
Combining Cryo-EM Density Map and Residue Contact for Protein Secondary Structure Topologies

Abstract
:1. Introduction
2. Results
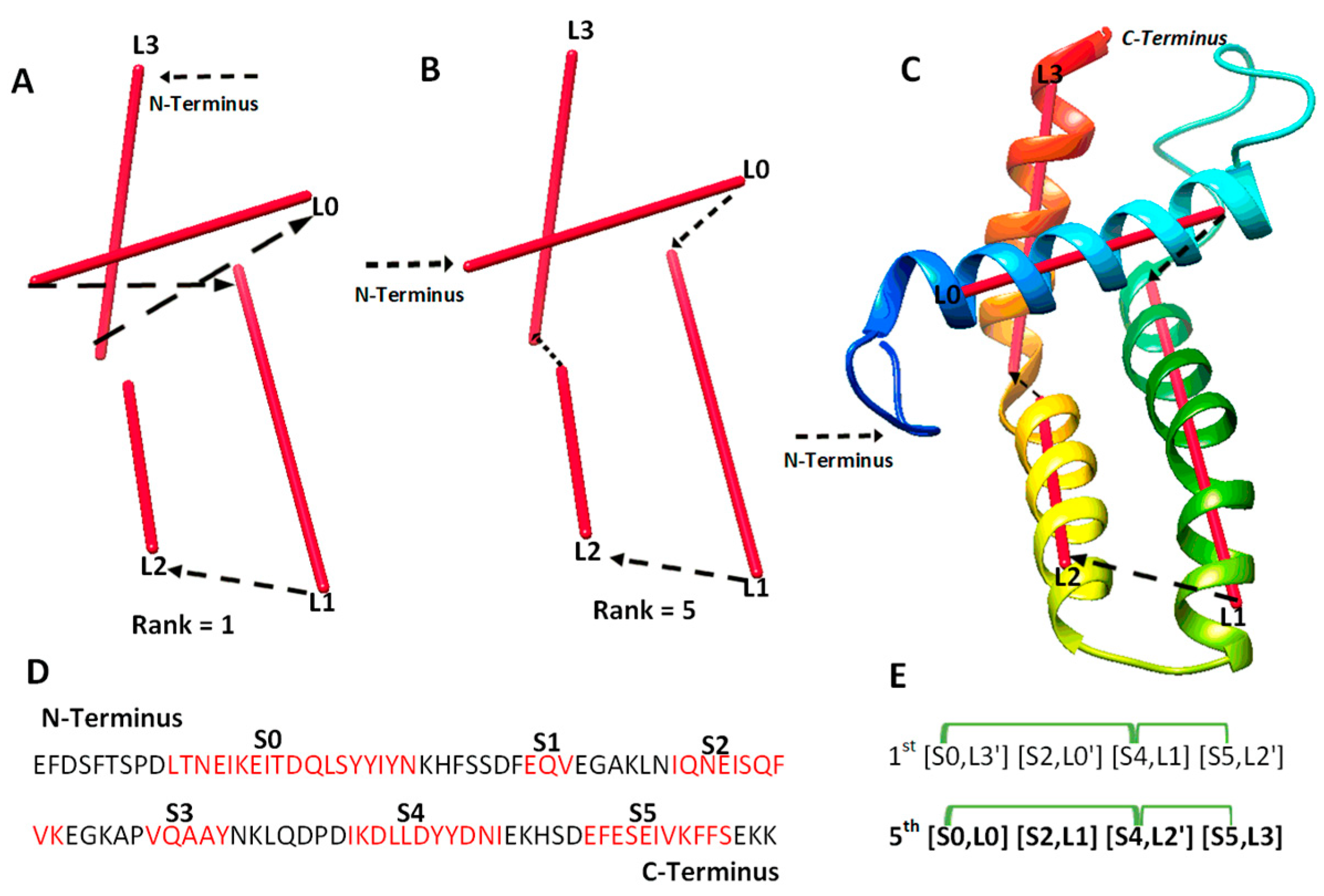
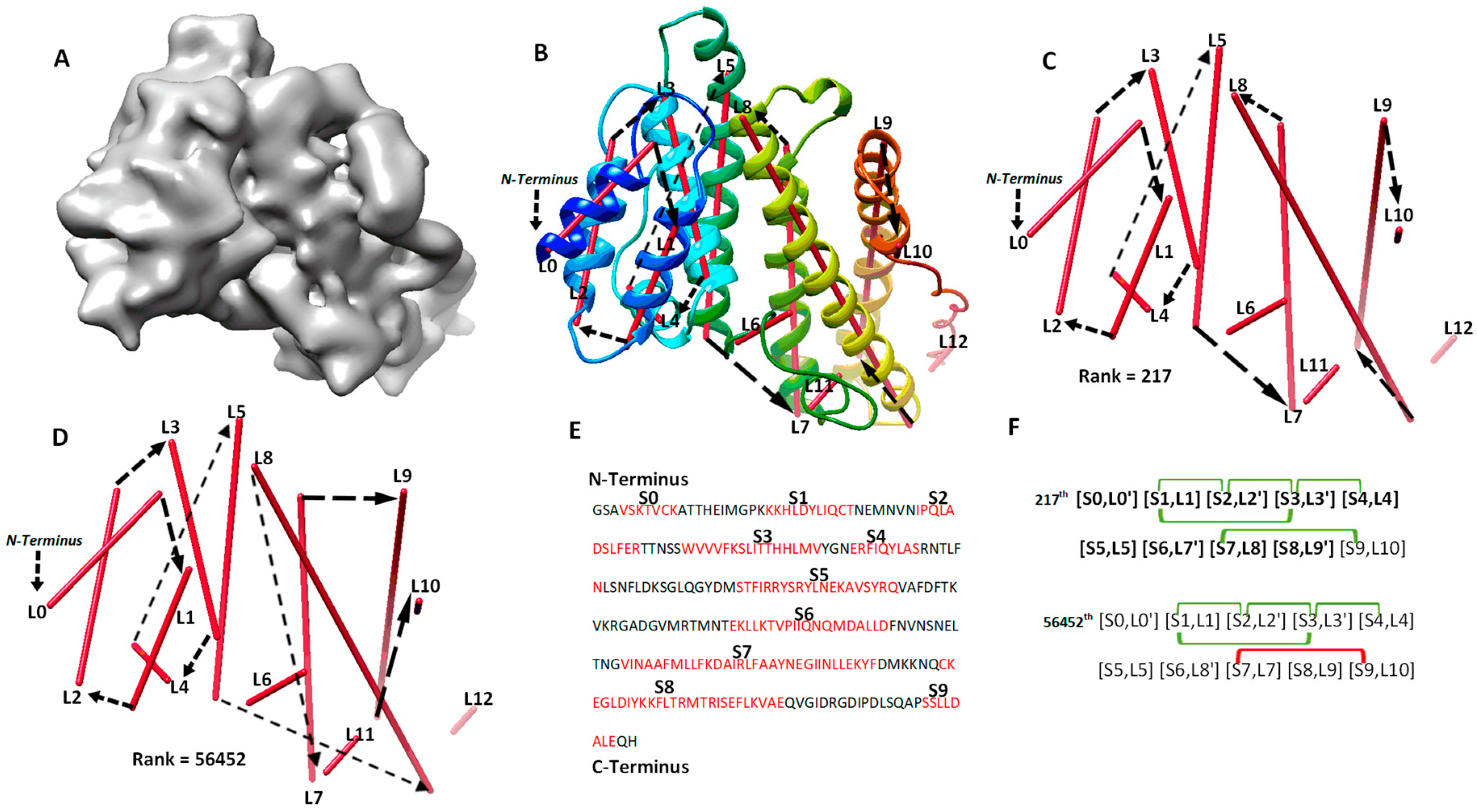
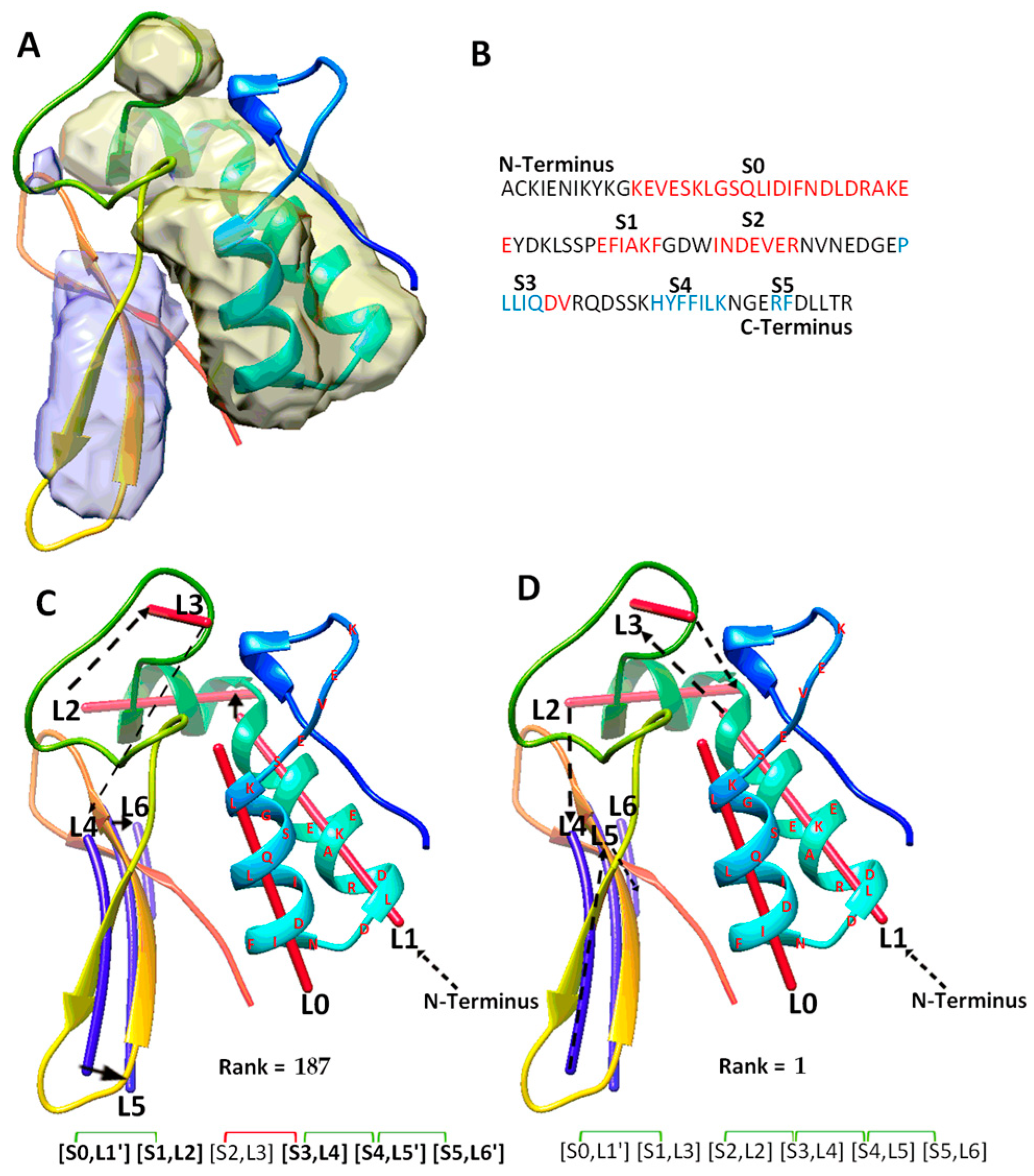
2.1. Evaluation of Topologies of Secondary Structures
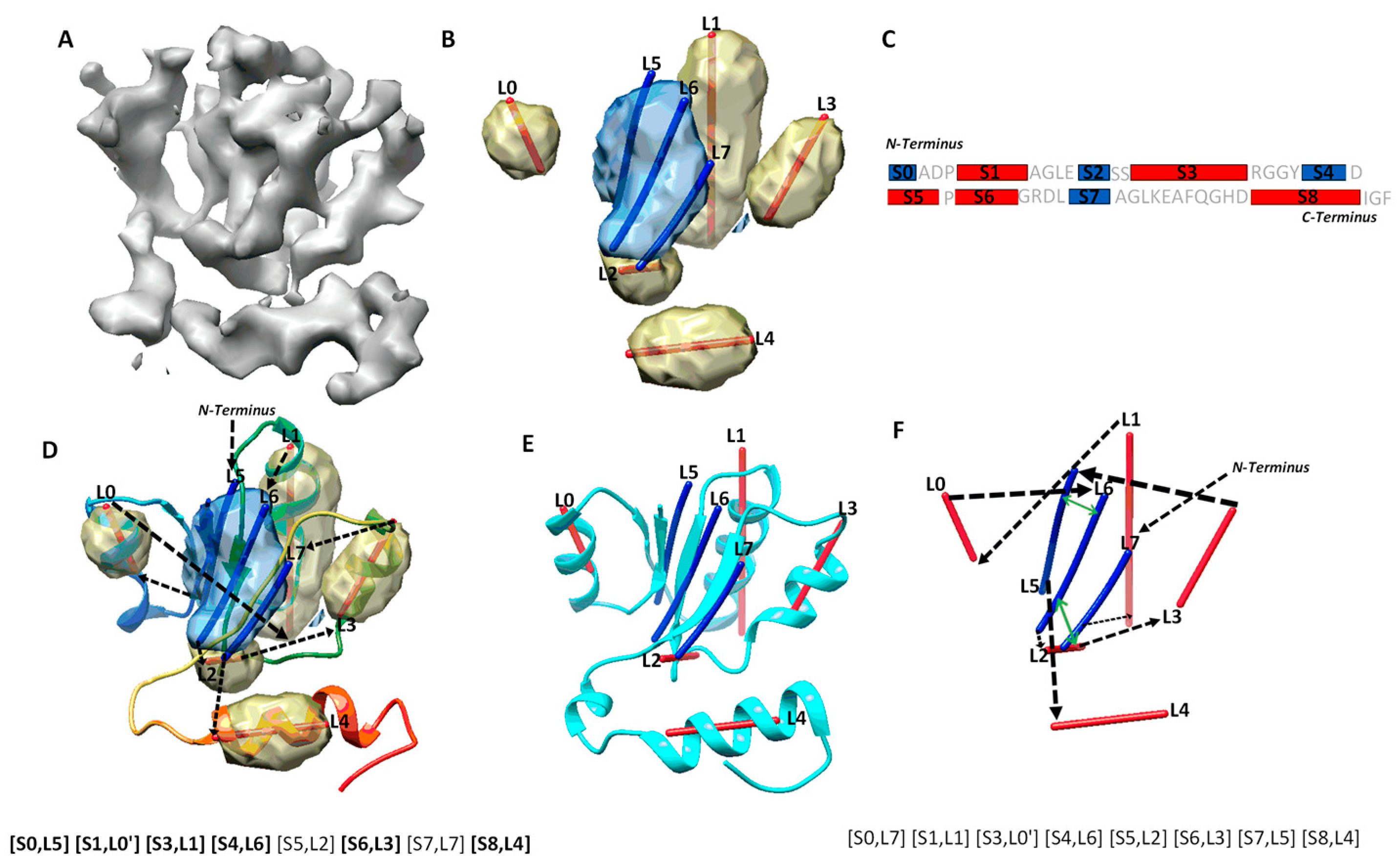
2.2. A Case Study for Secondary Structure Prediction and Evaluation of Topologies
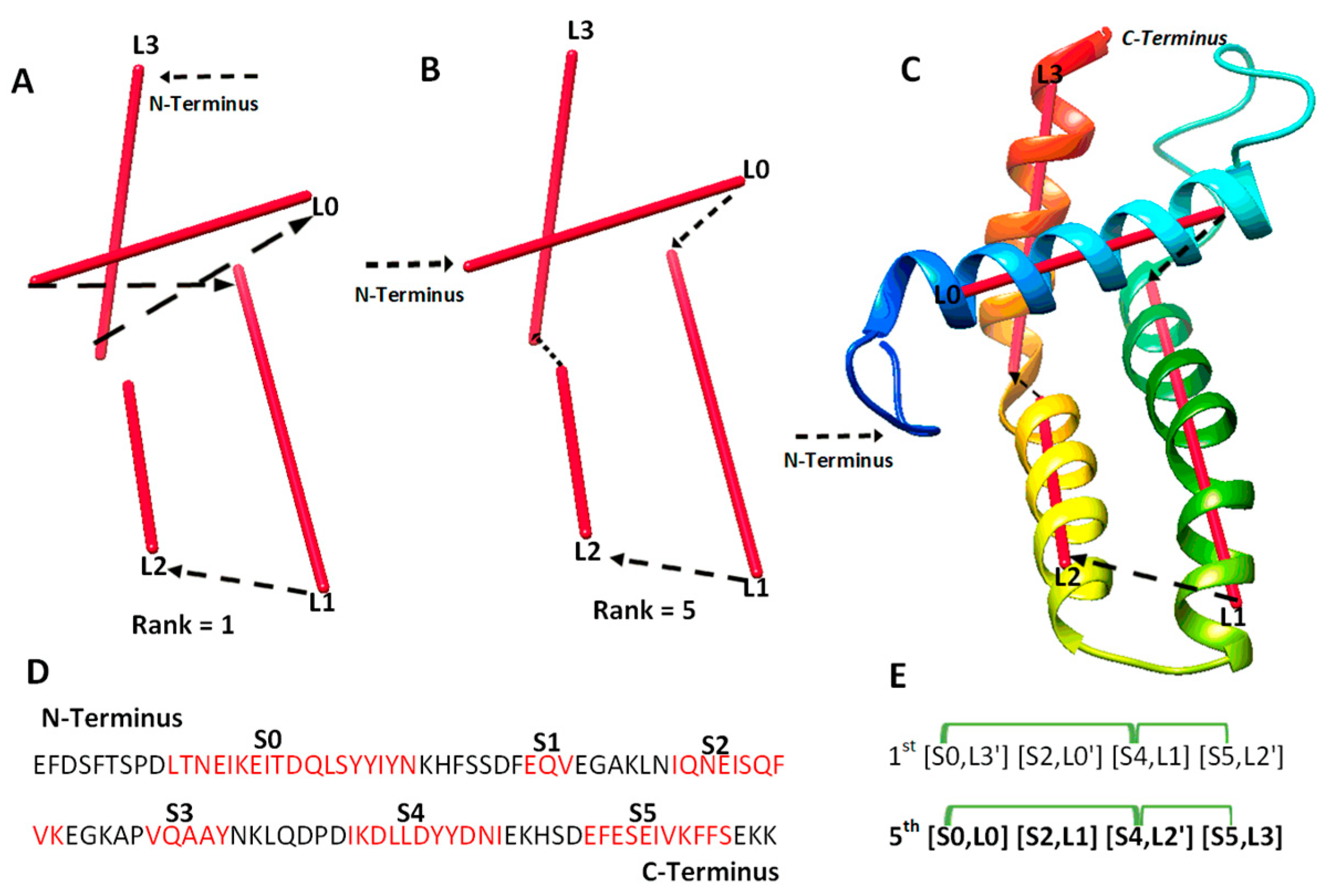
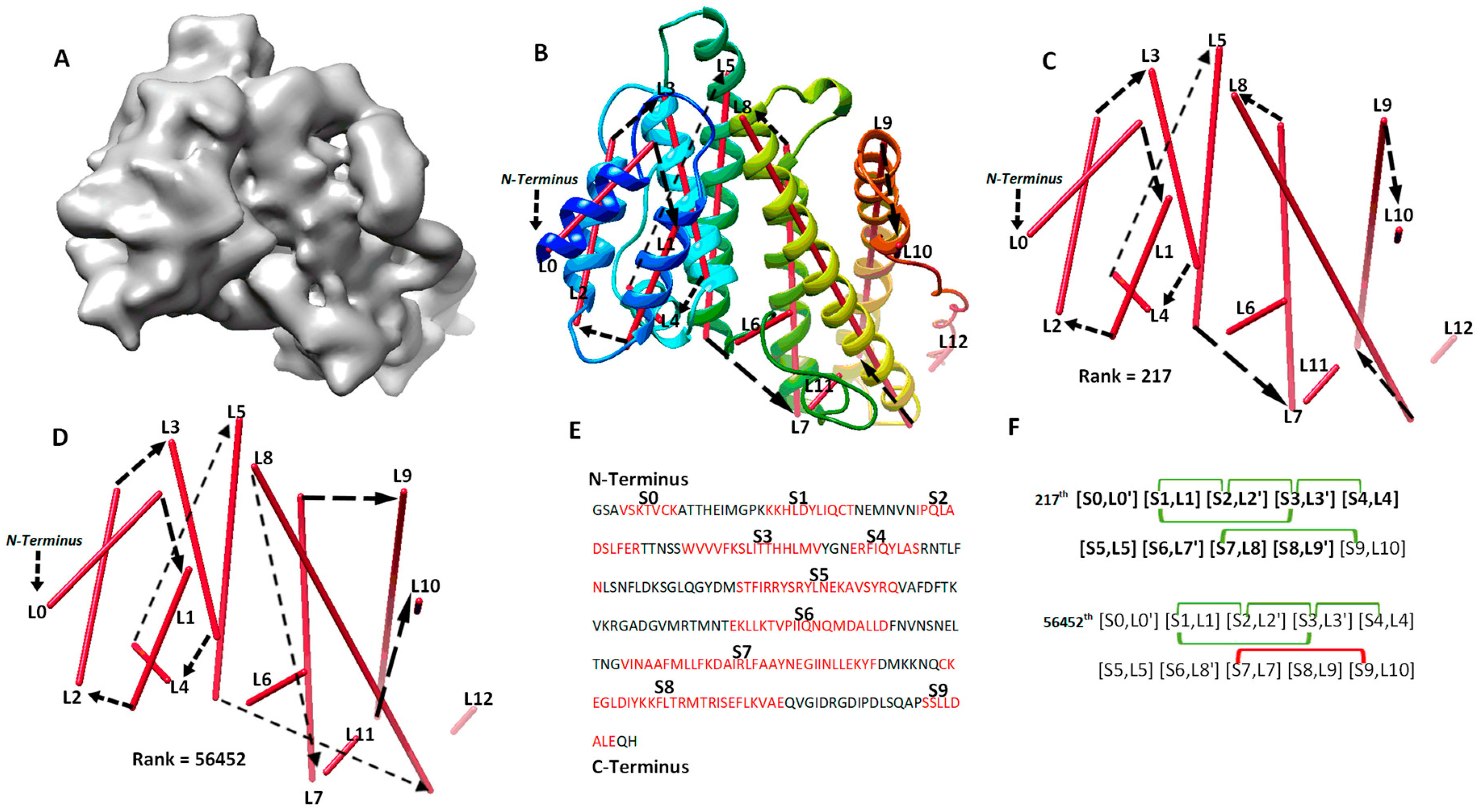
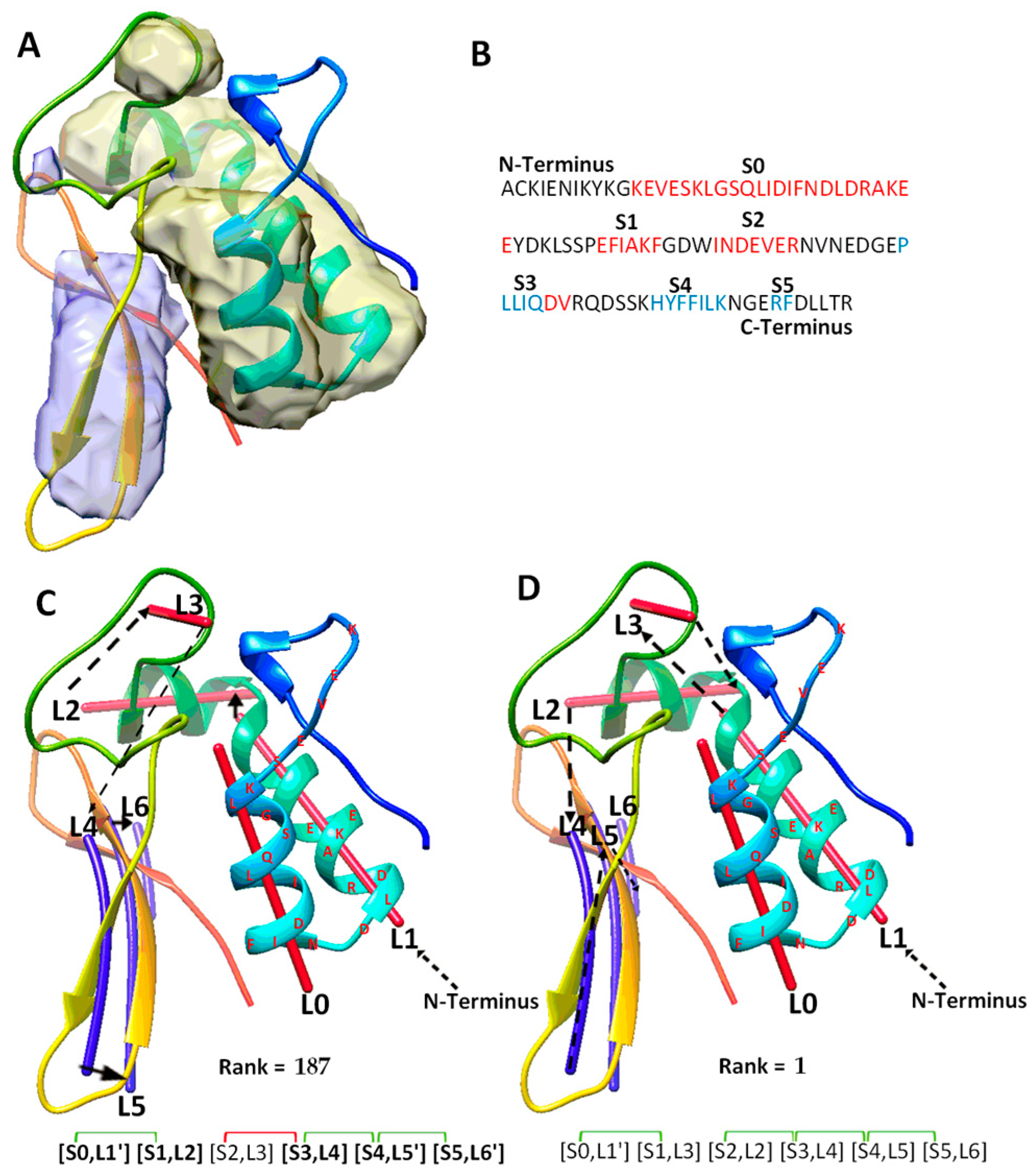
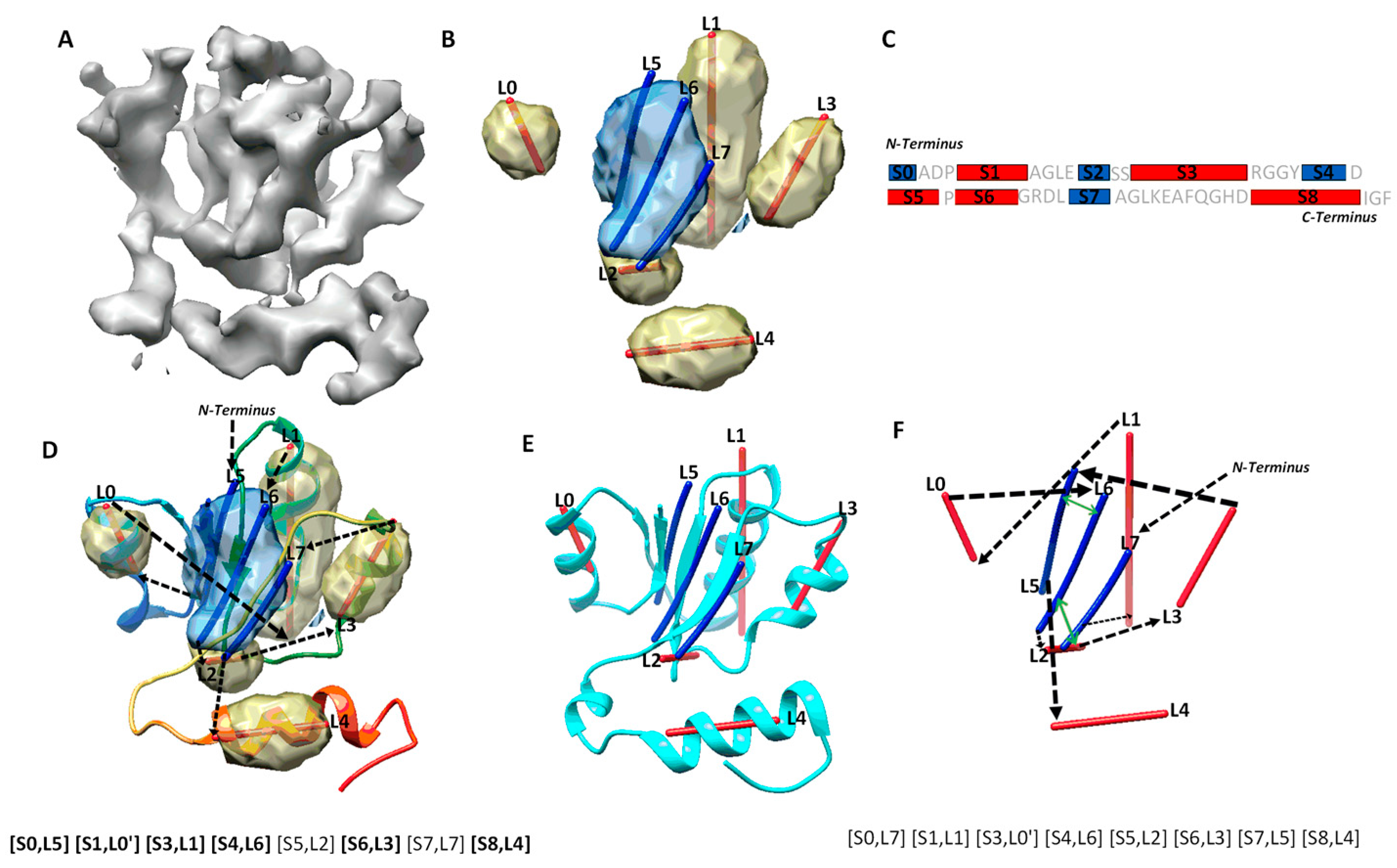
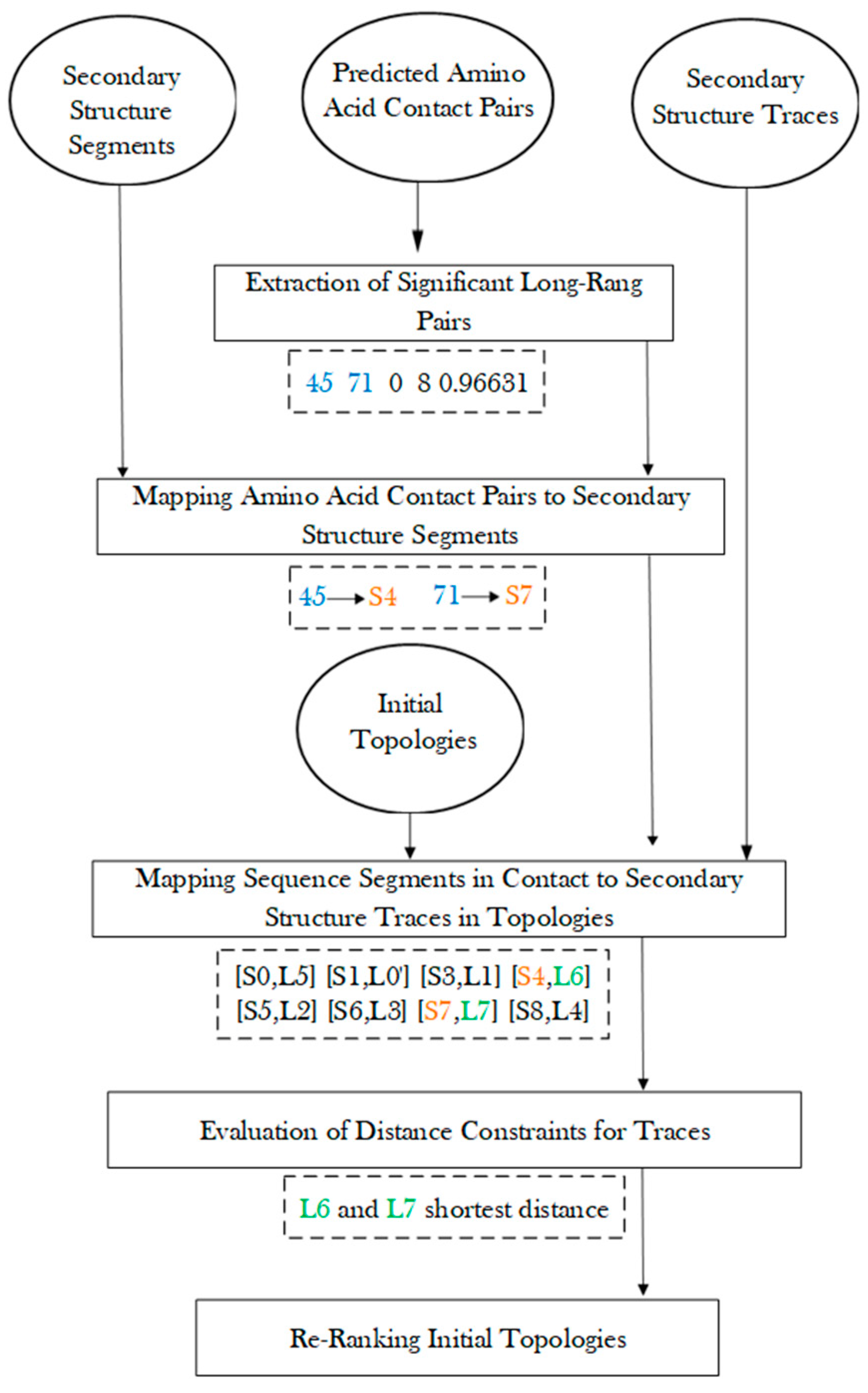
2.3. Secondary Structure Contact Pairs and Their Effect in Ranking Initial Topologies
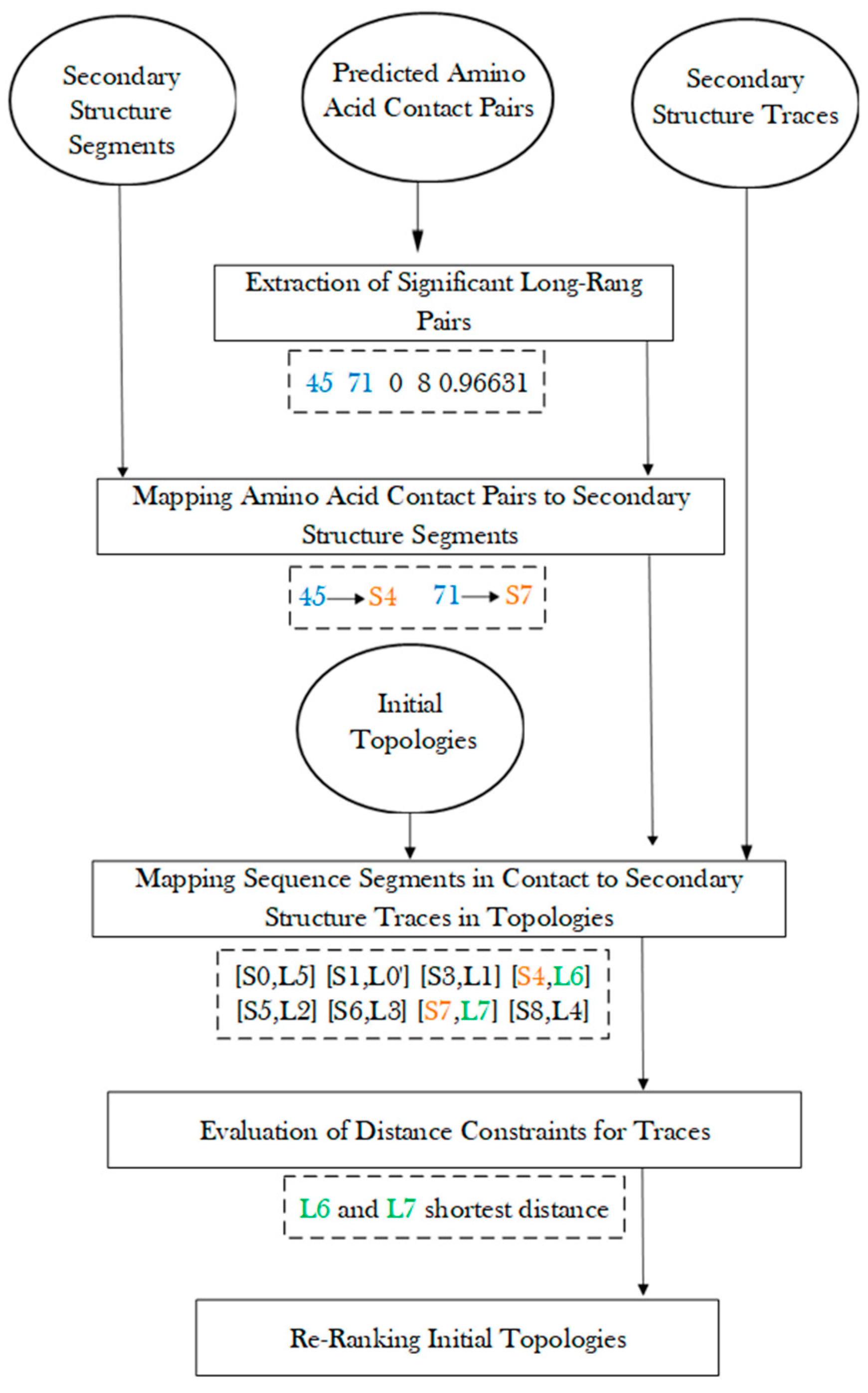
3. Methods
3.1. Preparation of Data
3.2. Protein Secondary Structure Contact
3.3. Secondary Structure Traces from Cryo-EM Density Maps
3.4. Generation of Initial Topologies
3.5. Re-Rank Topologies Using Secondary Structure Contact Pairs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Z.; Guo, F.; Wang, F.; Li, T.C.; Jiang, W. 2.9 Å Resolution Cryo-EM 3D Reconstruction of Close-Packed Virus Particles. Structure 2016, 24, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Jin, L.; Koh, S.B.; Atanasov, I.; Schein, S.; Wu, L.; Zhou, Z.H. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 2010, 329, 1038–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, W.L.; Yu, D.; Ouyang, Q.; Lu, Y.; Mao, Y. Nucleotide-Driven Triple-State Remodeling of the AAA-ATPase Channel in the Activated Human 26S Proteasome. bioRxiv 2017, 132613. