
Radical and Ionic Mechanisms in Rearrangements of o-Tolyl Aryl Ethers and Amines Initiated by the Grubbs–Stoltz Reagent, Et3SiH/KOtBu



Abstract
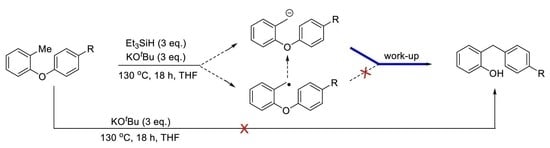
:
1. Introduction
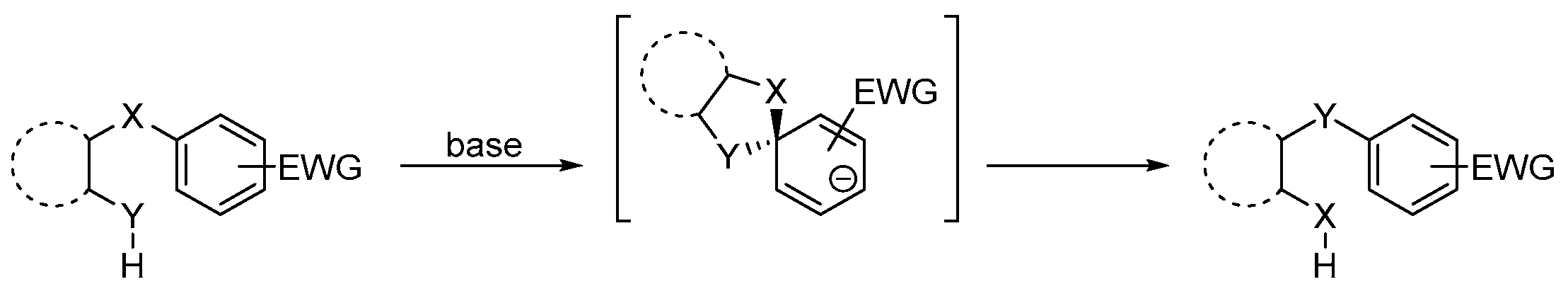
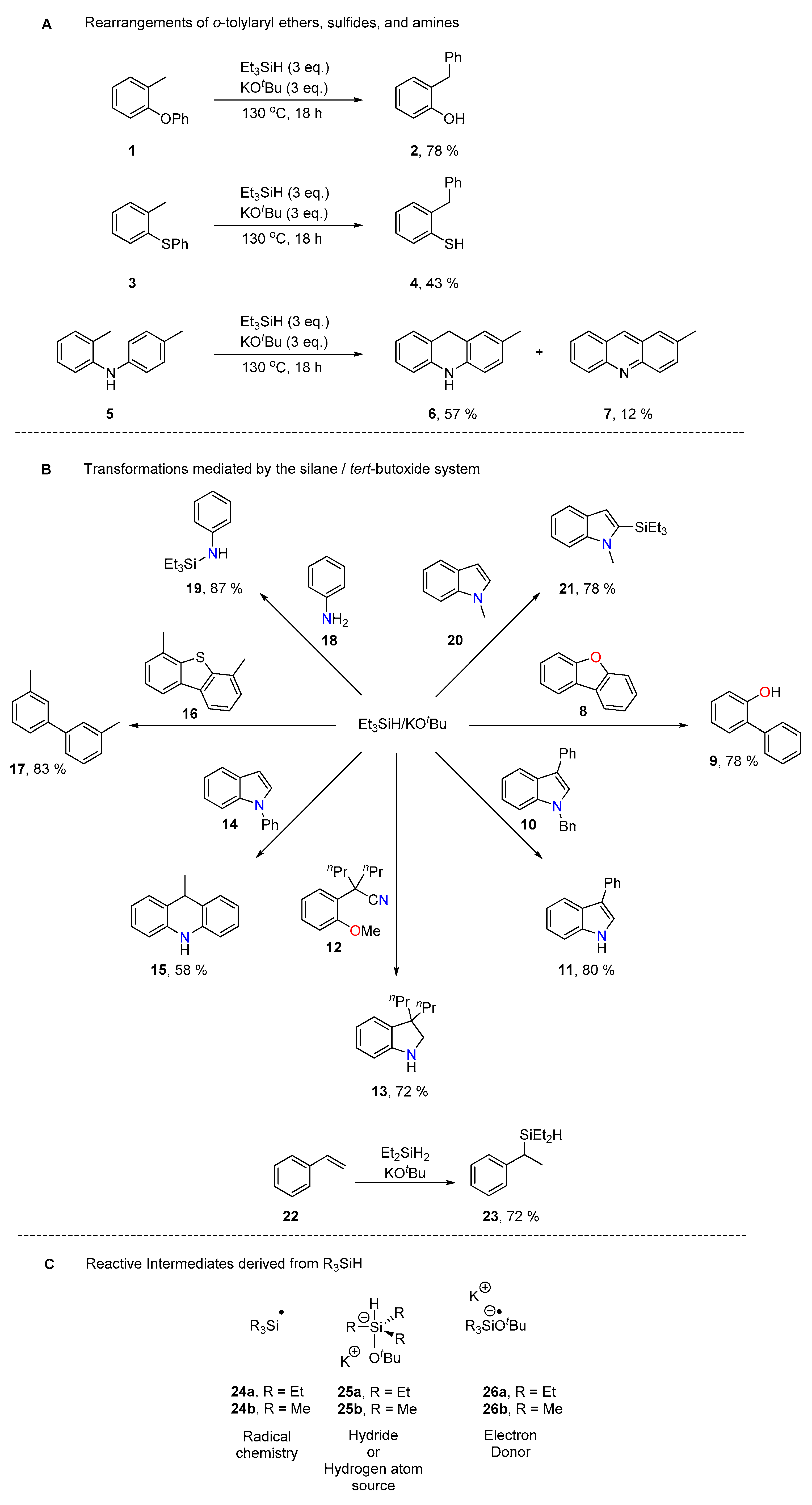
