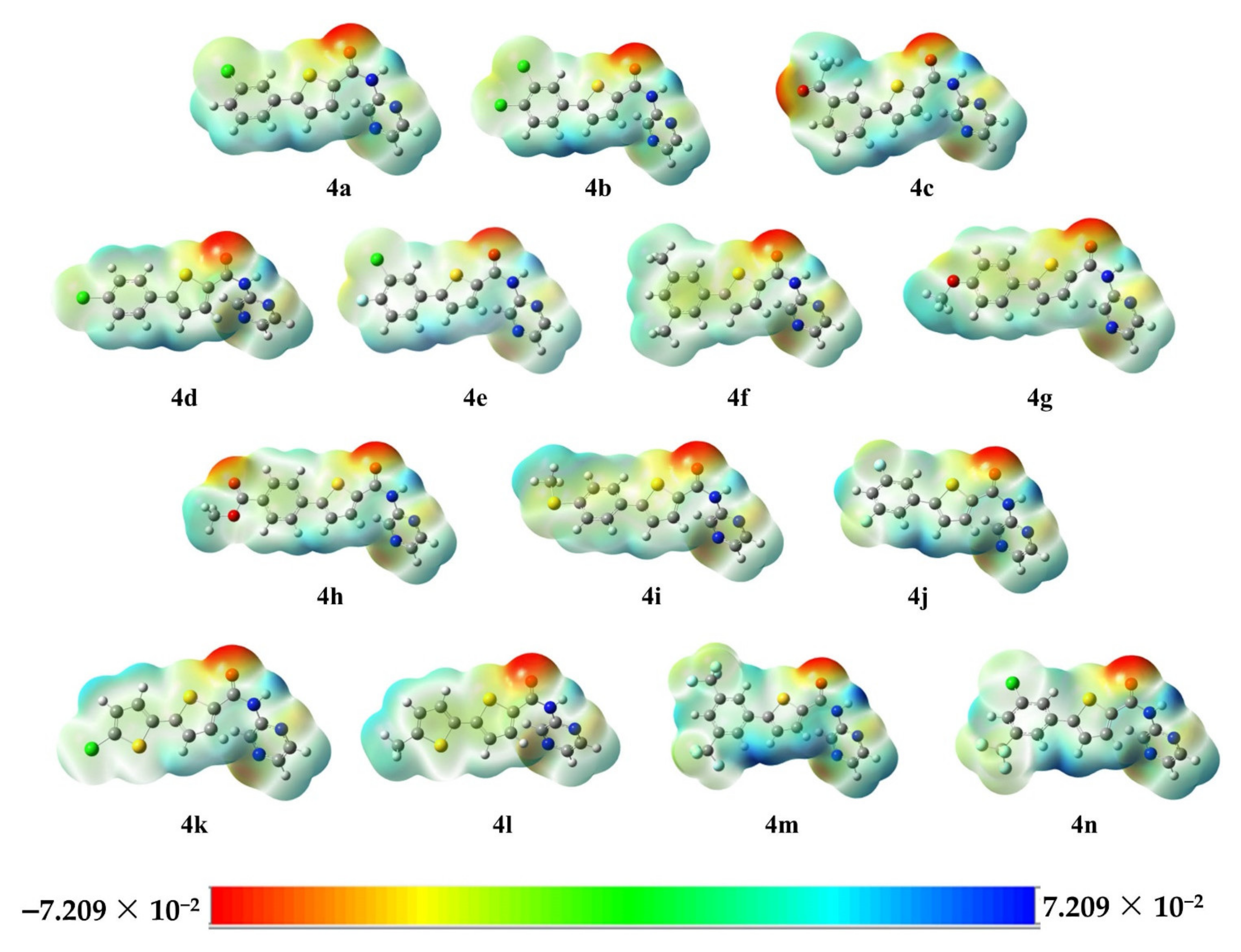
2.2.4. Molecular Electrostatic Potential (MESP) Analysis
2.2.5. Reactivity Descriptors
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedure for 5-Bromo-N-(pyrazin-2-yl)thiophene-2-carboxamide (3)
3.3. Synthetic Procedure for 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamide (4a–4n)
3.4. Characterization Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dubuisson, M.; Rees, J.-F.; Marchand-Brynaert, J. Discovery and validation of a new family of antioxidants: The aminopyrazine derivatives. Mini Rev. Med. Chem. 2004, 4, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Higasio, Y.S.; Shoji, T. Heterocyclic compounds such as pyrroles, pyridines, pyrollidins, piperdines, indoles, imidazol and pyrazins. Appl. Catal. A 2001, 221, 197–207. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Bharathi, E.V.; Sagar, M.V.P.; Pushpavalli, S.; Ray, P.; Pal-Bhadra, M. Design, synthesis and biological evaluation of imidazopyridine/pyrimidine-chalcone derivatives as potential anticancer agents. MedChemComm 2010, 1, 355–360. [Google Scholar] [CrossRef]
- Ghosh, P.; Rasul, M.; Chakraborty, M.; Mandal, A.; Saha, A. Microwave assisted one-pot synthesis of pyrazine derivatives of pentacyclic triterpenoids and their biological activity. Indian J. Chem. 2011, 50B, 1519–1523. [Google Scholar] [CrossRef]
- Meher, C.; Rao, A.; Omar, M. Piperazine-pyrazine and their multiple biological activities. Asian J. Pharm. Sci. 2013, 3, 43–60. [Google Scholar]
- Bonde, C.G.; Gaikwad, N.J. Synthesis and preliminary evaluation of some pyrazine containing thiazolines and thiazolidinones as antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Reddy, S.P. Synthesis of pyrazinamide Mannich bases and its antitubercular properties. Bioorg. Med. Chem. Lett. 2006, 16, 2113–2116. [Google Scholar] [CrossRef]
- Vergara, F.M.; Lima, C.H.d.S.; das Graças M. de O. Henriques, M.; Candéa, A.L.; Lourenço, M.C.; Ferreira, M.d.L.; Kaiser, C.R.; de Souza, M.V. Synthesis and antimycobacterial activity of N′-[(E)-(monosubstituted-benzylidene)]-2-pyrazinecarbohydrazide derivatives. Eur. J. Med. Chem. 2009, 44, 4954–4959. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, M.; Abdel-Rahman, H.M. Synthesis and anti-mycobacterial evaluation of some pyrazine-2-carboxylic acid hydrazide derivatives. Eur. J. Med. Chem. 2010, 45, 3384–3388. [Google Scholar] [CrossRef] [PubMed]
- Czerny, M.; Mayer, F.; Grosch, W. Sensory study on the character impact odorants of roasted Arabica coffee. J. Agric. Food Chem. 1999, 47, 695–699. [Google Scholar] [CrossRef]