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-C.; Lyu, M.; Morgan, C.E.; Bolla, J.R.; Robinson, C.V.; Edward, W.Y. A ‘Build and Retrieve’methodology to simultaneously solve cryo-EM structures of membrane proteins. Nat. Methods 2021, 18, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Adachi, N.; Moriya, T.; Oki, H.; Maru, T.; Kawasaki, M.; Suzuki, K.; Chen, S.; Ishii, R.; Yonemori, K. Cryo-EM structure of K+-bound hERG channel complexed with the blocker astemizole. Structure 2021, 29, 203–212.e204. [Google Scholar] [CrossRef] [PubMed]
- Schröder, G.F.; Brunger, A.T.; Levitt, M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure 2007, 15, 1630–1641. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.Y.; Trabuco, L.G.; Schreiner, E.; Schulten, K. Cryo-electron microscopy modeling by the molecular dynamics flexible fitting method. Biopolymers 2012, 97, 678–686. [Google Scholar] [CrossRef]
- Wriggers, W.; Birmanns, S. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J. Struct. Biol. 2001, 133, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Kulik, M.; Mori, T.; Sugita, Y. Multi-scale flexible fitting of proteins to cryo-EM density maps at medium resolution. Front. Mol. Biosci. 2021, 8, 631854. [Google Scholar] [CrossRef]
- Costa, M.G.; Fagnen, C.; Vénien-Bryan, C.; Perahia, D. A new strategy for atomic flexible fitting in cryo-EM maps by molecular dynamics with excited normal modes (MDeNM-EMfit). J. Chem. Inf. Modeling 2020, 60, 2419–2423. [Google Scholar] [CrossRef]
- Mori, T.; Kulik, M.; Miyashita, O.; Jung, J.; Tama, F.; Sugita, Y. Acceleration of cryo-EM flexible fitting for large biomolecular systems by efficient space partitioning. Structure 2019, 27, 161–174.e163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeysinghe, S.; Ju, T.; Baker, M.L.; Chiu, W. Shape modeling and matching in identifying 3D protein structures. Comput. -Aided Des. 2008, 40, 708–720. [Google Scholar] [CrossRef]