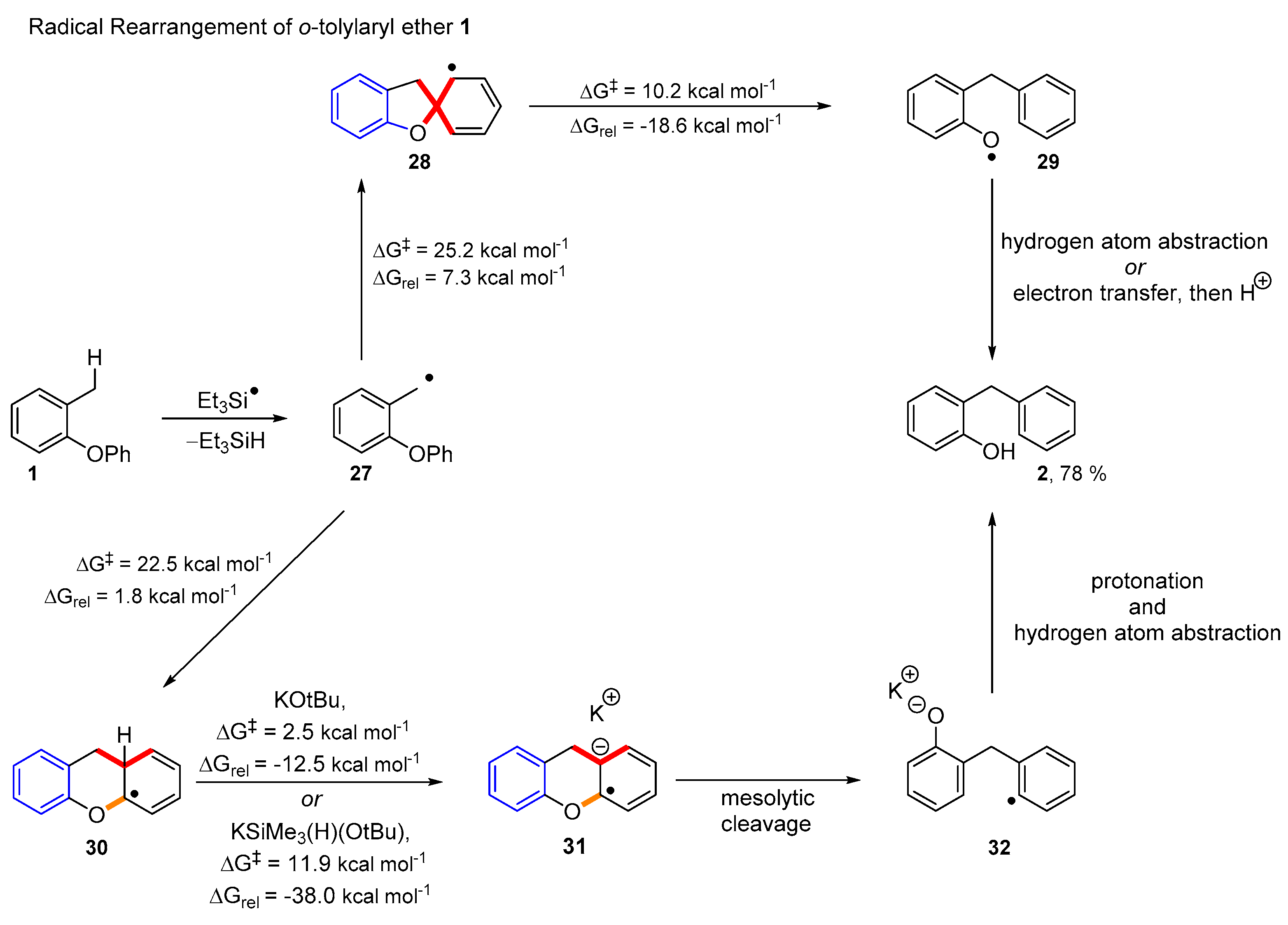
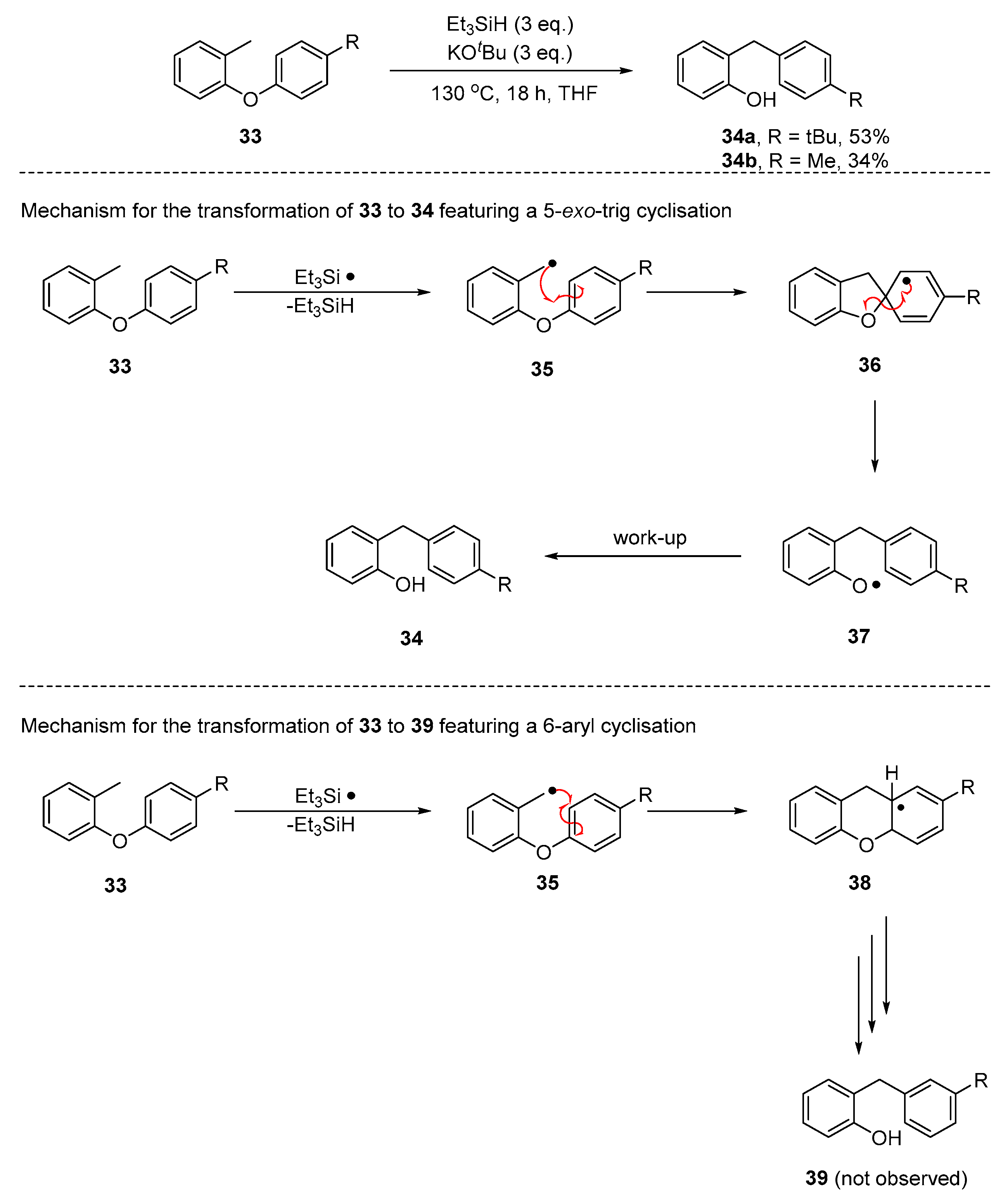