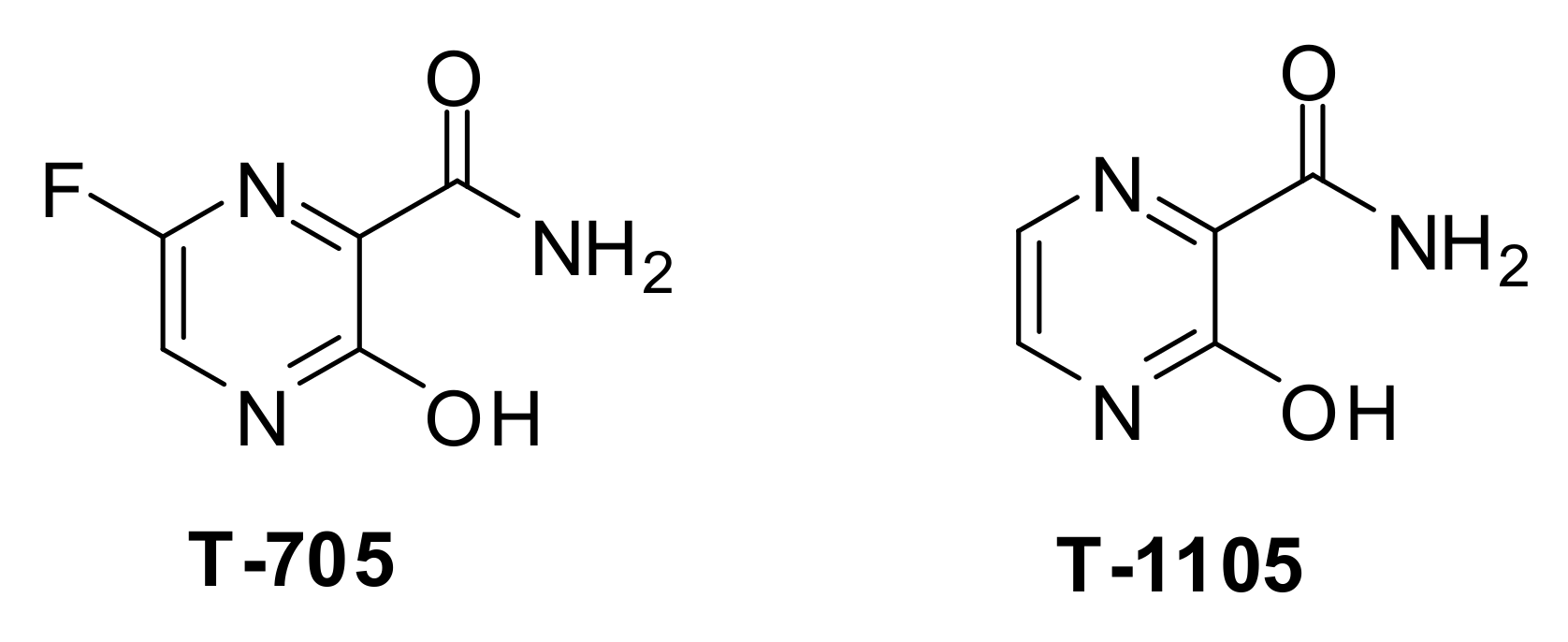
- Cai, L.; Sun, Y.; Song, Y.; Xu, L.; Bei, Z.; Zhang, D.; Dou, Y.; Wang, H. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication. Arch. Virol. 2017, 162, 2847–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.-S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.X.; Chen, X.P. Favipiravir: Pharmacokinetics and concerns about clinical trials for 2019-nCoV infection. Clin. Pharmacol. Ther. 2020, 108, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Mallesha, L.; Mohana, K.N. Synthesis, antimicrobial and antioxidant activities of 1-(1,4-benzodioxane-2-carbonyl) piperazine derivatives. Eur. J. Chem. 2011, 2, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, M.; Zitko, J.; Osicka, Z.; Kunes, J.; Vejsova, M.; Buchta, V.; Dohnal, J.; Jampilek, J.; Kralova, K. Synthesis, antimycobacterial, antifungal and photosynthesis-inhibiting activity of chlorinated N-phenylpyrazine-2-carboxamides. Molecules 2010, 15, 8567–8581. [Google Scholar] [CrossRef]
- Satoskar, R.S.; Bhandarkar, S. Pharmacology and Pharmacotherapeutics; Elsevier India: Kolkata, India, 2020. [Google Scholar]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Dorn, R.; Baums, D.; Kersten, P.; Regener, R. Nonlinear optical materials for integrated optics: Telecommunications and sensors. Adv. Mater. 1992, 4, 464–473. [Google Scholar] [CrossRef]
- Marder, S.R.; Perry, J.W. Molecular materials for second-order nonlinear optical applications. Adv. Mater. 1993, 5, 804–815. [Google Scholar] [CrossRef]
- Dini, D.; Calvete, M.J.; Hanack, M. Nonlinear optical materials for the smart filtering of optical radiation. Chem. Rev. 2016, 116, 13043–13233. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Progress in the enhancement of electro-optic coefficients and orientation stability for organic second-order nonlinear optical materials. Dyes Pigments 2020, 181, 108509. [Google Scholar] [CrossRef]
- Bureš, F. Fundamental aspects of property tuning in push–pull molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef] [Green Version]
- Meti, P.; Park, H.-H.; Gong, Y.-D. Recent developments in pyrazine functionalized π-conjugated materials for optoelectronic applications. J. Mater. Chem. C 2020, 8, 352–379. [Google Scholar] [CrossRef]
- Achelle, S.; Baudequin, C.; Plé, N. Luminescent materials incorporating pyrazine or quinoxaline moieties. Dyes Pigments 2013, 98, 575–600. [Google Scholar] [CrossRef] [Green Version]
- Hebbar, N.; Ramondenc, Y.; Plé, G.; Dupas, G.; Plé, N. Push–pull structures with a pyrazine core and hexatriene chain: Synthesis and light-emitting properties. Tetrahedron 2009, 65, 4190–4200. [Google Scholar] [CrossRef]