- Al-Nasr, K.; Ranjan, D.; Zubair, M.; He, J. Ranking Valid Topologies of the Secondary Structure Elements Using a Constraint Graph. J. Bioinform. Comput. Biol. 2011, 9, 415–430. [Google Scholar] [CrossRef] [Green Version]
- Lindert, S.; Alexander, N.; Wötzel, N.; Karakaş, M.; Stewart, P.L.; Meiler, J. EM-fold: De novo atomic-detail protein structure determination from medium-resolution density maps. Structure 2012, 20, 464–478. [Google Scholar] [CrossRef] [Green Version]
- Al Nasr, K.; Ranjan, D.; Zubair, M.; Chen, L.; He, J. Solving the Secondary Structure Matching Problem in Cryo-EM De Novo Modeling Using a Constrained K-Shortest Path Graph Algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 419–430. [Google Scholar] [CrossRef]
- Biswas, A.; Ranjan, D.; Zubair, M.; He, J. A Dynamic Programming Algorithm for Finding the Optimal Placement of a Secondary Structure Topology in Cryo-EM Data. J. Comput. Biol. 2015, 22, 837–843. [Google Scholar] [CrossRef]
- Biswas, A.; Ranjan, D.; Zubair, M.; Zeil, S.; Al Nasr, K.; He, J. An effective computational method incorporating multiple secondary structure predictions in topology determination for cryo-EM images. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 578–586. [Google Scholar] [CrossRef]
- Al Nasr, K.; Yousef, F.; Jones, C.; Jebril, R. Geometry Analysis for Protein Secondary Structures Matching Problem. In Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017. [Google Scholar]
- Al Nasr, K.; Yousef, F.; Jebril, R.; Jones, C. Analytical approaches to improve accuracy in solving the protein topology problem. Molecules 2018, 23, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Sazzed, S.; Alshammari, M.; Sun, J.; He, J. A Tool for Segmentation of Secondary Structures in 3D Cryo-EM Density Map Components Using Deep Convolutional Neural Networks. Front. Bioinform. 2021, 51. [Google Scholar] [CrossRef]