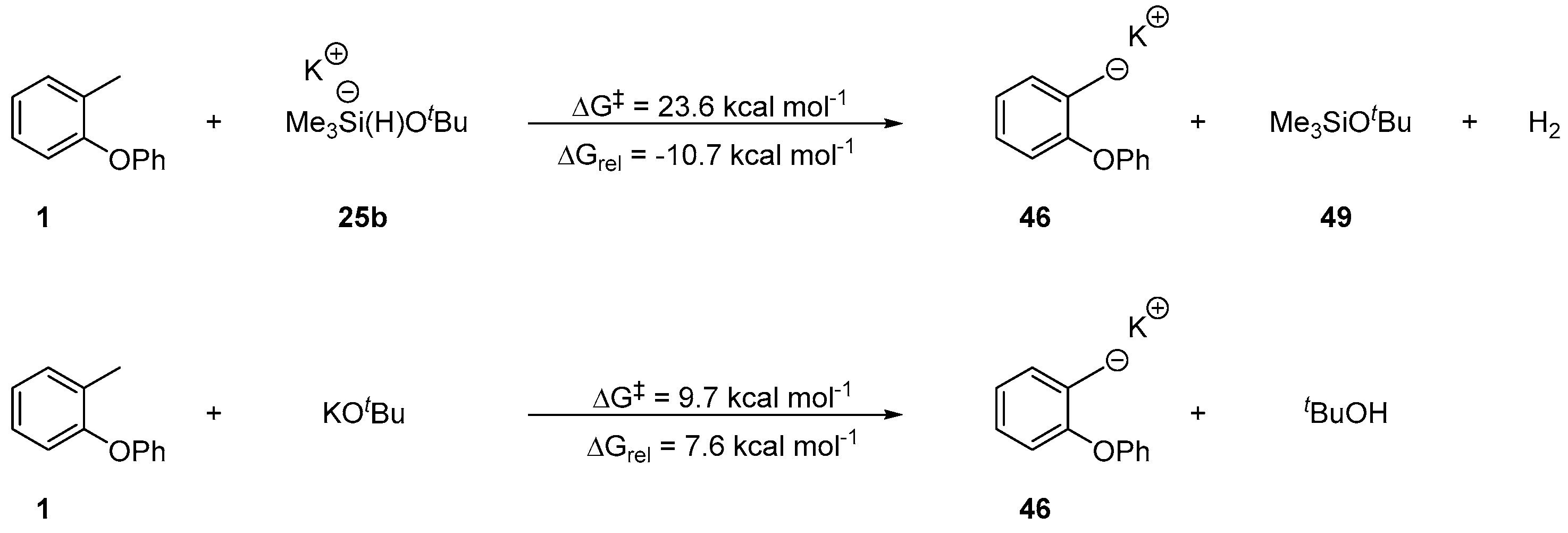
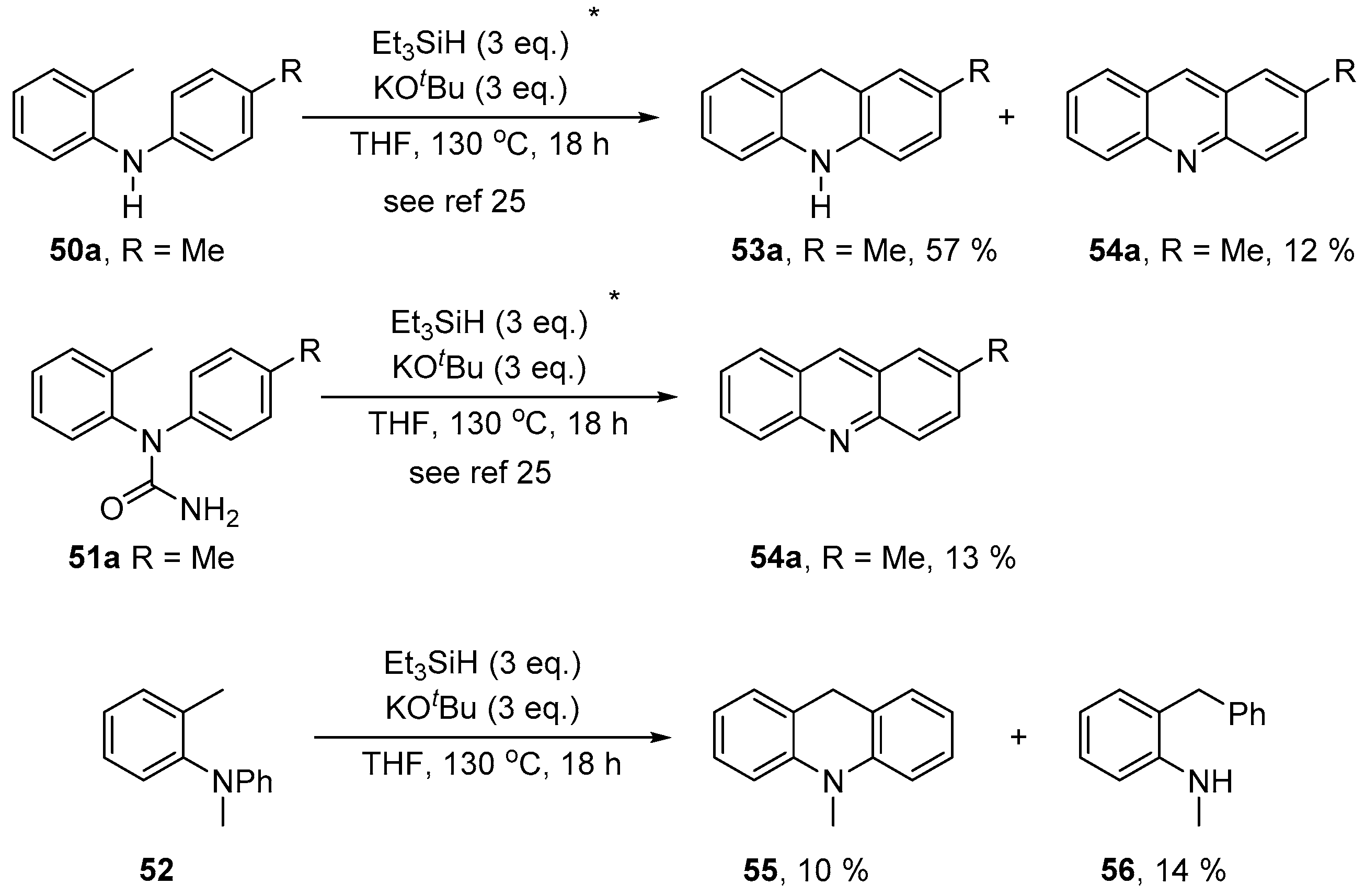
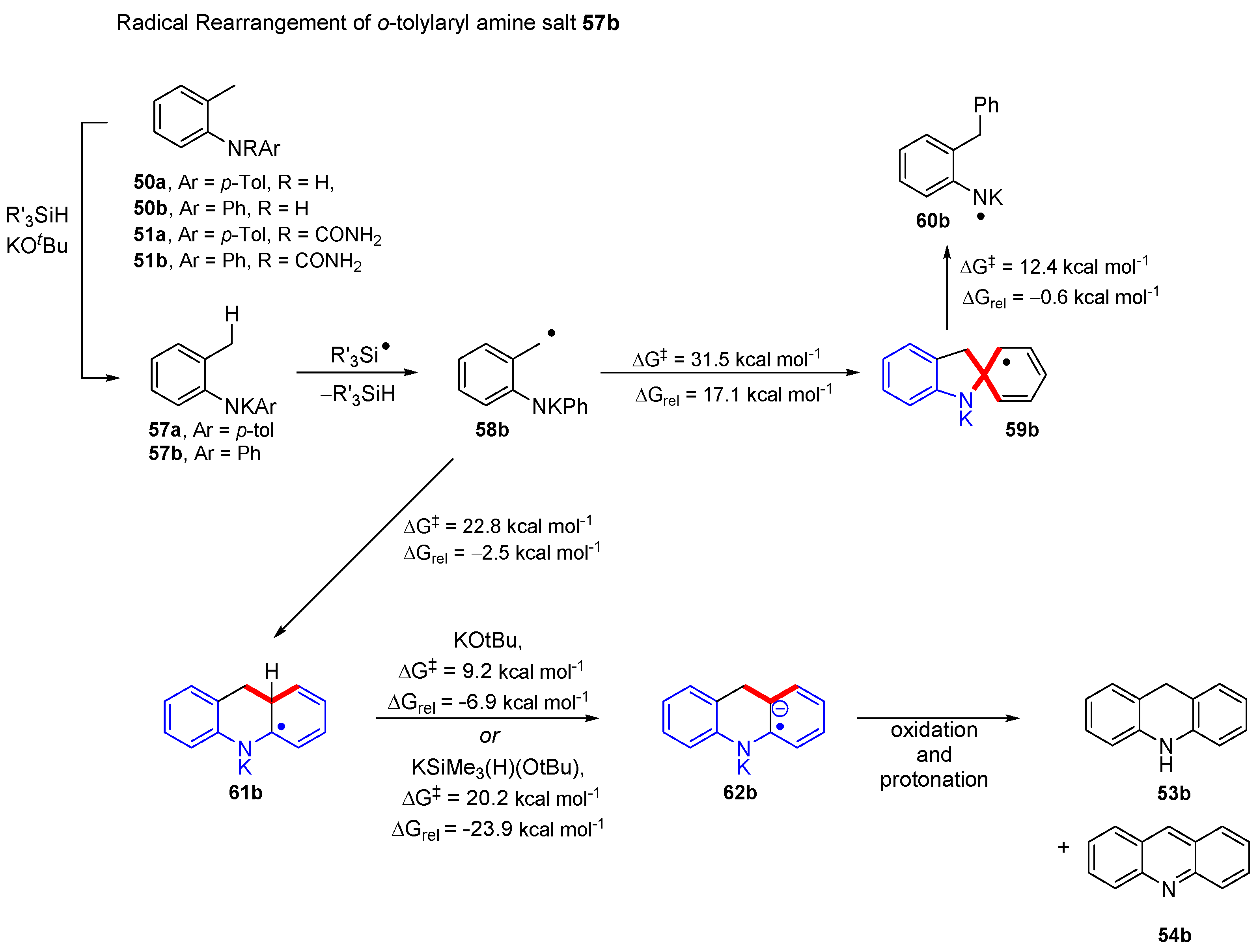