- Coe, B.J.; Fielden, J.; Foxon, S.P.; Asselberghs, I.; Clays, K.; Brunschwig, B.S. Two-dimensional, pyrazine-based nonlinear optical chromophores with ruthenium (II) ammine electron donors. Inorg. Chem. 2010, 49, 10718–10726. [Google Scholar] [CrossRef]
- Dokládalová, L.; Bureš, F.; Kuznik, W.; Kityk, I.V.; Wojciechowski, A.; Mikysek, T.; Almonasy, N.; Ramaiyan, M.; Padělková, Z.; Kulhánek, J. Dicyanobenzene and dicyanopyrazine derived X-shaped charge-transfer chromophores: Comparative and structure–property relationship study. Org. Biomol. Chem. 2014, 12, 5517–5527. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, X.T.; Guo, J.F.; Ren, A.M.; Wang, D. Theoretical investigation of two-photon absorption and fluorescence properties of cypridina luciferin-based derivatives: 2, 3, 5-trisubstituted pyrazine compounds. J. Phys. Org. Chem. 2013, 26, 822–833. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Bi, Z.; Xu, R.; Li, M.; Shen, X.; Tang, G.; Han, K. Theoretical study on the spectroscopic and third-order nonlinear optical properties of two-dimensional charge-transfer pyrazine derivatives. Mol. Phys. 2017, 115, 3164–3171. [Google Scholar] [CrossRef]
- Chen, M.; Nie, H.; Song, B.; Li, L.; Sun, J.Z.; Qin, A.; Tang, B.Z. Triphenylamine-functionalized tetraphenylpyrazine: Facile preparation and multifaceted functionalities. J. Mater. Chem. C 2016, 4, 2901–2908. [Google Scholar] [CrossRef] [Green Version]
- Imran, H.M.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Altaf, A.A.; Ahmed, G.; Malik, A.; Kausar, S.; Khan, S.U.-D.; Ahmad, A. Synthesis of halogenated [1,1′-biphenyl]-4-yl benzoate and [1,1′:3′,1″-terphenyl]-4′-yl benzoate by palladium catalyzed cascade C–C coupling and structural analysis through computational approach. J. Mol. Struct. 2020, 1222, 1–9. [Google Scholar] [CrossRef]
- Lewars, E. Introduction to the theory and applications of molecular and quantum mechanics. In Computational Chemistry; Springer: Berlin, Germany, 2003; Volume 318. [Google Scholar]
- Malik, A.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Zahoor, A.F.; Ahmad, G.; Altaf, A.A.; Shah, S.A.A.; Sultan, S.; Zakaria, Z.A. Suzuki–miyaura reactions of (4-bromophenyl)-4,6-dichloropyrimidine through commercially available palladium catalyst: Synthesis, optimization and their structural aspects identification through computational studies. Processes 2020, 8, 1342. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0.16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Schlegel, H.B. Geometry optimization. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 790–809. [Google Scholar] [CrossRef]