- Si, D.; He, J. Tracing Beta Strands Using StrandTwister from Cryo-EM Density Maps at Medium Resolutions. Structure 2014, 22, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Baker, M.L.; Ludtke, S.J.; Chiu, W. Bridging the information gap: Computational tools for intermediate resolution structure interpretation. J. Mol. Biol. 2001, 308, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Dal Palu, A.; He, J.; Pontelli, E.; Lu, Y. Identification of Alpha-Helices from Low Resolution Protein Density Maps. In Proceedings of the Computational Systems Bioinformatics Conference(CSB) (2006), Stanford, CA, USA, 14–18 August 2006; pp. 89–98. [Google Scholar]
- Baker, M.L.; Ju, T.; Chiu, W. Identification of secondary structure elements in intermediate-resolution density maps. Structure 2007, 15, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusu, M.; Wriggers, W. Evolutionary bidirectional expansion for the tracing of alpha helices in cryo-electron microscopy reconstructions. J. Struct. Biol. 2012, 177, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, D.; Ji, S.; Nasr, K.A.; He, J. A Machine Learning Approach for the Identification of Protein Secondary Structure Elements from Electron Cryo-Microscopy Density Maps. Biopolymers 2012, 97, 698–708. [Google Scholar] [CrossRef]
- Si, D.; He, J. Beta-sheet Detection and Representation from Medium Resolution Cryo-EM Density Maps. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics (2013), Washington, DC, USA, 22–25 September 2013; pp. 764–770. [Google Scholar]
- Li, R.; Si, D.; Zeng, T.; Ji, S.; He, J. Deep convolutional neural networks for detecting secondary structures in protein density maps from cryo-electron microscopy. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016. [Google Scholar]
