
Evaluation of Sample Preparation Methods for Non-Target Screening of Organic Micropollutants in Urban Waters Using High-Resolution Mass Spectrometry


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparison of the Cartridge Retention Ability
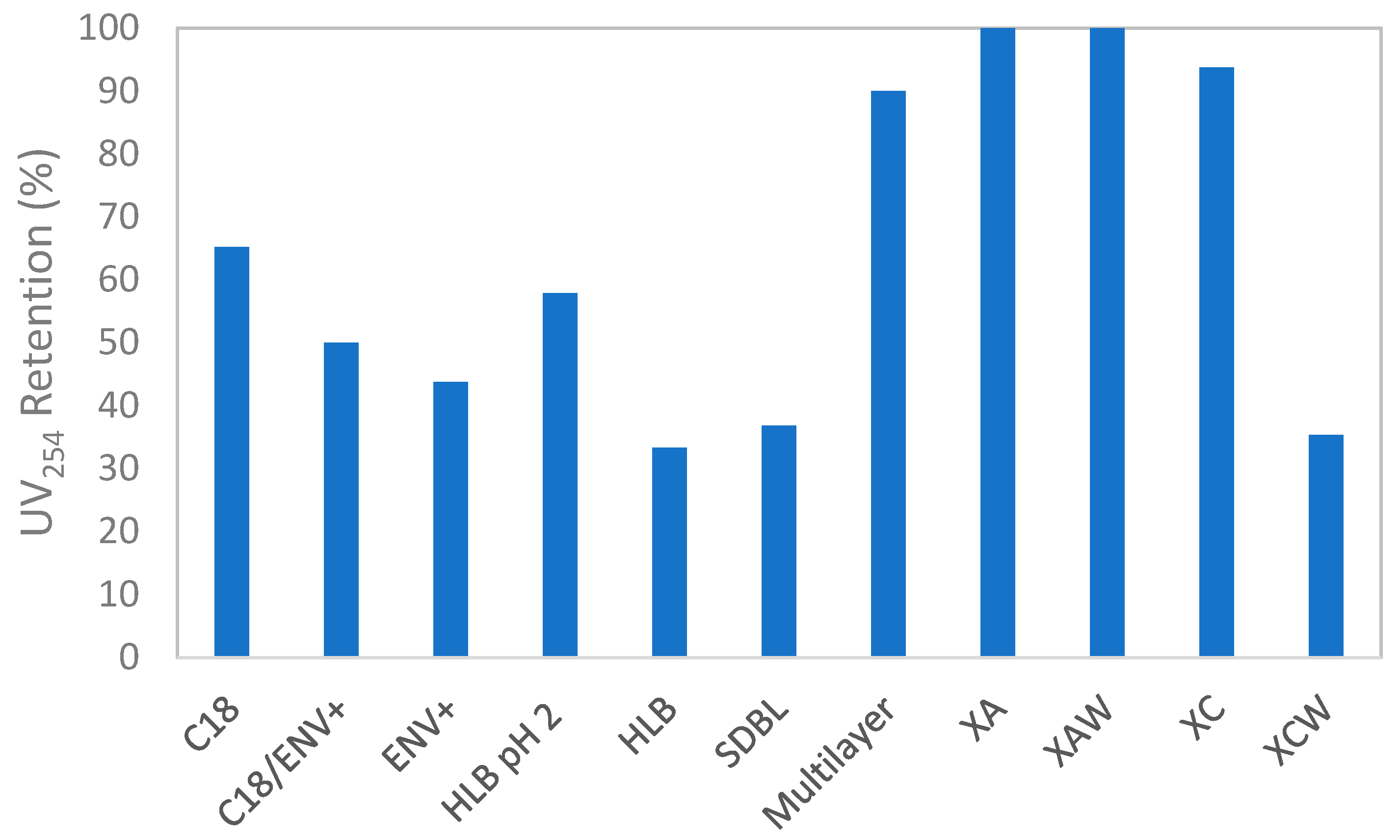
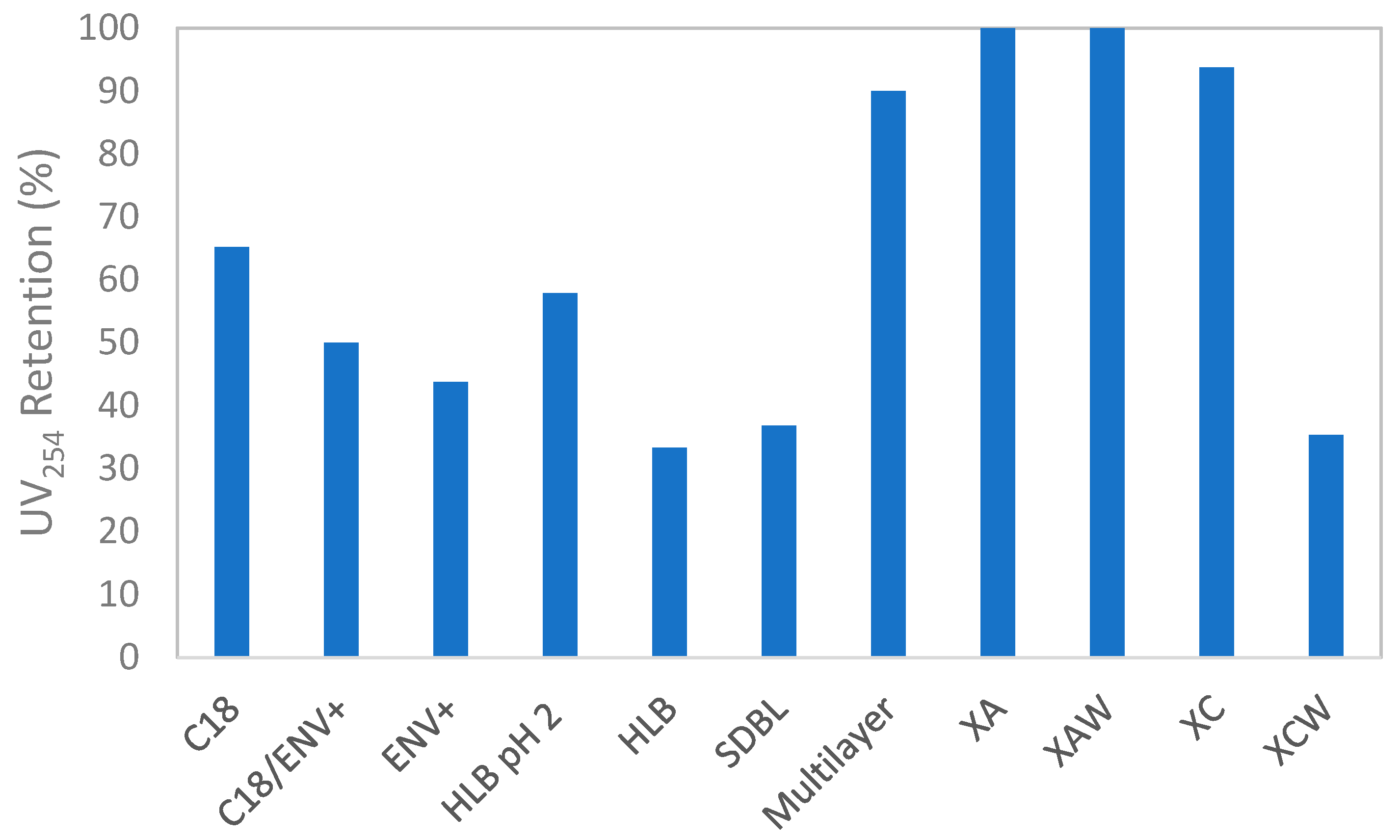
2.1.1. Discrimination of Cartridges Based on Optical Properties
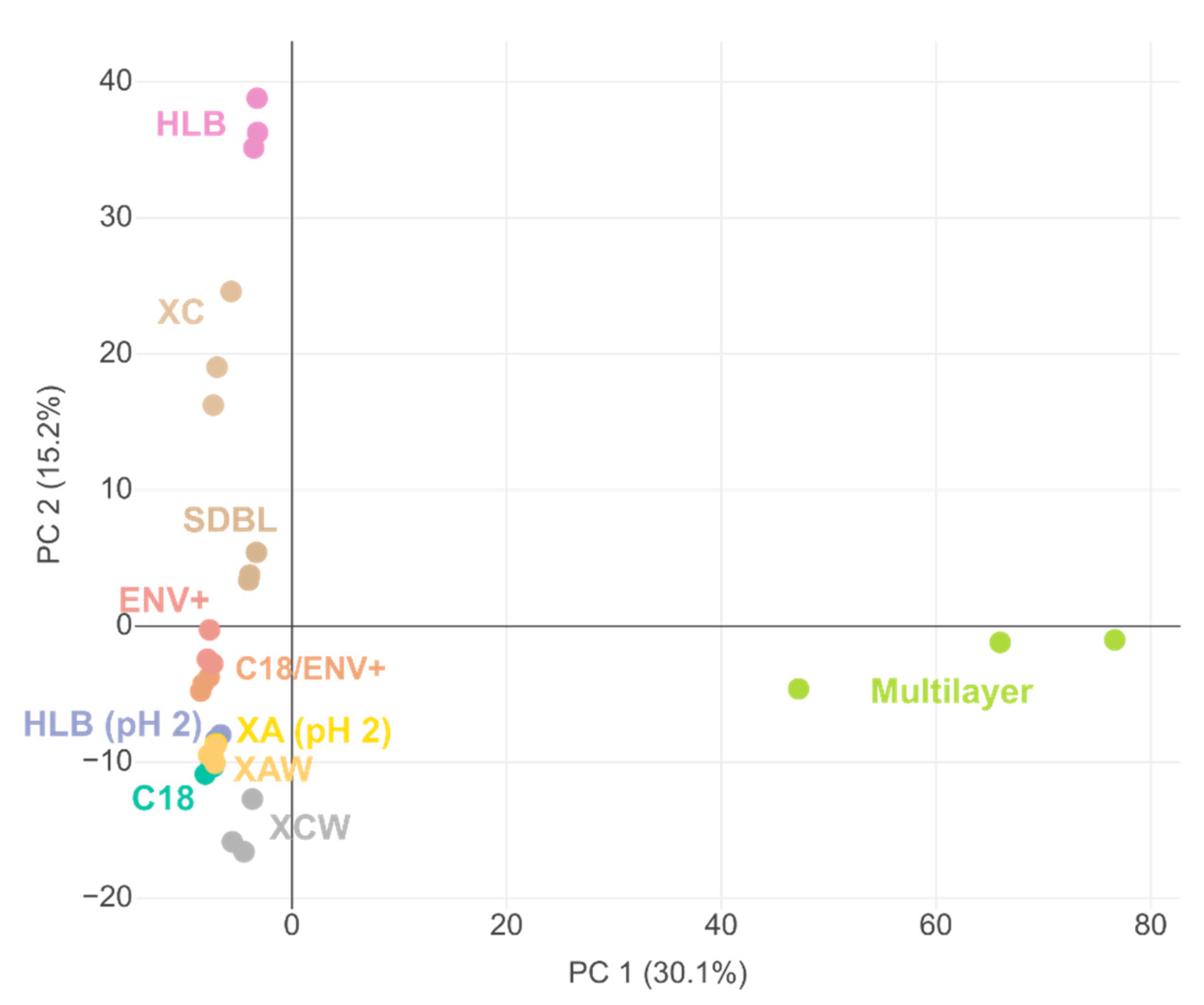
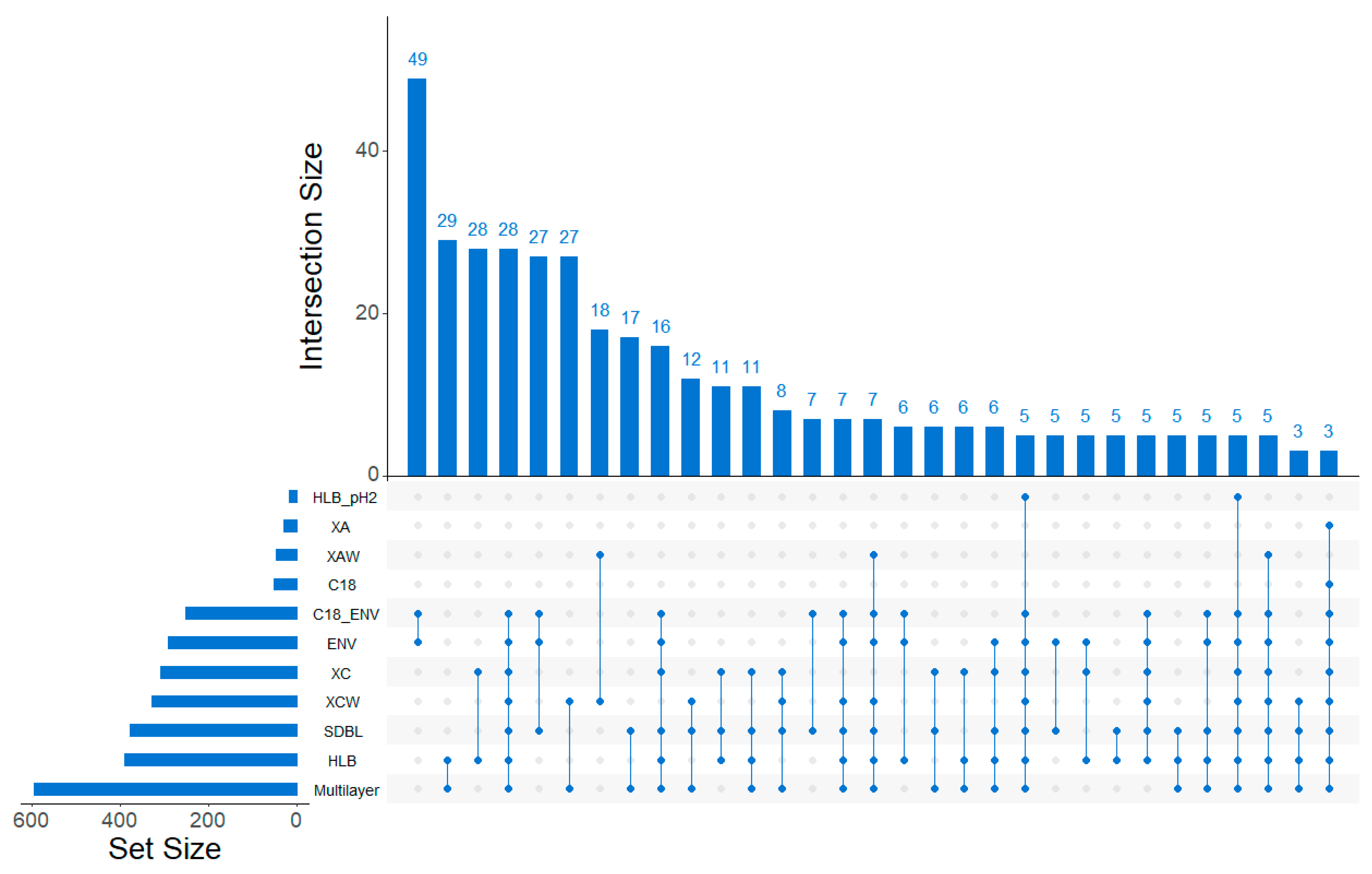
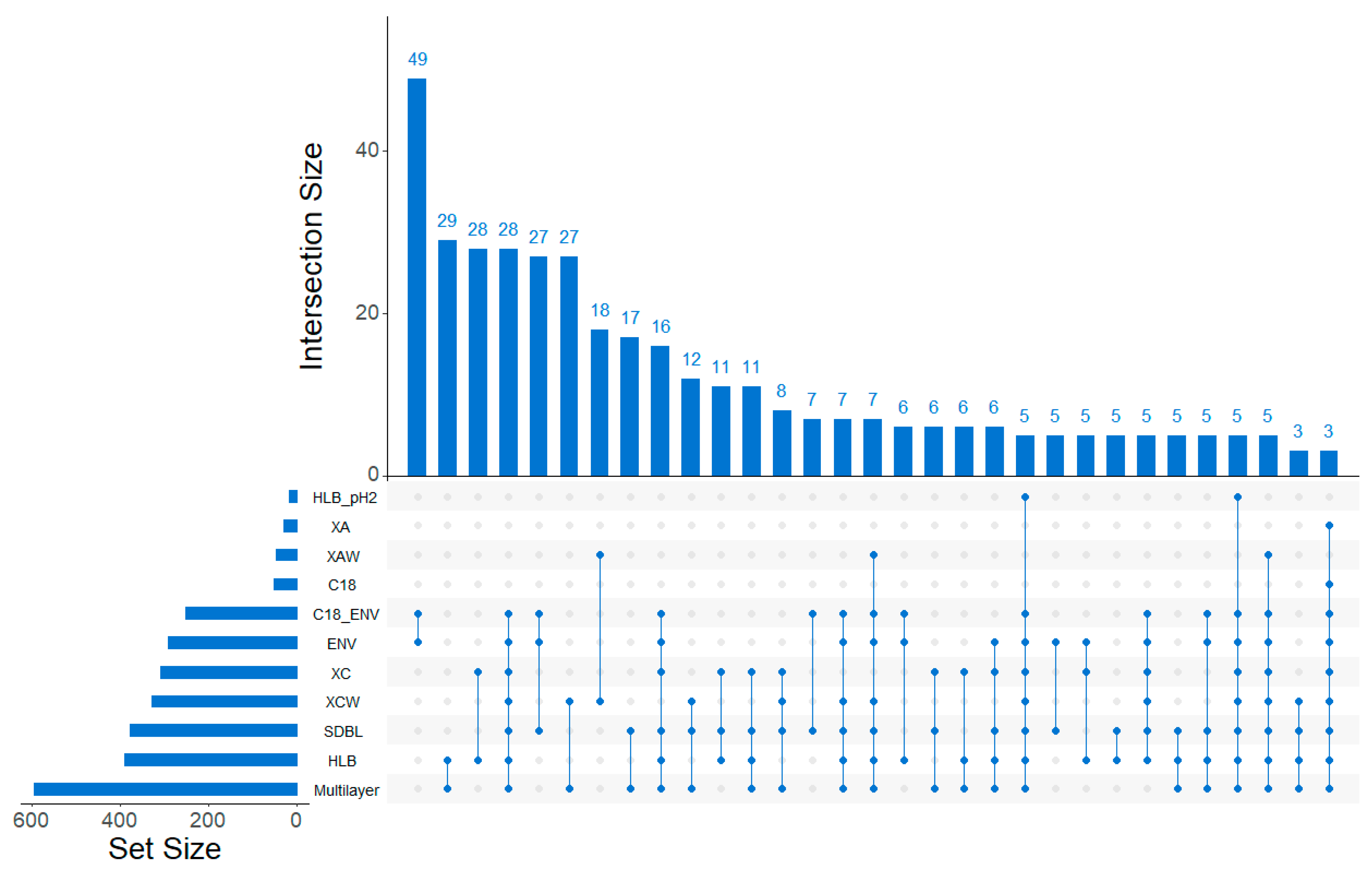
2.1.2. Discrimination of Cartridges Based on HRMS Features
2.2. Characterization of the Cartridge Retention Ability with Different Matrices
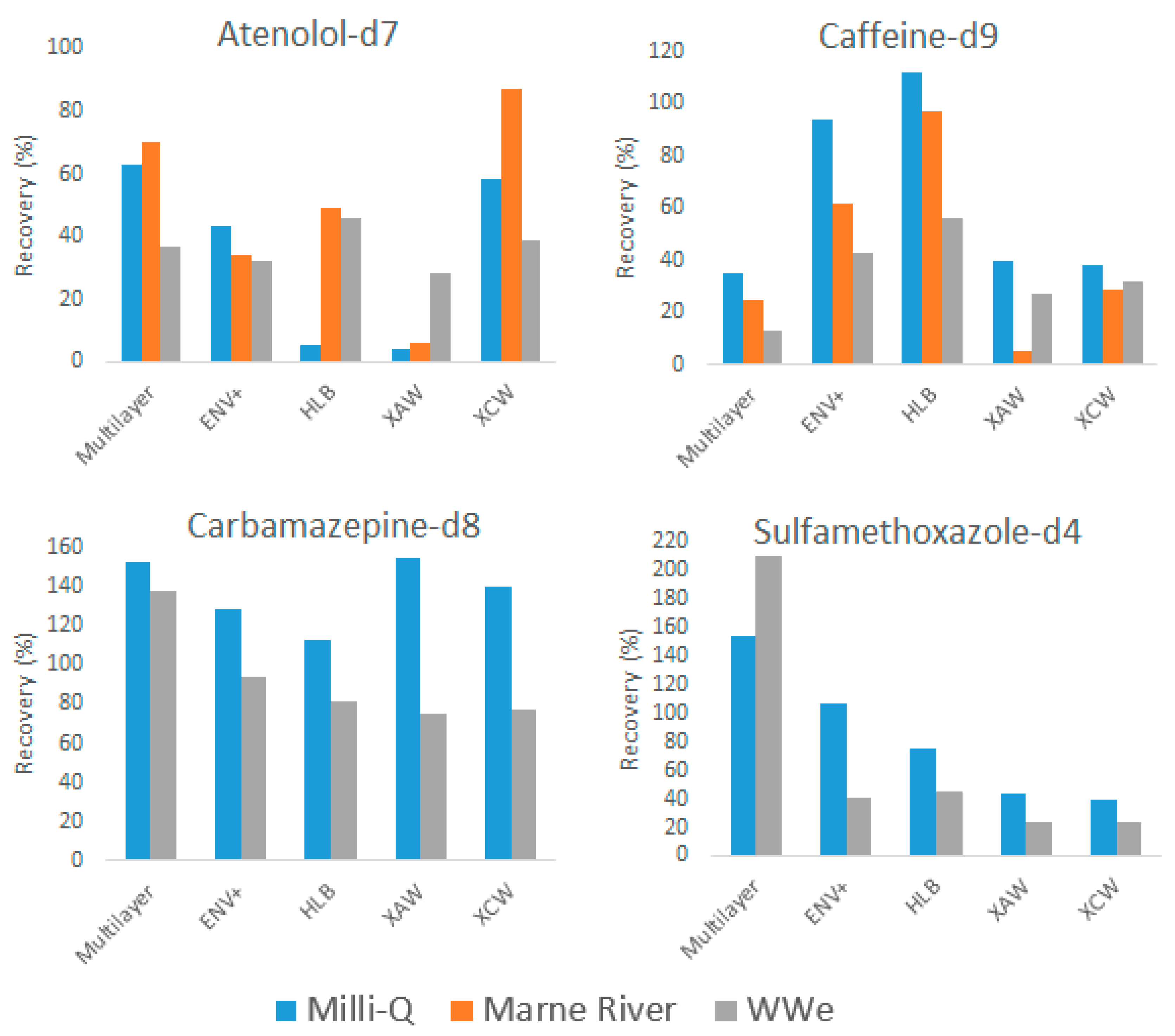
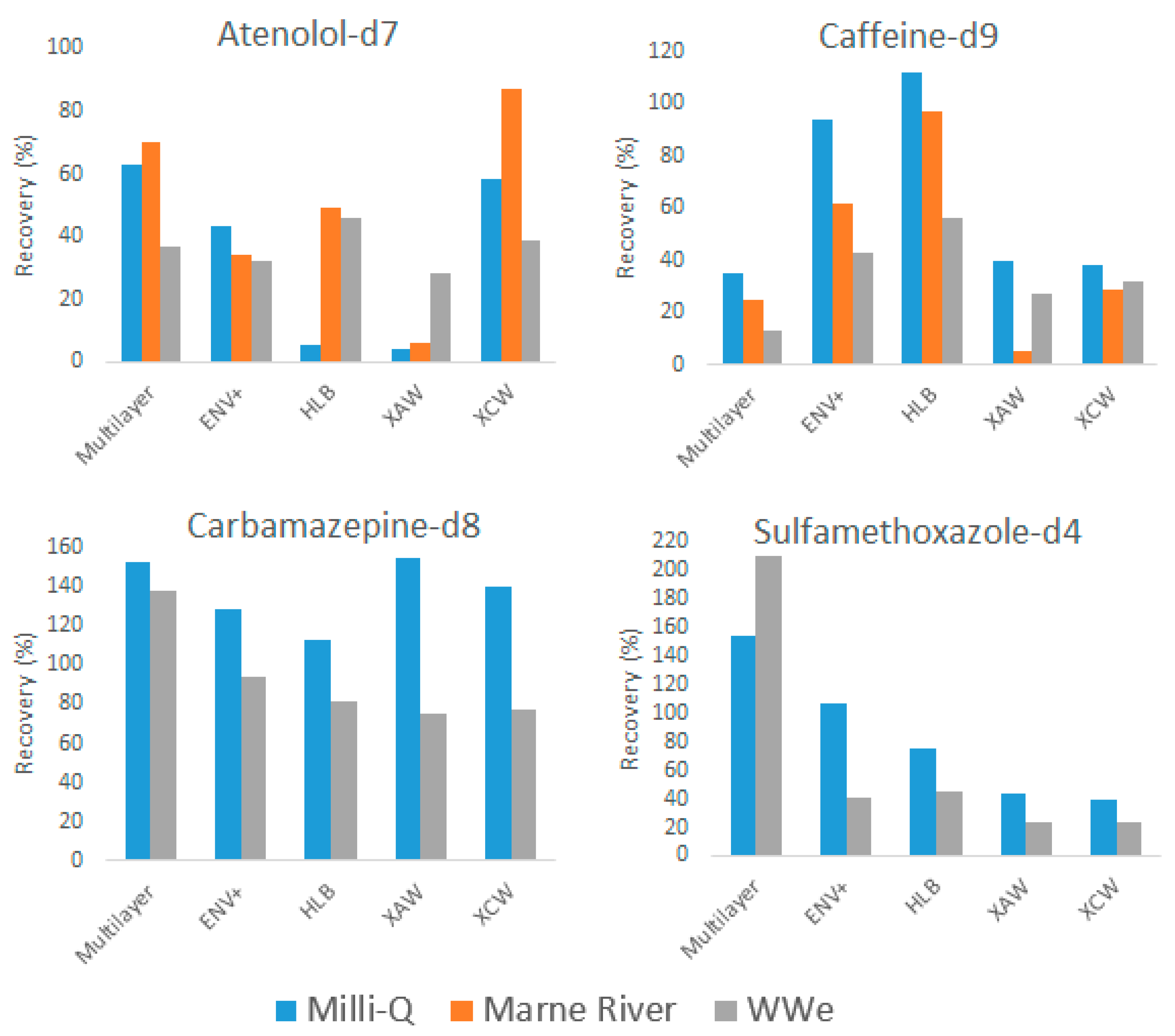
2.2.1. Recoveries of Internal Standards
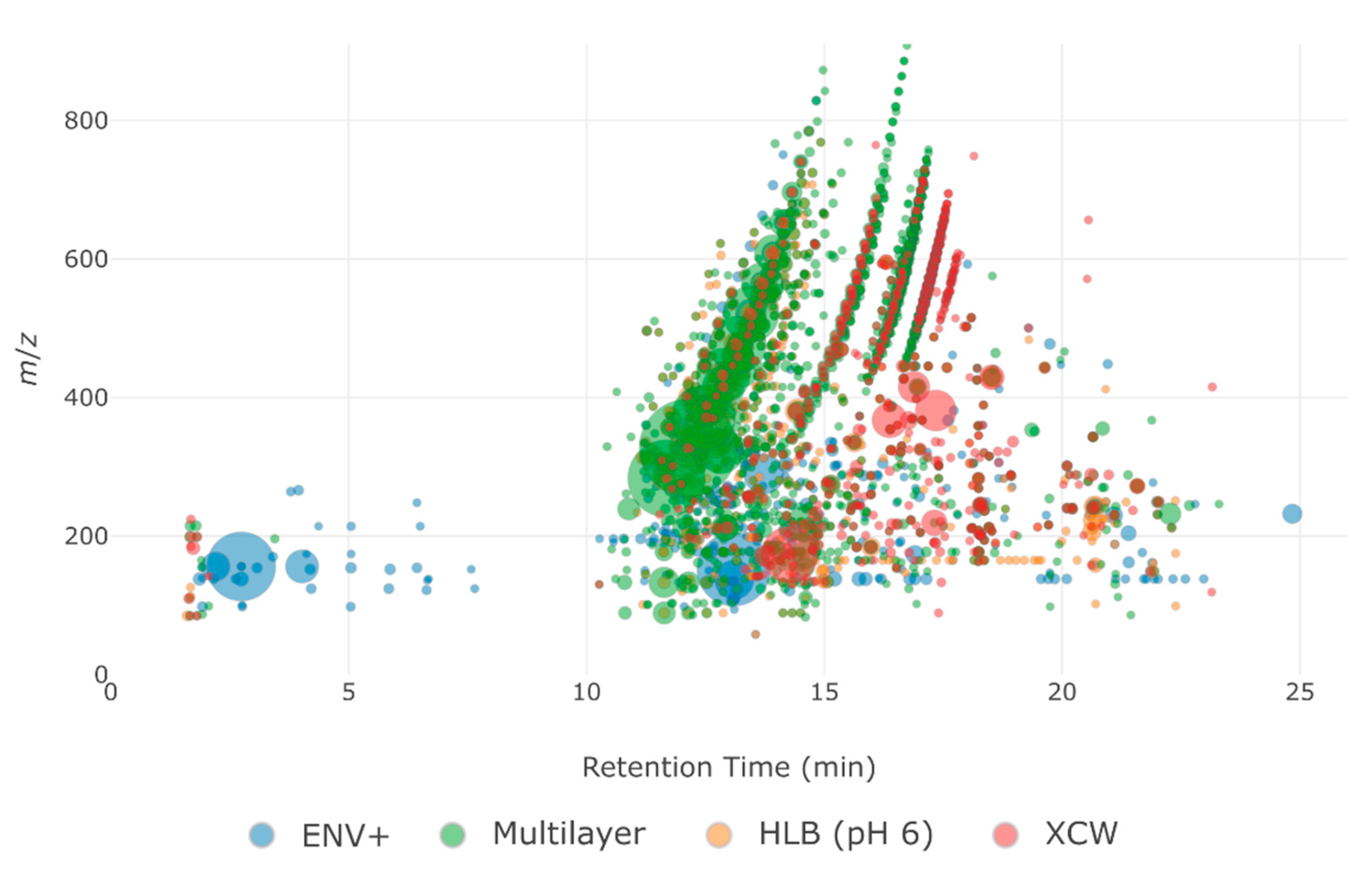
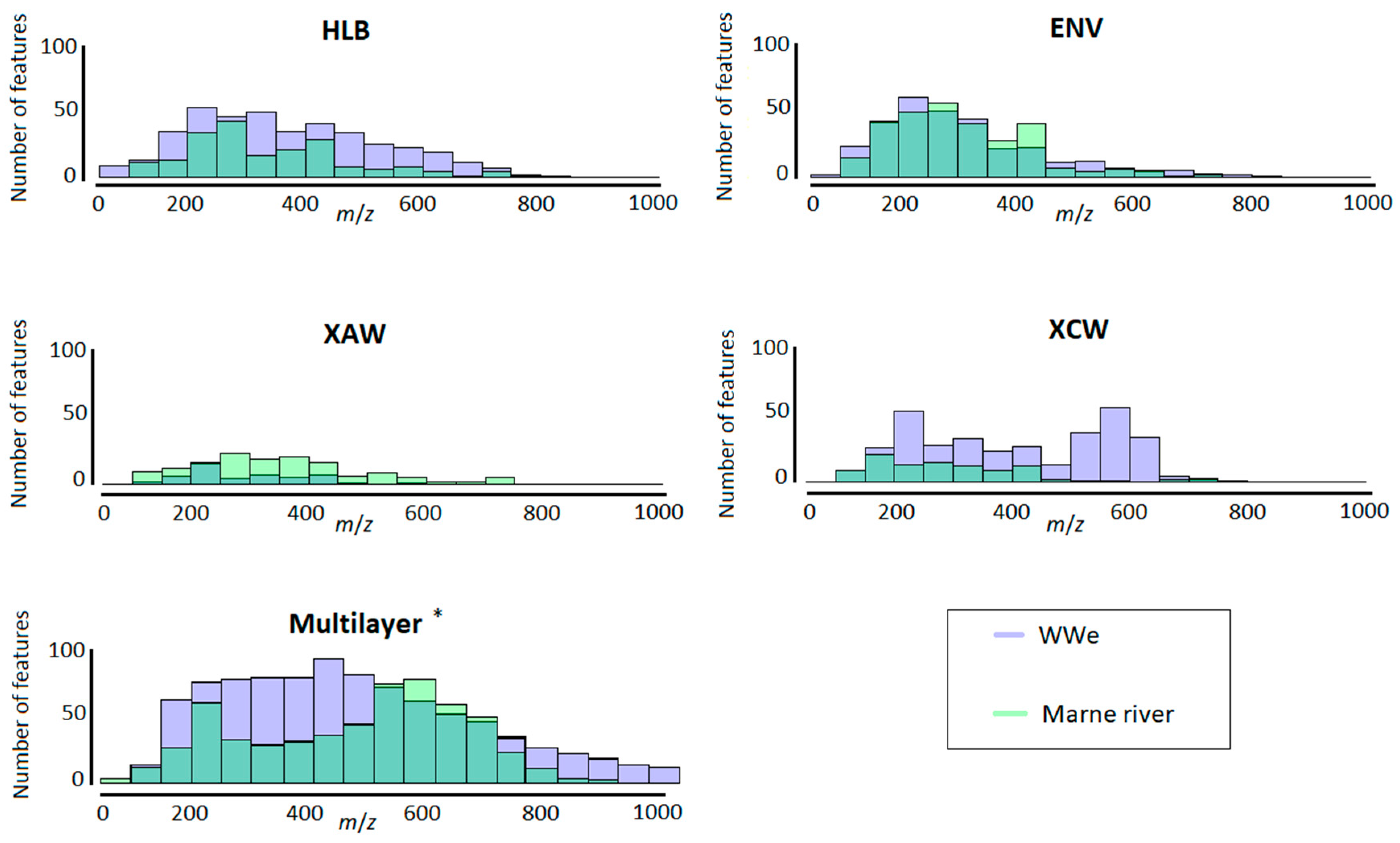
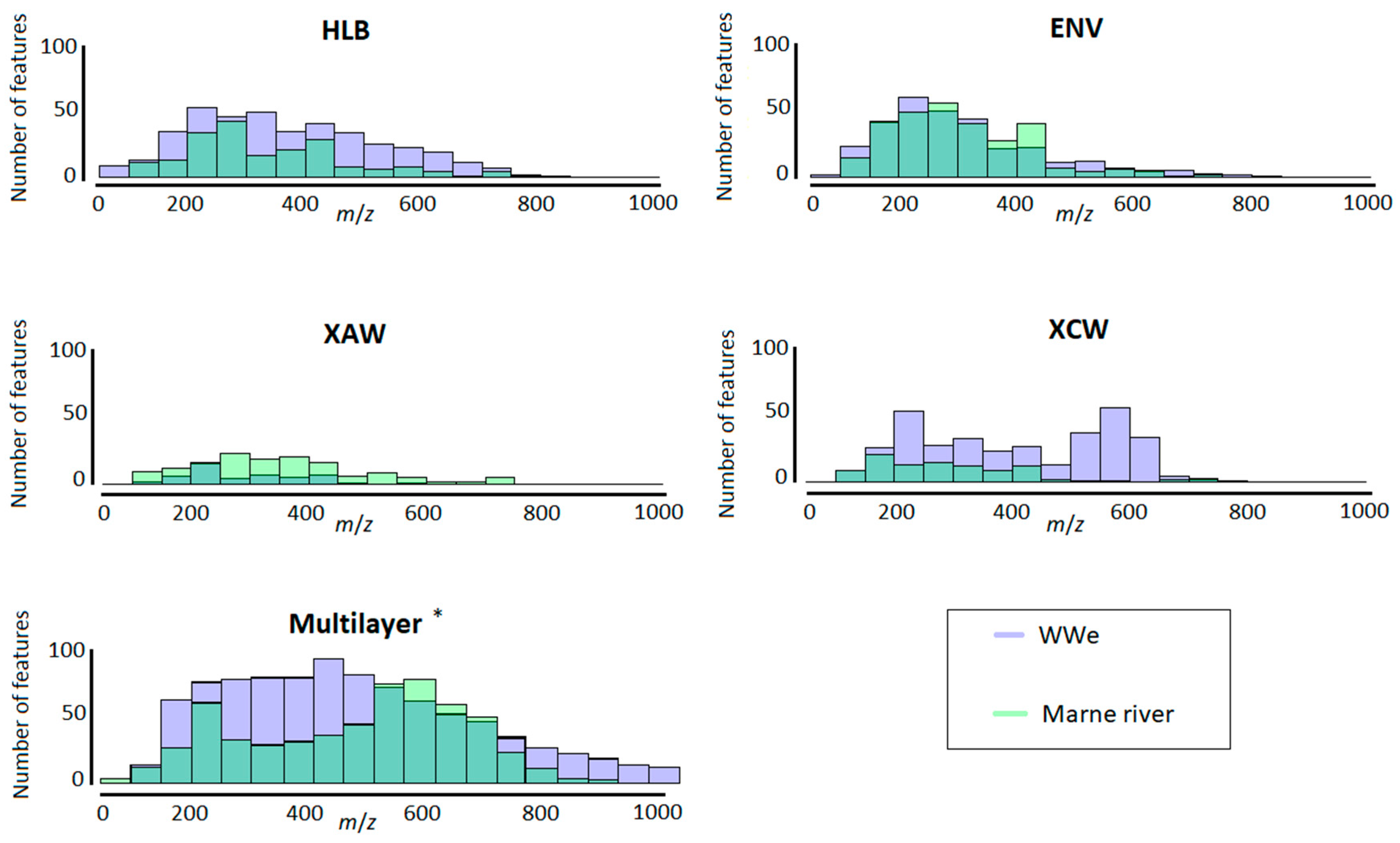