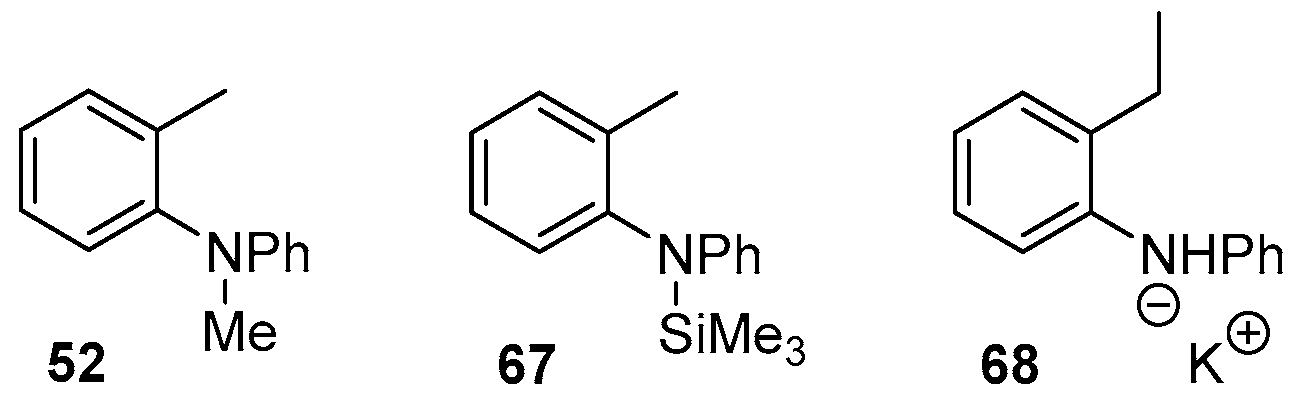
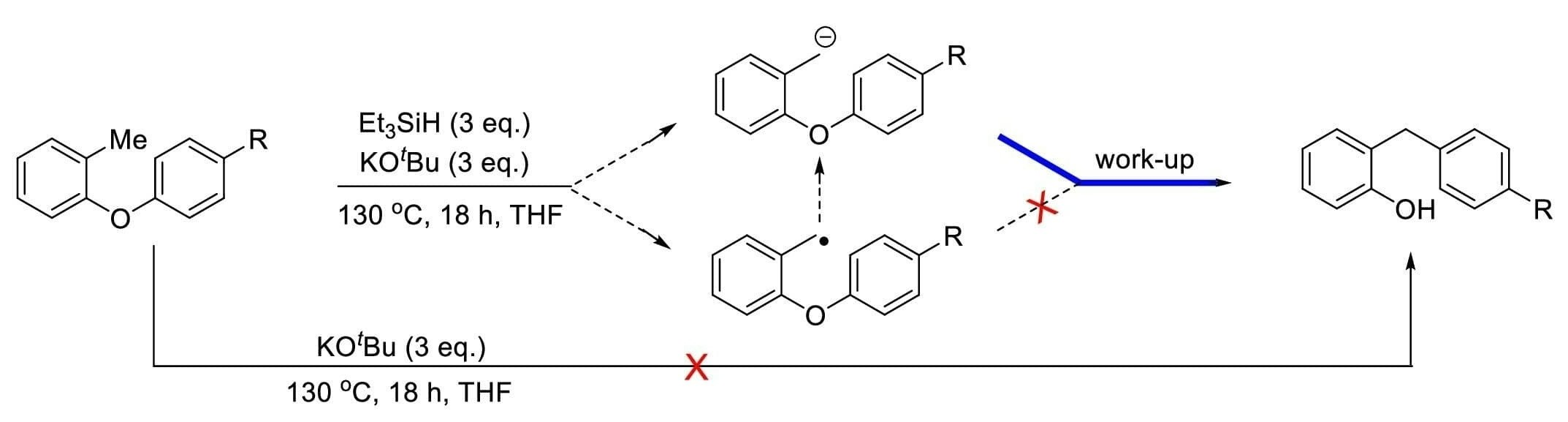