- Maddhuri, S.V.S.; Terashi, G.; Kihara, D. Protein secondary structure detection in intermediate-resolution cryo-EM maps using deep learning. Nat. Methods 2019, 16, 911–917. [Google Scholar] [CrossRef]
- Cuff, J.A.; Clamp, M.E.; Siddiqui, A.S.; Finlay, M.; Barton, G.J. JPred: A consensus secondary structure prediction server. Bioinformatics 1998, 14, 892–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Wu, J.; Lan, H.; Zheng, L.; Pei, J.; Wang, S.; Liu, W.; Huang, J. When Homologous Sequences Meet Structural Decoys: Accurate Contact Prediction by tFold in CASP14. Proteins Struct. Funct. Bioinform. 2021. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Bell, E.W.; Zheng, W.; Zhou, X.; Yu, D.-J.; Zhang, Y. Deducing high-accuracy protein contact-maps from a triplet of coevolutionary matrices through deep residual convolutional networks. PLoS Comput. Biol. 2021, 17, e1008865. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Zheng, W.; Zhou, X.; Bell, E.W.; Yu, D.J.; Zhang, Y. Protein inter-residue contact and distance prediction by coupling complementary coevolution features with deep residual networks in CASP14. Proteins Struct. Funct. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics 2018, 34, 4039–4045. [Google Scholar] [CrossRef]
- Seemayer, S.; Gruber, M.; Söding, J. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics 2014, 30, 3128–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Singh, T.; Kosciolek, T.; Tetchner, S. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 2015, 31, 999–1006. [Google Scholar] [CrossRef]