2.2.2. Range and Properties of Retained Features
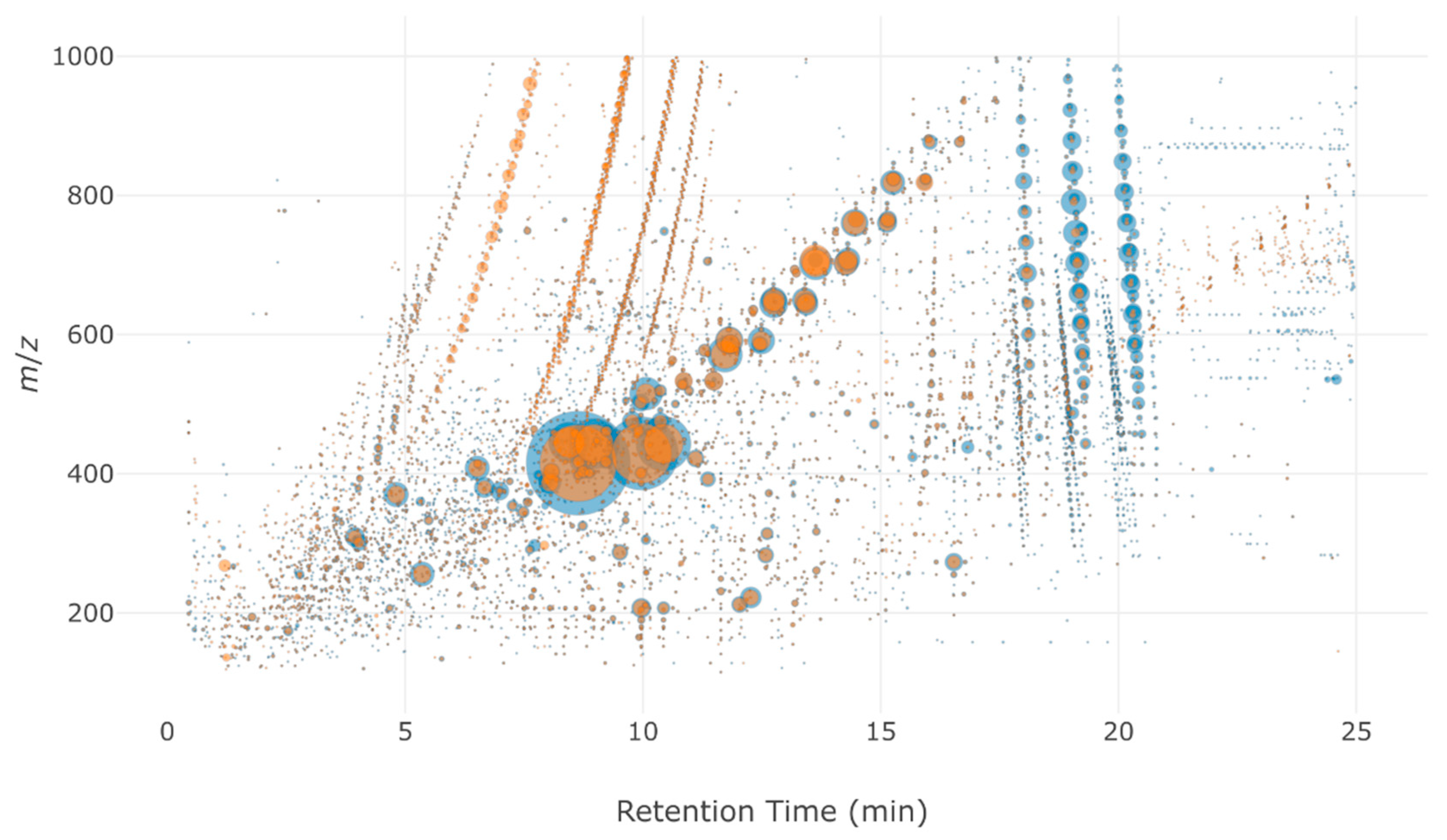
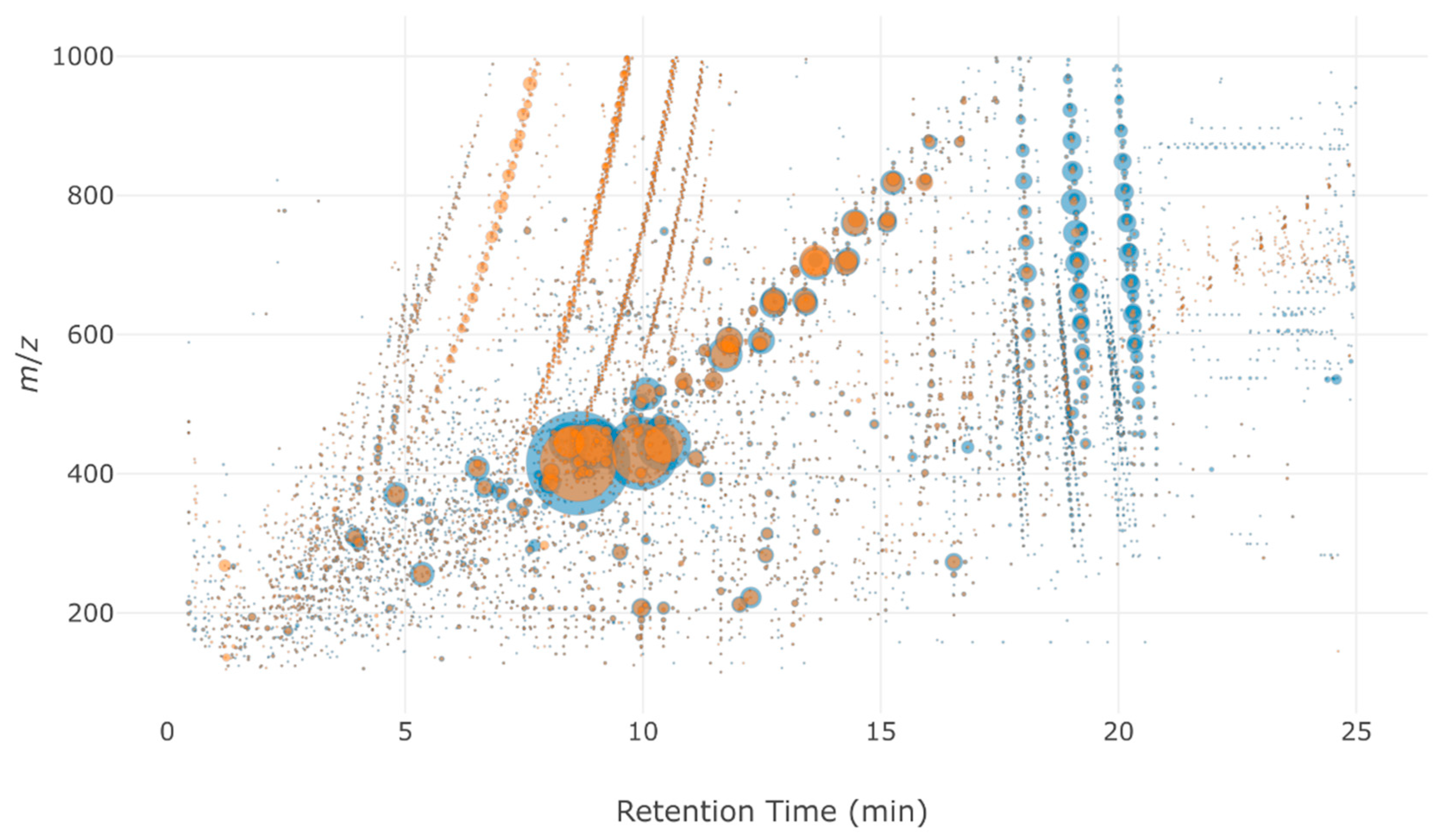
2.3. Application: Evaluating a Disinfection Treatment by Performic Acid
3. Materials and Methods
3.1. Chemicals and Standards
3.2. Sample Collection and Preliminary Preparation
3.3. SPE Cartridges
3.3.1. Selection of Cartridges
3.3.2. Extraction Protocol
3.4. UV-Visible and 3D Fluorescence Analyses
3.5. LC-HRMS Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- European Parliament and of the Council, EU Directive 2000/60/CE, Water Framework Directive on Water, Last Revised August 2018. 2000. Available online: https://eur-lex.europa.eu/legal-content/FR/TXT/?uri=celex%3A32000L0060 (accessed on 18 November 2021).
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go? Environ. Sci. Technol. 2017, 51, 11505–11512. [Google Scholar] [CrossRef] [Green Version]