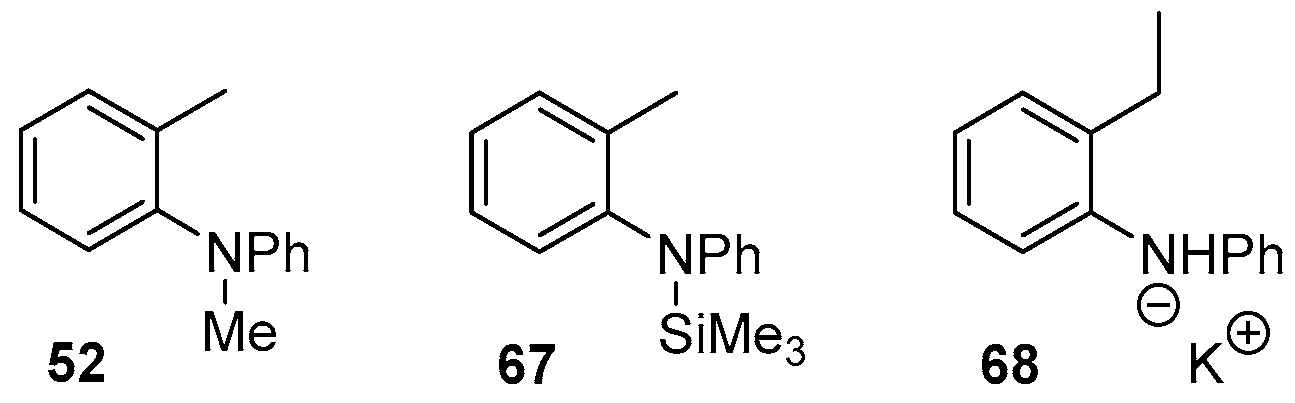
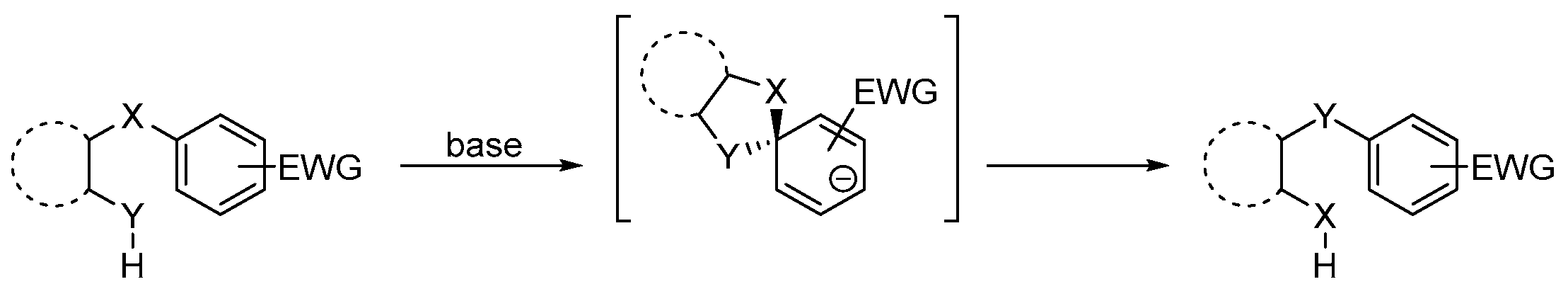
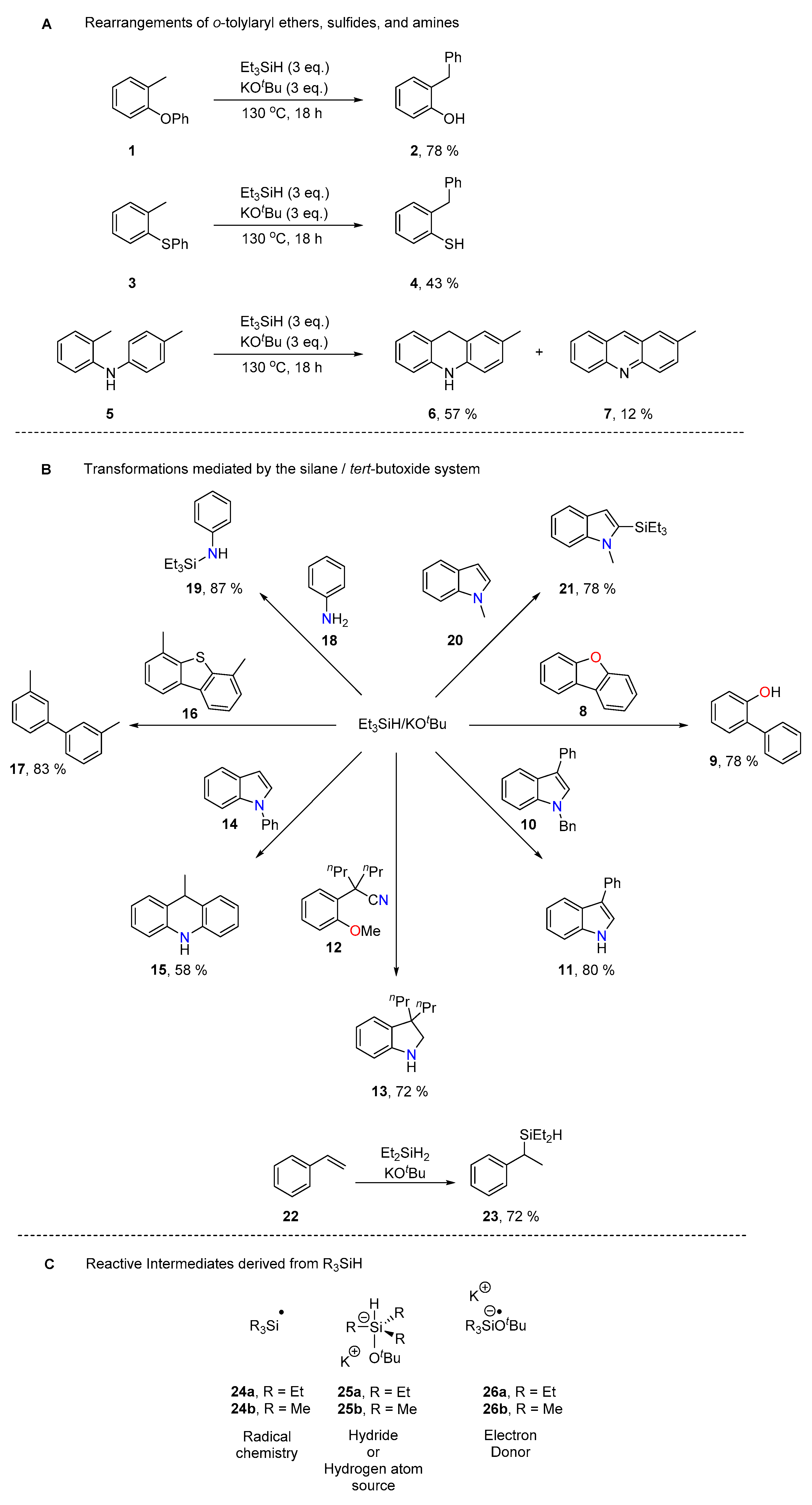