- Jones, D.T.; Buchan, D.W.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2012, 28, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, B.; Hou, J.; Cheng, J. DNCON2: Improved protein contact prediction using two-level deep convolutional neural networks. Bioinformatics 2018, 34, 1466–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Li, J.; Wang, Z.; Eickholt, J.; Deng, X. The MULTICOM toolbox for protein structure prediction. BMC Bioinform. 2012, 13, 65. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mortuza, S.; Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Zhang, Y. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat. Commun. 2021, 12, 5011. [Google Scholar] [CrossRef]
- Liu, J.; Wu, T.; Guo, Z.; Hou, J.; Cheng, J. Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.; He, J. Combine Cryo-EM Density Map and Residue Contact for Protein Structure Prediction: A Case Study. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics (2020), Virtual Event, USA, 21–24 September 2020; pp. 1–6. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Al Nasr, K.; Liu, C.; Rwebangira, M.; Burge, L.; He, J. Intensity-based skeletonization of CryoEM gray-scale images using a true segmentation-free algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 1289–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32 (Suppl. 2), W500–W502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintilie, G.; Zhang, J.; Chiu, W.; Gossard, D. Identifying components in 3D density maps of protein nanomachines by multi-scale segmentation. In Proceedings of the 2009 IEEE/NIH Life Science Systems and Applications Workshop (2009), Bethesda, MD, USA, 9–10 April 2009; pp. 44–47. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a Case | b Secondary Structure Contact and Number of Significant Long-Range Pairs |
|---|---|