- González-Gaya, B.; Lopez-Herguedas, N.; Bilbao, D.; Mijangos, L.; Iker, A.M.; Etxebarria, N.; Irazola, M.; Prieto, A.; Olivares, M.; Zuloaga, O. Suspect and Non-Target Screening: The Last Frontier in Environmental Analysis. Anal. Methods 2021, 13, 1876–1904. [Google Scholar] [CrossRef]
- Rodriguez-Aller, M.; Gurny, R.; Veuthey, J.-L.; Guillarme, D. Coupling Ultra High-Pressure Liquid Chromatography with Mass Spectrometry: Constraints and Possible Applications. J. Chromatogr. A 2013, 1292, 2–18. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.P.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S.; et al. Non-Target Screening with High-Resolution Mass Spectrometry: Critical Review Using a Collaborative Trial on Water Analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef]
- Hernández, F.; Pozo, Ó.J.; Sancho, J.V.; López, F.J.; Marín, J.M.; Ibáñez, M. Strategies for Quantification and Confirmation of Multi-Class Polar Pesticides and Transformation Products in Water by LC–MS2 Using Triple Quadrupole and Hybrid Quadrupole Time-of-Flight Analyzers. TrAC Trends Anal. Chem. 2005, 24, 596–612. [Google Scholar] [CrossRef]
- Cortéjade, A.; Kiss, A.; Cren, C.; Vulliet, E.; Buleté, A. Development of an Analytical Method for the Targeted Screening and Multi-Residue Quantification of Environmental Contaminants in Urine by Liquid Chromatography Coupled to High Resolution Mass Spectrometry for Evaluation of Human Exposures. Talanta 2016, 146, 694–706. [Google Scholar] [CrossRef]
- Ibáñez, M.; Sancho, J.V.; Hernández, F.; McMillan, D.; Rao, R. Rapid Non-Target Screening of Organic Pollutants in Water by Ultraperformance Liquid Chromatography Coupled to Time-of-Light Mass Spectrometry. TrAC Trends Anal. Chem. 2008, 27, 481–489. [Google Scholar] [CrossRef]