- Singh, R.K.; Singh, A.K.; Siddiqui, S.; Arshad, M.; Jafri, A. Synthesis, molecular structure, spectral analysis and cytotoxic activity of two new aroylhydrazones. J. Mol. Struct. 2017, 1135, 82–97. [Google Scholar] [CrossRef]
- Gökce, H.; Öztürk, N.; Kazıcı, M.; Göreci, Ç.Y.; Güneş, S. Structural, spectroscopic, electronic, nonlinear optical and thermodynamic properties of a synthesized Schiff base compound: A combined experimental and theoretical approach. J. Mol. Struct. 2017, 1136, 288–302. [Google Scholar] [CrossRef]
- Kanaani, A.; Ajloo, D.; Grivani, G.; Ghavami, A.; Vakili, M. Tautomeric stability, molecular structure, NBO, electronic and NMR analyses of salicylideneimino-ethylimino-pentan-2-one. J. Mol. Struct. 2016, 1112, 87–96. [Google Scholar] [CrossRef]
- O’Brien, S.E.; Browne, H.L.; Bradshaw, T.D.; Westwell, A.D.; Stevens, M.F.; Laughton, C.A. Antitumor benzothiazoles. Frontier molecular orbital analysis predicts bioactivation of 2-(4-aminophenyl) benzothiazoles to reactive intermediates by cytochrome P4501A1. Org. Biomol. Chem. 2003, 1, 493–497. [Google Scholar] [CrossRef]
- Arshad, M.N.; Bibi, A.; Mahmood, T.; Asiri, A.M.; Ayub, K. Synthesis, crystal structures and spectroscopic properties of triazine-based hydrazone derivatives; a comparative experimental-theoretical study. Molecules 2015, 20, 5851–5874. [Google Scholar] [CrossRef] [Green Version]
- Belletête, M.; Beaupré, S.; Bouchard, J.; Blondin, P.; Leclerc, M.; Durocher, G. Theoretical and experimental investigations of the spectroscopic and photophysical properties of fluorene-phenylene and fluorene-thiophene derivatives: Precursors of light-emitting polymers. J. Phys. Chem. B 2000, 104, 9118–9125. [Google Scholar] [CrossRef]
- Peng, W.; Peiwang, Z.; Chuanguang, W.; Cheng, Y. Theoretical investigation and molecular design of pyrazine derivatives with large hyperpolarizabilities (β). J. Mol. Struct. Theor. Chem 1999, 459, 155–162. [Google Scholar] [CrossRef]
- Ceylan, Ü.; Tarı, G.Ö.; Gökce, H.; Ağar, E. Spectroscopic (FT–IR and UV–Vis) and theoretical (HF and DFT) investigation of 2-Ethyl-N-[(5-nitrothiophene-2-yl) methylidene] aniline. J. Mol. Struct. 2016, 1110, 1–10. [Google Scholar] [CrossRef]
- Mathammal, R.; Sangeetha, K.; Sangeetha, M.; Mekala, R.; Gadheeja, S. Molecular structure, vibrational, UV, NMR, HOMO-LUMO, MEP, NLO, NBO analysis of 3, 5 di tert butyl 4 hydroxy benzoic acid. J. Mol. Struct. 2016, 1120, 1–14. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef]