| 6810-5y5x-H (3SD) | (S4, S5)-(β, α)-1; (S4, S7)-(β, β)-4 |
| 9534-5gpn-Ae (1SD) | (S0, S1)-(α, α)-1; (S1, S2)-(α, α)-1; (S2, S3)-(α, α)-3 |
| 8518-5u8s-A (3SD) | (S1, S2)-(α, α)-1; (S4, S7)-(β, β)-9; (S4, S9)-(β, β)-3; (S5, S6)-(β, β)-1; (S7, S8)-(β, α)-4; (S8, S9)-(α, β)-1 |
| 3948-6esg-B (1SD) | (S0, S1)-(α, α)-3; (S1, S2)-(α, α)-9; (S2, S3)-(α, β)-5 |
| 2620-4uje-BH (3SD) | (S2, S3)-(β, β)-22; (S2, S5)-(β, β)-10; (S3,S4)-(β, α)-1; (S4, S5)-(α, β)-1; (S9, S10)-(α, β)-4; |
| 8357-5t4o-L (2SD) | (S0, S5)-(α, α)-2; (S0, S1)-(α, α)-1; (S1, S2)-(α, α)-1; (S6, S8)-(β, β)-24; (S6, S9)-(β, β)-14; (S7, S8)-(α, β)-1; (S9, S10)-(β, β)-5 |
| 3LTJ (2SD) | (S3, S5)-(α, α)-1; (S5, S7)-(α, α)-1; (S8, S9)-(α, α)-1 |
| 2XB5 (3SD) | (S0, S1)-(α, α)-8; (S0, S3)-(α, α)-2; (S1, S2)-(α, β)-1; (S4, S9)-(α, α)-6; (S4, S6)-(α, α)-1; (S4, S7)-(α, α)-1; (S6, S7)-(α, α)-5; (S7, S9)-(α, α)-3 |
| 1HG5 (2SD) | (S1, S3)-(α, α)-5; (S1, S2)-(α, α)-1; (S2, S3)-(α, α)-1; (S3, S4)-(α, α)-2; (S7, S9)-(α, α)-2 |
| 3ACW (3SD) | (S2, S6)-(α, α)-1; (S5, S6)-(α, α)-1; (S6, S7)-(α, α)-4; (S7,S10)-(α, α)-1; (S10, S11)-(α,α)-1 |
| 1Z1L (3SD) | (S3, S8)-(α, α)-1; (S4, S5)-(α, α)-7; (S5, S9)-(α, α)-7; (S5, S11)-(α, α)-1; (S8, S9)-(α, α)-4; (S9, S11)-(α, α)-3; (S10, S11)-(α,α)-3; (S12, S13)-(α,α)-2; (S3, S4)-(α, α)-1; (S9, S10) (α, α)-2; (S13, S14)-(α,α)-6 |
| T1029 (3SD) | (S1, S6)-(α, α)-4; (S2, S3)-(β, β)-9; (S3, S4)-(β, β)-11; (S4, S5)-(β, β)-12 |
| T1031 (3SD) | (S0, S1)-(α, α)-1; (S2, S3)-(α, β)-1; (S3, S4)-(β, β)-18; (S4, S5)-(β, β)-6 |
| T1033 (3SD) | (S0, S4)-(α, α)-2; (S2, S3)-(α, α)-7; (S3, S4)-(α, α)-6; (S4, S5)-(α, α)-2 |
| Case a | #a.a.b | True Struct. c | Seq Pred. d | Image Detect. e | Max Pairs f | Rank of Maximum-Match Topology | |
|---|---|---|---|---|---|---|---|
| No_C g | With_C h | ||||||