- Diaz, R.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Qualitative Validation of a Liquid Chromatography–Quadrupole-Time of Flight Mass Spectrometry Screening Method for Organic Pollutants in Waters. J. Chromatogr. A 2013, 1276, 47–57. [Google Scholar] [CrossRef]
- Deeb, A.A.; Stephan, S.; Schmitz, O.J.; Schmidt, T.C. Suspect Screening of Micropollutants and Their Transformation Products in Advanced Wastewater Treatment. Sci. Total Environ. 2017, 601–602, 1247–1253. [Google Scholar] [CrossRef]
- Schollée, J.E.; Bourgin, M.; von Gunten, U.; McArdell, C.S.; Hollender, J. Non-Target Screening to Trace Ozonation Transformation Products in a Wastewater Treatment Train Including Different Post-Treatments. Water Res. 2018, 142, 267–278. [Google Scholar] [CrossRef]
- Singer, H.P.; Wössner, A.E.; McArdell, C.S.; Fenner, K. Rapid Screening for Exposure to “Non-Target” Pharmaceuticals from Wastewater Effluents by Combining HRMS-Based Suspect Screening and Exposure Modeling. Environ. Sci. Technol. 2016, 50, 6698–6707. [Google Scholar] [CrossRef]
- Krauss, M.; Singer, H.; Hollender, J. LC–High Resolution MS in Environmental Analysis: From Target Screening to the Identification of Unknowns. Anal. Bioanal. Chem. 2010, 397, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.; Lucke, T.; Balsaa, P.; Hinnenkamp, V.; Brüggen, S.; Dünnbier, U.; Liebmann, D.; Fink, A.; Götz, S.; Geiss, S.; et al. Non-Target Screening in Water Analysis—Guideline for the Application of LC-ESI-HRMS for Screening; Water Chemistry Society, Division of the Gesellschaft Deutscher Chemiker: Mülheim an der Ruhr, Germany, 2019; Available online: http://www.wasserchemische-gesellschaft.de (accessed on 11 April 2020).