- Guo, L.; Safi, Z.S.; Kaya, S.; Shi, W.; Tüzün, B.; Altunay, N.; Kaya, C. Anticorrosive effects of some thiophene derivatives against the corrosion of iron: A computational study. Front. Chem. 2018, 6, 155. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J. Local reactivity descriptors from degenerate frontier molecular orbitals. Chem. Phys. Lett. 2009, 478, 310–322. [Google Scholar] [CrossRef]
- Mubarik, A.; Rasool, N.; Hashmi, M.A.; Mansha, A.; Zubair, M.; Shaik, M.R.; Sharaf, M.A.; Awwad, E.M.; Abdelgawad, A. Computational Study of Structural, Molecular Orbitals, Optical and Thermodynamic Parameters of Thiophene Sulfonamide Derivatives. Crystals 2021, 11, 211. [Google Scholar] [CrossRef]
- Leggio, A.; Bagalà, J.; Belsito, E.L.; Comandè, A.; Greco, M.; Liguori, A. Formation of amides: One-pot condensation of carboxylic acids and amines mediated by TiCl4. Chem. Cent. J. 2017, 11, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, G.; Rasool, N.; Rizwan, K.; Imran, I.; Zahoor, A.F.; Zubair, M.; Sadiq, A.; Rashid, U. Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl) thiophene-2-carboxamide analogs. Bioorg. Chem. 2019, 92, 103216. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, G.; Rasool, N.; Ikram, H.M.; Gul Khan, S.; Mahmood, T.; Ayub, K.; Zubair, M.; Al-Zahrani, E.; Ali Rana, U.; Akhtar, M.N. Efficient synthesis of novel pyridine-based derivatives via Suzuki cross-coupling reaction of commercially available 5-bromo-2-methylpyridin-3-amine: Quantum mechanical investigations and biological activities. Molecules 2017, 22, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, H.; Rasool, N.; Ahmad, G.; Chotana, G.; Musharraf, S.; Zubair, M.; Rana, U.; Haq, M.Z.U.; Jaafar, H. Selective C-arylation of 2, 5-dibromo-3-hexylthiophene via suzuki cross coupling reaction and their pharmacological aspects. Molecules 2015, 20, 5202–5214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, G.; Rasool, N.; Rizwan, K.; Altaf, A.A.; Rashid, U.; Mahmood, T.; Ayub, K. Role of Pyridine Nitrogen in Palladium-Catalyzed Imine Hydrolysis: A Case Study of (E)-1-(3-bromothiophen-2-yl)-N-(4-methylpyridin-2-yl) methanimine. Molecules 2019, 24, 2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, G.; Rasool, N.; Qamar, M.U.; Alam, M.M.; Kosar, N.; Mahmood, T.; Imran, M. Facile synthesis of 4-aryl-N-(5-methyl-1H-pyrazol-3-yl) benzamides via Suzuki Miyaura reaction: Antibacterial activity against clinically isolated NDM-1-positive bacteria and their Docking Studies. Arab. J. Chem. 