| 6810-5y5x-H(5 Å) | 104/100 | 5/3 | 5/4 | 5/3 | 6(4/2) | 1 | 1 |
| 9534-5gpn-Ae(5.4 Å) | 116/88 | 4/0 | 4/0 | 4/0 | 4(4/0) | 2 | 2 |
| 8518-5u8s-A(6.1 Å) | 208/208 | 6/2 | 5/3 + 2 | 5/3 + 2 | 7(5/2) | 142 | 116 |
| 3948-6esg-B(5.4 Å) | 102/78 | 3/0 | 3/1 | 3/0 | 3(3/0) | 5 | 5 |
| 2620-4uje-BH(6.9 Å) | 194/191 | 7/3 + 3 | 5/3 + 3 | 4/3 + 3 | 10(4/6) | NA | - |
| 8357-5t4o-L(6.9 Å) | 177/160 | 9/0 | 7/4 | 8/2 | 7(7/0) | 2 | 2 |
| 3LTJ(8 Å) | 201/191 | 16/0 | 12/0 | 12/0 | 12(12/0) | NA | - |
| 2XB5(8 Å) | 207/207 | 12/0 | 9/1 | 10/3 | 9(9/0) | NA | - |
| 1HG5(8 Å) | 289/263 | 11/0 | 10/0 | 13/0 | 9(9/0) | 1022 | 217 |
| 3ACW(8 Å) | 293/284 | 15/0 | 12/1 | 12/2 | 12(12/0) | 2072 | 1141 |
| 1Z1L(8 Å) | 345/338 | 23/0 | 15/0 | 15/0 | 13(13/0) | NA | - |
| T1029(8 Å) | 125/125 | 6/4 | 3/5 | 6/4 | 7(3/4) | 437 | 117 |
| T1031(8 Å) | 95/95 | 4/3 | 3 */3 | 4/3 | 5(2/3) | 56 | 187 |
| T1033(8 Å) | 100/100 | 3/0 | 6/0 | 4/0 | 4(4/0) | 5 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, M.; He, J. Combining Cryo-EM Density Map and Residue Contact for Protein Secondary Structure Topologies. Molecules 2021, 26, 7049. https://doi.org/10.3390/molecules26227049
Alshammari M, He J. Combining Cryo-EM Density Map and Residue Contact for Protein Secondary Structure Topologies. Molecules. 2021; 26(22):7049. https://doi.org/10.3390/molecules26227049
Chicago/Turabian StyleAlshammari, Maytha, and Jing He. 2021. "Combining Cryo-EM Density Map and Residue Contact for Protein Secondary Structure Topologies" Molecules 26, no. 22: 7049. https://doi.org/10.3390/molecules26227049
APA StyleAlshammari, M., & He, J. (2021). Combining Cryo-EM Density Map and Residue Contact for Protein Secondary Structure Topologies. Molecules, 26(22), 7049. https://doi.org/10.3390/molecules26227049