- Hohrenk, L.L.; Itzel, F.; Baetz, N.; Tuerk, J.; Vosough, M.; Schmidt, T.C. Comparison of Software Tools for Liquid Chromatography–High-Resolution Mass Spectrometry Data Processing in Nontarget Screening of Environmental Samples. Anal. Chem. 2020, 92, 1898–1907. [Google Scholar] [CrossRef]
- Pluskal, T.; Uehara, T.; Yanagida, M. Highly Accurate Chemical Formula Prediction Tool Utilizing High-Resolution Mass Spectra, MS/MS Fragmentation, Heuristic Rules, and Isotope Pattern Matching. Anal. Chem. 2012, 84, 4396–4403. [Google Scholar] [CrossRef]
- Hufsky, F.; Böcker, S. Mining Molecular Structure Databases: Identification of Small Molecules Based on Fragmentation Mass Spectrometry Data. Mass Spectrom. Rev. 2017, 36, 624–633. [Google Scholar] [CrossRef]
- Hug, C.; Ulrich, N.; Schulze, T.; Brack, W.; Krauss, M. Identification of Novel Micropollutants in Wastewater by a Combination of Suspect and Nontarget Screening. Environ. Pollut. 2014, 184, 25–32. [Google Scholar] [CrossRef]
- Alygizakis, N.A.; Gago-Ferrero, P.; Hollender, J.; Thomaidis, N.S. Untargeted Time-Pattern Analysis of LC-HRMS Data to Detect Spills and Compounds with High Fluctuation in Influent Wastewater. J. Hazard. Mater. 2019, 361, 19–29. [Google Scholar] [CrossRef]
- Krauss, M.; Hug, C.; Bloch, R.; Schulze, T.; Brack, W. Prioritising Site-Specific Micropollutants in Surface Water from LC-HRMS Non-Target Screening Data Using a Rarity Score. Environ. Sci. Eur. 2019, 31, 45. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Schulz, W.; Ruck, W.K.L.; Weber, W.H. A New Approach to Data Evaluation in the Non-Target Screening of Organic Trace Substances in Water Analysis. Chemosphere 2011, 85, 1211–1219. [Google Scholar] [CrossRef]
- Smith, R.M. Before the Injection—Modern Methods of Sample Preparation for Separation Techniques. J. Chromatogr. A 2003, 1000, 3–27. [Google Scholar] [CrossRef]
- Chon, K.; Chon, K.; Cho, J. Characterization of Size Fractionated Dissolved Organic Matter from River Water and Wastewater Effluent Using Preparative High Performance Size Exclusion Chromatography. Org. Geochem. 2017, 103, 105–112. [Google Scholar] [CrossRef]
- Fontanals, N.; Marcé, R.M.; Borrull, F. New Materials in Sorptive Extraction Techniques for Polar Compounds. J. Chromatogr. A 2007, 1152, 14–31. [Google Scholar] [CrossRef]
- Nürenberg, G.; Schulz, M.; Kunkel, U.; Ternes, T.A. Development and Validation of a Generic Nontarget Method Based on Liquid Chromatography – High Resolution Mass Spectrometry Analysis for the Evaluation of Different Wastewater Treatment Options. J. Chromatogr. A 2015, 1426, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Moschet, C.; Piazzoli, A.; Singer, H.; Hollender, J. Alleviating the Reference Standard Dilemma Using a Systematic Exact Mass Suspect Screening Approach with Liquid Chromatography-High Resolution Mass Spectrometry. Anal. Chem. 2013, 85, 10312–10320. [Google Scholar] [CrossRef]
- Kern, S.; Fenner, K.; Singer, H.P.; Schwarzenbach, R.P.; Hollender, J. Identification of Transformation Products of Organic Contaminants in Natural Waters by Computer-Aided Prediction and High-Resolution Mass Spectrometry. Environ. Sci. Technol. 2009, 43, 7039–7046. [Google Scholar] [CrossRef]
- Bastian, S.; Youngjoon, J.; Sarit, K.; Amy, H.L.; Pradeep, D.; Jake, O.; Maria Jose, G.R.; Sara, G.G.; Jochen, M.F.; Kevin, T.V.; et al. An Assessment of Quality Assurance/Quality Control Efforts in High Resolution Mass Spectrometry Non-Target Workflows for Analysis of Environmental Samples. TrAC Trends Anal. Chem. 2020, 133, 116063. [Google Scholar] [CrossRef]
- Drotleff, B.; Lämmerhofer, M. Guidelines for Selection of Internal Standard-Based Normalization Strategies in Untargeted Lipidomic Profiling by LC-HR-MS/MS. Anal. Chem. 2019, 91, 9836–9843. [Google Scholar] [CrossRef]
- Köppe, T.; Jewell, K.S.; Dietrich, C.; Wick, A.; Ternes, T.A. Application of a Non-Target Workflow for the Identification of Specific Contaminants Using the Example of the Nidda River Basin. Water Res. 2020, 178, 115703. [Google Scholar] [CrossRef]
- Peter, K.T.; Wu, C.; Tian, Z.; Kolodziej, E.P. Application of Nontarget High Resolution Mass Spectrometry Data to Quantitative Source Apportionment. Environ. Sci. Technol. 2019, 53, 12257–12268. [Google Scholar] [CrossRef]
- Albergamo, V.; Schollée, J.E.; Schymanski, E.L.; Helmus, R.; Timmer, H.; Hollender, J.; de Voogt, P. Nontarget Screening Reveals Time Trends of Polar Micropollutants in a Riverbank Filtration System. Environ. Sci. Technol. 2019. [Google Scholar] [CrossRef]
- Samanipour, S.; Baz-Lomba, J.A.; Reid, M.J.; Ciceri, E.; Rowland, S.; Nilsson, P.; Thomas, K.V. Assessing Sample Extraction Efficiencies for the Analysis of Complex Unresolved Mixtures of Organic Pollutants: A Comprehensive Non-Target Approach. Anal. Chim. Acta 2018, 1025, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogenboom, A.C.; van Leerdam, J.A.; de Voogt, P. Accurate Mass Screening and Identification of Emerging Contaminants in Environmental Samples by Liquid Chromatography–Hybrid Linear Ion Trap Orbitrap Mass Spectrometry. J. Chromatogr. A 2009, 1216, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Schymanski, E.L.; Singer, H.P.; Longrée, P.; Loos, M.; Ruff, M.; Stravs, M.A.; Ripollés Vidal, C.; Hollender, J. Strategies to Characterize Polar Organic Contamination in Wastewater: Exploring the Capability of High Resolution Mass Spectrometry. Environ. Sci. Technol. 2014, 48, 1811–1818. [Google Scholar] [CrossRef]
- Ruff, M.; Mueller, M.S.; Loos, M.; Singer, H.P. Quantitative Target and Systematic Non-Target Analysis of Polar Organic Micro-Pollutants along the River Rhine Using High-Resolution Massspectrometry e Identification of Unknown Sources and Compounds. Water Res. 2015, 87, 145–154. [Google Scholar] [CrossRef]
- Gago-Ferrero, P.; Schymanski, E.L.; Bletsou, A.A.; Aalizadeh, R.; Hollender, J.; Thomaidis, N.S. Extended Suspect and Non-Target Strategies to Characterize Emerging Polar Organic Contaminants in Raw Wastewater with LC-HRMS/MS. Environ. Sci. Technol. 2015, 49, 12333–12341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmi, J.; Pellinen, J.; Rantalainen, A.-L. Critical Evaluation of Screening Techniques for Emerging Environmental Contaminants Based on Accurate Mass Measurements with Time-of-Flight Mass Spectrometry. J. Mass Spectrom. 2012, 47, 303–312. [Google Scholar] [CrossRef]
- Díaz, R.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Target and Non-Target Screening Strategies for Organic Contaminants, Residues and Illicit Substances in Food, Environmental and Human Biological Samples by UHPLC-QTOF-MS. Anal. Methods 2012, 4, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Guillossou, R.; Le Roux, J.; Goffin, A.; Mailler, R.; Varrault, G.; Vulliet, E.; Morlay, C.; Nauleau, F.; Guérin, S.; Rocher, V.; et al. Fluorescence Excitation/Emission Matrices as a Tool to Monitor the Removal of Organic Micropollutants from Wastewater Effluents by Adsorption onto Activated Carbon. Water Res. 2021, 190, 116749. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, W.; Sun, P.; Lai, Q.; Mulcahy, D.; Guo, S. Rapid Determination of Nine Haloacetic Acids in Wastewater Effluents Using Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry. Anal. Lett. 2013, 46, 569–588. [Google Scholar] [CrossRef]
- Wickramasekara, S.; Hernández-Ruiz, S.; Abrell, L.; Arnold, R.; Chorover, J. Natural Dissolved Organic Matter Affects Electrospray Ionization during Analysis of Emerging Contaminants by Mass Spectrometry. Anal. Chim. Acta 2012, 717, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sgroi, M.; Roccaro, P.; Korshin, G.V.; Greco, V.; Sciuto, S.; Anumol, T.; Snyder, S.A.; Vagliasindi, F.G.A. Use of Fluorescence EEM to Monitor the Removal of Emerging Contaminants in Full Scale Wastewater Treatment Plants. J. Hazard. Mater. 2017, 323, 367–376. [Google Scholar] [CrossRef]
- Coble, P.G. Characterization of Marine and Terrestrial DOM in Seawater Using Excitation-Emission Matrix Spectroscopy. Mar. Chem. 1996, 51, 325–346. [Google Scholar] [CrossRef]
- Parlanti, E.; Wörz, K.; Geoffroy, L.; Lamotte, M. Dissolved Organic Matter Fluorescence Spectroscopy as a Tool to Estimate Biological Activity in a Coastal Zone Submitted to Anthropogenic Inputs. Org. Geochem. 2000, 31, 1765–1781. [Google Scholar] [CrossRef]
- Chen, J.; LeBoeuf, E.J.; Dai, S.; Gu, B. Fluorescence Spectroscopic Studies of Natural Organic Matter Fractions. Chemosphere 2003, 50, 639–647. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, J.; Ma, J.; Qiang, Z. Fluorescence Spectroscopic Characterization of DOM Fractions Isolated from a Filtered River Water after Ozonation and Catalytic Ozonation. Chemosphere 2008, 71, 911–921. [Google Scholar] [CrossRef]
- Jeanneau, L.; Faure, P.; Jardé, E. Influence of Natural Organic Matter on the Solid-Phase Extraction of Organic Micropollutants: Application to the Water-Extract from Highly Contaminated River Sediment. J. Chromatogr. A 2007, 1173, 1–9. [Google Scholar] [CrossRef]
- Lacey, C.; McMahon, G.; Bones, J.; Barron, L.; Morrissey, A.; Tobin, J.M. An LC–MS Method for the Determination of Pharmaceutical Compounds in Wastewater Treatment Plant Influent and Effluent Samples. Talanta 2008, 75, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open Source Software for Rapid Proteomics Tools Development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- PatRoon, Version 1. 2019. Available online: https://github.com/rickhelmus/patRoon (accessed on 18 November 2021).