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Mahmood, N.; Rasool, N.; Ikram, H.M.; Hashmi, M.A.; Mahmood, T.; Zubair, M.; Ahmad, G.; Rizwan, K.; Rashid, T.; Rashid, U. Synthesis of 3, 4-Biaryl-2, 5-Dichlorothiophene through Suzuki Cross-Coupling and Theoretical Exploration of Their Potential Applications as Nonlinear Optical Materials. Symmetry 2018, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, K.; Rasool, N.; Rehman, R.; Mahmood, T.; Ayub, K.; Rasheed, T.; Ahmad, G.; Malik, A.; Khan, S.A.; Akhtar, M.N. Facile synthesis of N-(4-bromophenyl)-1-(3-bromothiophen-2-yl) methanimine derivatives via Suzuki cross-coupling reaction: Their characterization and DFT studies. Chem. Cent. J. 2018, 12, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Ikram, H.M.; Rasool, N.; Hashmi, M.A.; Anjum, M.A.; Ali, K.G.; Zubair, M.; Ahmad, G.; Mahmood, T. Density functional theory-supported studies of structural and electronic properties of substituted-phenol derivatives synthesized by efficient O-or C-arylation via Chan--Lam or Suzuki cross-coupling reactions. Turk. J. Chem. 2019, 43, 1306–1321. [Google Scholar] [CrossRef]
- Abbas, M.; Rizwan, K.; Rasool, N.; Hashmi, M.A.; Ahmad, G.; Rashid, U.; Shah, S.A.A. Palladium (0) catalyzed synthesis of thiophene based 1, 3, 4-oxadiazoles their reactivities and potential nonlinear optical properties. Chiang Mai J. Sci. 2020, 47, 1255–1264. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 4a | ||||
|---|---|---|---|---|
| Carbon No. | Carbon Type | 1H NMR (δ, ppm), Experimental | 1H NMR (δ, ppm), Computed | Δδ, ppm |
| 2 | C | – | – | – |
| 3 | CH | 9.39 | 10.55 | −1.16 |
| 4 | N | – | – | – |
| 5 | CH | 8.45 | 8.43 | 0.02 |
| 6 | CH | 8.45 | 8.42 | 0.03 |
| 2′ | C | – | – | – |
| 3′ | CH | 7.84 | 7.24 | 0.60 |
| 4′ | CH | 7.72 | 6.80 | 0.92 |
| 5′ | C | – | – | – |
| 1″ | C | – | – | – |
| 2″ | CH | 8.26 | 7.45 | 0.81 |
| 3″ | C | – | – | – |
| 4″ | CH | 7.50 | 7.37 | 0.13 |
| 5″ | CH | 7.45 | 7.39 | 0.06 |
| 6″ | CH | 7.70 | 7.25 | 0.45 |
| Compounds | EHOMO (eV) | ELUMO (eV) | ELUMO−EHOMO (eV) | α (a.u.) | βo (a.u.) |
|---|---|---|---|---|---|
| 4a | −6.781 | −1.958 | 4.823 | 310.96 | 2700.54 |
| 4b | −6.770 | −2.006 | 4.763 | 325.22 | 4139.08 |
| 4c | −6.771 | −2.008 | 4.763 | 338.09 | 2571.25 |
| 4d | −6.702 | −1.914 | 4.788 | 311.83 | 3958.52 |
| 4e | −6.756 | −1.973 | 4.784 | 310.60 | 4004.42 |
| 4f | −6.345 | −2.005 | 4.340 | 354.10 | 6120.41 |
| 4g | −6.229 | −1.770 | 4.459 | 330.84 | 8398.11 |
| 4h | −6.818 | −2.082 | 4.736 | 345.57 | 1363.79 |
| 4i | −6.027 | −1.815 | 4.212 | 349.43 | 9139.57 |
| 4j | −6.870 | −1.977 | 4.893 | 293.78 | 1926.82 |
| 4k | −6.475 | −2.103 | 4.373 | 310.21 | 6520.24 |