- FactoMineR, Version 2.4. 2020. Available online: https://cran.r-project.org/web/packages/FactoMineR/index.html (accessed on 18 November 2021).
- Factoextra, Versions 1.0.7. 2020. Available online: https://cran.r-project.org/web/packages/factoextra/index.html (accessed on 18 November 2021).
- Conway, J.; Gehlenborg, N. pSetR: A More Scalable Alternative to Venn and Euler Diagrams for Visualizing Intersecting Sets, Version 1.4.0. 2019. Available online: https://cran.r-project.org/web/packages/UpSetR/index.html (accessed on 18 November 2021).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | pH | SPE Cartridge | Elution Solvents | Ref |
|---|---|---|---|---|
| Surface water and wastewater (influent and effluent) | N.A. a | HLB | N.A. a | [8] |
| Surface water, groundwater and Drinking water | 2 | HLB, MCX | Acetonitrile (HLB) Acetonitrile, Acetonitrile + 5% ammonia (MCX) | [34] |
| Surface water, wastewater influent and effluent | 6.7 | Multilayer (HLB, ENV+, X-AW, X-CW) | Methanol/Ethyl acetate (50:50, v/v) + 2% ammonia and methanol/ Ethyl acetate (50:50, v/v) + 1.7% formic acid | [11,26,27,35,36,37] |
| Landfill leachate and groundwater | 7 and 3 | ENV+ | Methanol | [21] |
| Wastewater effluent | 2 | MCX and Strata X in series | Methanol + 5% ammonia | [38] |
| Wastewater (influent and effluent) | N.A. a | HLB | Methanol | [39] |
| Surface water | N.A. a | Multilayer (HR-X, HR-XAW, HR-XCW) | Ethyl acetate; methanol; methanol + 2% ammonia and methanol + 1% formic acid | [5] |
| Wastewater effluent | N.A. a | MAX and MCX in series | Methanol/ethyl acetate/formic acid (69:29:2, v/v) (MAX) methanol/ethyl acetate/ammonia solution (67.5:27.5:5, v/v) (MCX) | [10] |
| Riverbank filtration system | N.A. a | HLB | Methanol | [32] |
| Index | C18 | C18 ENV+ | ENV+ | HLB pH 2 | HLB | Multilayer | SDBL | XA | XAW | XC | XCW |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Region I FRI | 84 | 85 | 64 | 70 | 58 | 71 | 58 | 94 | 89 | 55 | 49 |
| Region II FRI | 76 | 69 | 67 | 68 | 58 | 86 | 48 | 96 | 90 | 59 | 47 |
| Region III FRI | 72 | 46 | 46 | 55 | 55 | 73 | 27 | 92 | 86 | 50 | 28 |
| Region IV FRI | 68 | 62 | 64 | 65 | 45 | 80 | 41 | 93 | 86 | 53 | 45 |
| Region V FRI | 70 | 47 | 50 | 56 | 53 | 55 | 28 | 93 | 86 | 48 | 28 |
| Number of Features | Sum of Detected Areas | Average m/z | Weighted Average m/z | Average Retention (% ACN) | Weighted Average Retention (% ACN) | |
|---|---|---|---|---|---|---|
| Marne River | ||||||
| ENV | 315 | 4.25 × 106 | 306.6403 | 300.0973 | 43 | 41 |
| HLB | 403 | 4.20 × 106 | 367.9942 | 379.1581 | 44 | 43 |
| XAW | 49 | 8.69 × 105 | 295.4186 | 323.9237 | 52 | 52 |
| XCW | 350 | 3.43 × 106 | 404.9278 | 366.7695 | 48 | 46 |
| Multilayer | 594 | 5.84 × 106 | 470.6467 | 441.9003 | 46 | 44 |
| WWe | ||||||
| ENV | 301 | 2.72 × 107 | 306.7059 | 339.2997 | 37 | 33 |
| HLB | 201 | 1.75 × 107 | 337.8578 | 311.8444 | 35 | 27 |
| XAW | 149 | 9.03 × 107 | 345.6214 | 384.1570 | 34 | 22 |
| XCW | 100 | 8.32 × 106 | 297.6594 | 367.0206 | 28 | 23 |
| Multilayer | 7515 | 4.89 × 107 | 461.3586 | 416.1775 | 47 | 35 |
| Before PFA | After 30 ppm PFA | |||
|---|---|---|---|---|
| Number of Markers | Sum of Detected Areas | Number of Markers | Sum of Detected Areas | |
| Zone 1 m/z: 0–500 Retention Time: 0–10 min | 4861 | 2.51 × 108 | 3781 | 1.85 × 108 |
| Zone 2 m/z: 500–1000 Retention Time: 0–10 min | 996 | 0.35 × 108 | 2301 | 1.36 × 108 |
| Zone 3 m/z: 0–500 Retention Time: 10–25 min | 2795 | 1.43 × 108 | 1728 | 0.75 × 108 |
| Zone 4 m/z: 500–1000 Retention Time: 10–25 min | 2770 | 2.93 × 107 | 3557 | 1.95 × 108 |
| Cartridge | Sample | Conditioning | Washing | Eluting |
|---|---|---|---|---|
| X-A | 1 L pH 2–3 Marne | 10 mL MeOH 10 mL Milli-Q | 10 mL MeOH | 10 mL MeOH + 5% formic acid |
| X-AW | 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL Milli-Q | 10 mL MeOH | 5 mL MeOH + 5% ammonia 5 mL MeOH + 5% formic acid |
| X-C | 1 L pH 6–7 Marne | 10 mL MeOH 10 mL Milli-Q pH = 2 | 10 mL MeOH + 0.1 M HCl | 5 mL MeOH + 0.1M HCl 5 mL MeOH + 5% ammonia |
| X-CW | 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL Milli-Q | 10 mL MeOH | 5 mL MeOH + 5% formic acid 5 mL MeOH + 5% ammonia |
| HLB | 1 L pH 2–3 Marne 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL AcEt 5 mL DCM 10 mL Milli-Q | No washing | 5 mL MeOH 5 mL AcEt 5 mL DCM |
| ENV+ | 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL Milli-Q | 5 mL Milli-Q /MeOH (95/5, v/v) | 5 mL MeOH 5 mL acetone + 5% ammonia |
| C18 | 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL Milli-Q | No washing | 5 mL MeOH 5 mL acetone + 5% ammonia |
| C18/ENV+ | 1 L pH 6–7 Marne | 10 mL MeOH 10 mL Milli-Q | No washing | 5mL MeOH 5mL acetone + 5% ammonia |
| SDBL | 1 L pH 6–7 Marne | 10 mL MeOH 10 mL Milli-Q pH = 4 | No washing | 5 mL MeOH 5 mL acetone + 5% ammonia |
| Multilayer | 1 L pH 6–7 Marne, MQ, WWe | 10 mL MeOH 10 mL Milli-Q | No washing | 6 mL AcEt/MeOH (50/50, v/v) + 1.43% ammonia 3 mL AcEt/MeOH (50/50, v/v) + 1.7% formic acid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, N.; Caupos, E.; Soares Peirera, C.; Le Roux, J.; Bressy, A.; Moilleron, R. Evaluation of Sample Preparation Methods for Non-Target Screening of Organic Micropollutants in Urban Waters Using High-Resolution Mass Spectrometry. Molecules 2021, 26, 7064. https://doi.org/10.3390/molecules26237064
Huynh N, Caupos E, Soares Peirera C, Le Roux J, Bressy A, Moilleron R. Evaluation of Sample Preparation Methods for Non-Target Screening of Organic Micropollutants in Urban Waters Using High-Resolution Mass Spectrometry. Molecules. 2021; 26(23):7064. https://doi.org/10.3390/molecules26237064
Chicago/Turabian StyleHuynh, Nina, Emilie Caupos, Caroline Soares Peirera, Julien Le Roux, Adèle Bressy, and Régis Moilleron. 2021. "Evaluation of Sample Preparation Methods for Non-Target Screening of Organic Micropollutants in Urban Waters Using High-Resolution Mass Spectrometry" Molecules 26, no. 23: 7064. https://doi.org/10.3390/molecules26237064
APA StyleHuynh, N., Caupos, E., Soares Peirera, C., Le Roux, J., Bressy, A., & Moilleron, R. (2021). Evaluation of Sample Preparation Methods for Non-Target Screening of Organic Micropollutants in Urban Waters Using High-Resolution Mass Spectrometry. Molecules, 26(23), 7064. https://doi.org/10.3390/molecules26237064