| 4l | −6.231 | −2.000 | 4.231 | 325.48 | 9048.28 |
| 4m | −6.985 | −2.048 | 4.937 | 315.57 | 1103.20 |
| 4n | −6.919 | −2.113 | 4.806 | 322.84 | 1416.47 |
| Compounds | Calculated through Koopman’s Theorem | Calculated Directly from Charged Molecules | ||
|---|---|---|---|---|
| I (eV) | A (eV) | I (eV) | A (eV) | |
| 4a | 6.78 | 1.96 | 6.16 | 0.47 |
| 4b | 6.77 | 2.01 | 6.23 | 0.50 |
| 4c | 6.77 | 2.01 | 7.19 | 0.06 |
| 4d | 6.70 | 1.91 | 6.16 | 0.44 |
| 4e | 6.76 | 1.97 | 6.23 | 0.50 |
| 4f | 6.35 | 2.00 | 5.75 | 0.83 |
| 4g | 6.23 | 1.77 | 5.88 | 0.28 |
| 4h | 6.82 | 2.08 | 7.20 | 0.42 |
| 4i | 6.03 | 1.81 | 5.67 | 0.37 |
| 4j | 6.87 | 1.98 | 7.29 | 0.52 |
| 4k | 6.48 | 2.10 | 6.21 | 0.75 |
| 4l | 6.23 | 2.00 | 5.79 | 0.73 |
| 4m | 6.99 | 2.05 | 6.61 | 0.58 |
| 4n | 6.92 | 2.11 | 6.36 | 0.60 |
| Compounds | η (eV) | σ (eV−1) | μ (eV) | ω (eV) |
|---|---|---|---|---|
| 4a | 2.84 | 0.35 | −3.31 | 1.93 |
| 4b | 2.86 | 0.34 | −3.36 | 1.97 |
| 4c | 3.56 | 0.28 | −3.62 | 1.84 |
| 4d | 2.86 | 0.34 | −3.30 | 1.90 |
| 4e | 2.86 | 0.34 | −3.36 | 1.97 |
| 4f | 2.46 | 0.40 | −3.29 | 2.20 |
| 4g | 2.80 | 0.35 | −3.08 | 1.69 |
| 4h | 3.39 | 0.29 | −3.81 | 2.14 |
| 4i | 2.65 | 0.37 | −3.02 | 1.72 |
| 4j | 3.38 | 0.29 | −3.90 | 2.25 |
| 4k | 2.73 | 0.36 | −3.48 | 2.21 |
| 4l | 2.53 | 0.39 | −3.26 | 2.10 |
| 4m | 3.01 | 0.33 | −3.59 | 2.14 |
| 4n | 2.88 | 0.34 | −3.48 | 2.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, G.; Rasool, N.; Mubarik, A.; Zahoor, A.F.; Hashmi, M.A.; Zubair, M.; Bilal, M.; Hussien, M.; Akhtar, M.S.; Haider, S. Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations. Molecules 2021, 26, 7309. https://doi.org/10.3390/molecules26237309
Ahmad G, Rasool N, Mubarik A, Zahoor AF, Hashmi MA, Zubair M, Bilal M, Hussien M, Akhtar MS, Haider S. Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations. Molecules. 2021; 26(23):7309. https://doi.org/10.3390/molecules26237309
Chicago/Turabian StyleAhmad, Gulraiz, Nasir Rasool, Adeel Mubarik, Ameer Fawad Zahoor, Muhammad Ali Hashmi, Muhammad Zubair, Muhammad Bilal, Mohamed Hussien, Muhammad Saeed Akhtar, and Sajjad Haider. 2021. "Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations" Molecules 26, no. 23: 7309. https://doi.org/10.3390/molecules26237309
APA StyleAhmad, G., Rasool, N., Mubarik, A., Zahoor, A. F., Hashmi, M. A., Zubair, M., Bilal, M., Hussien, M., Akhtar, M. S., & Haider, S. (2021). Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl)thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations. Molecules, 26(23), 7309. https://doi.org/10.3390/molecules26237309