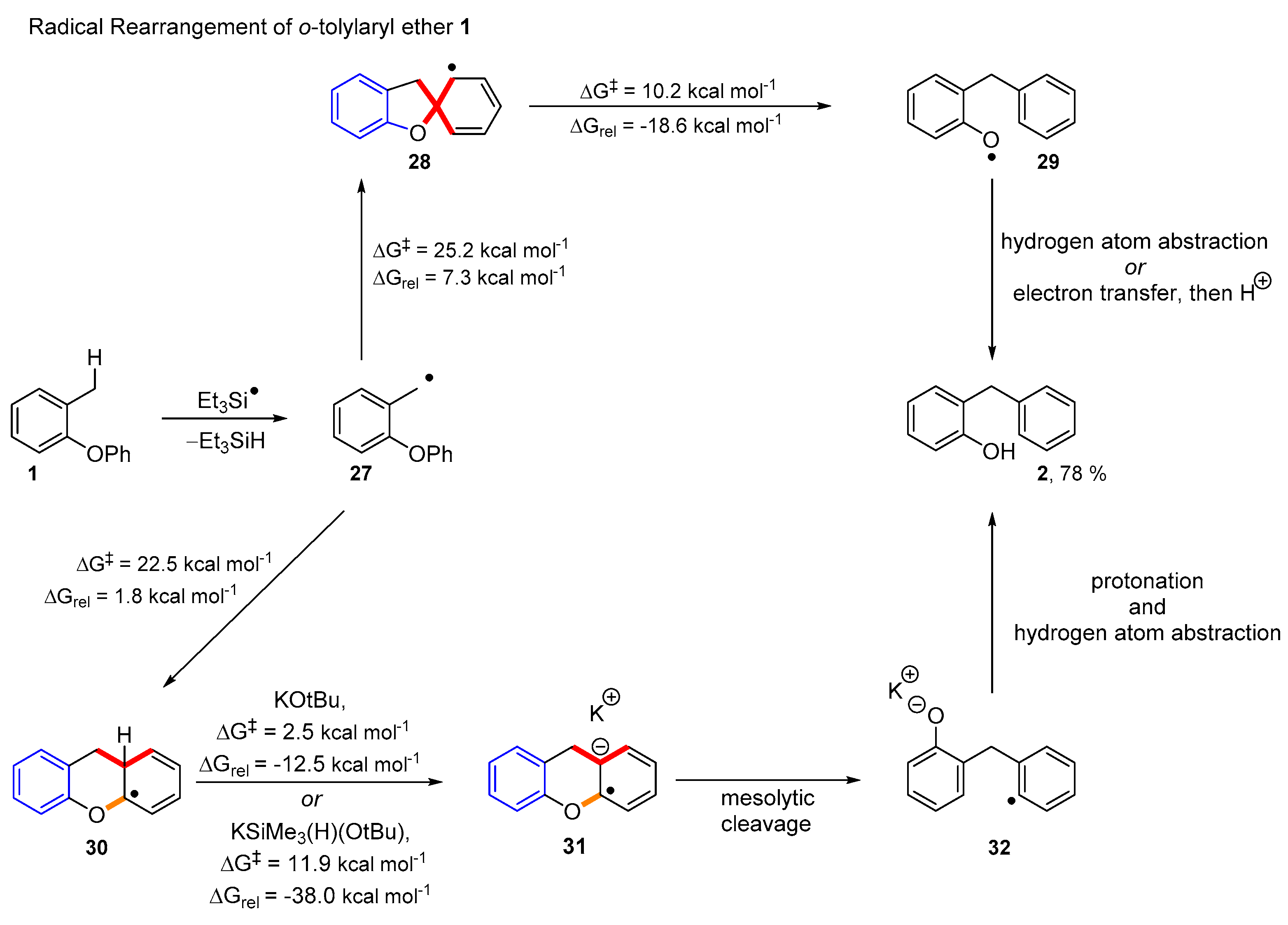
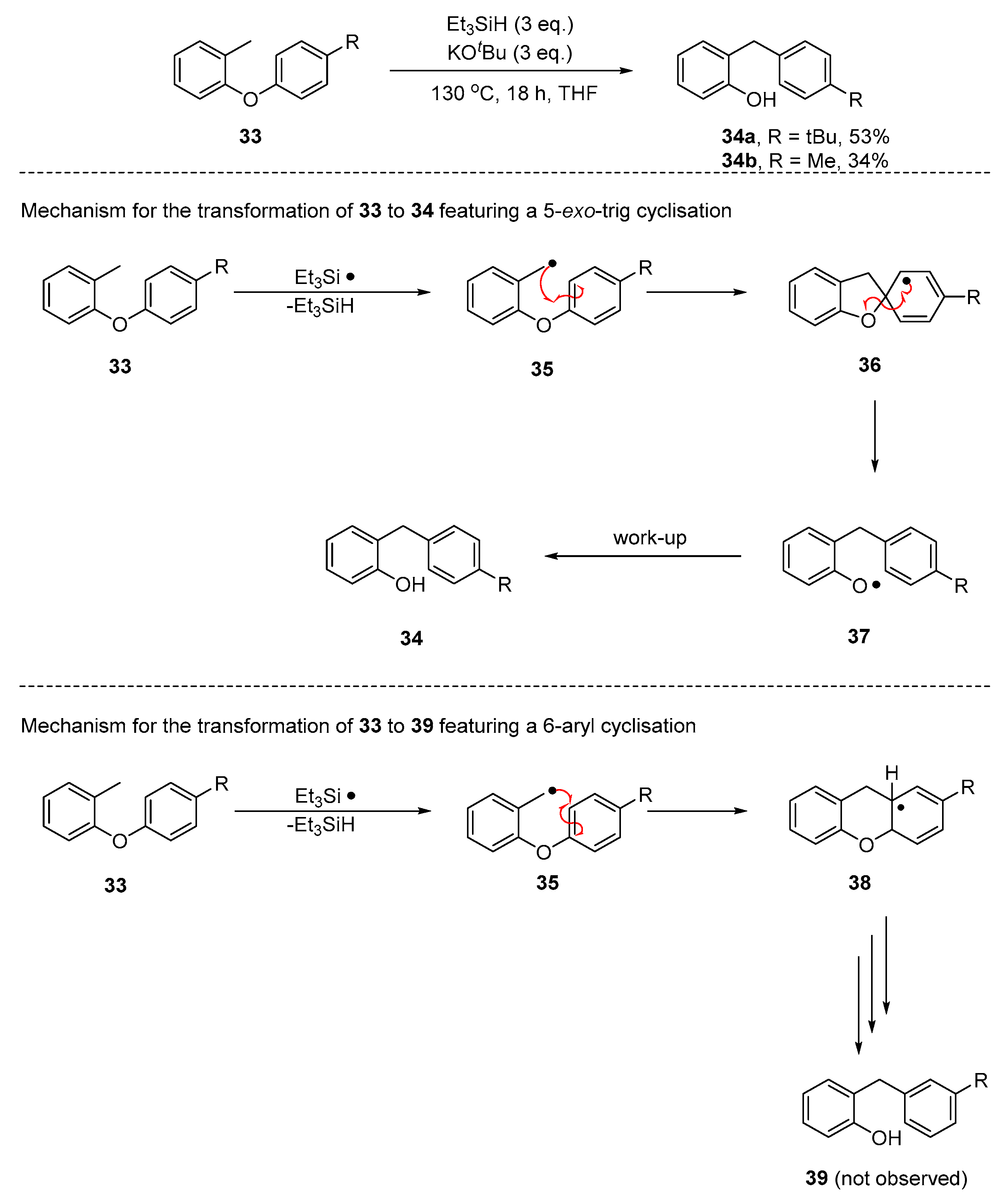
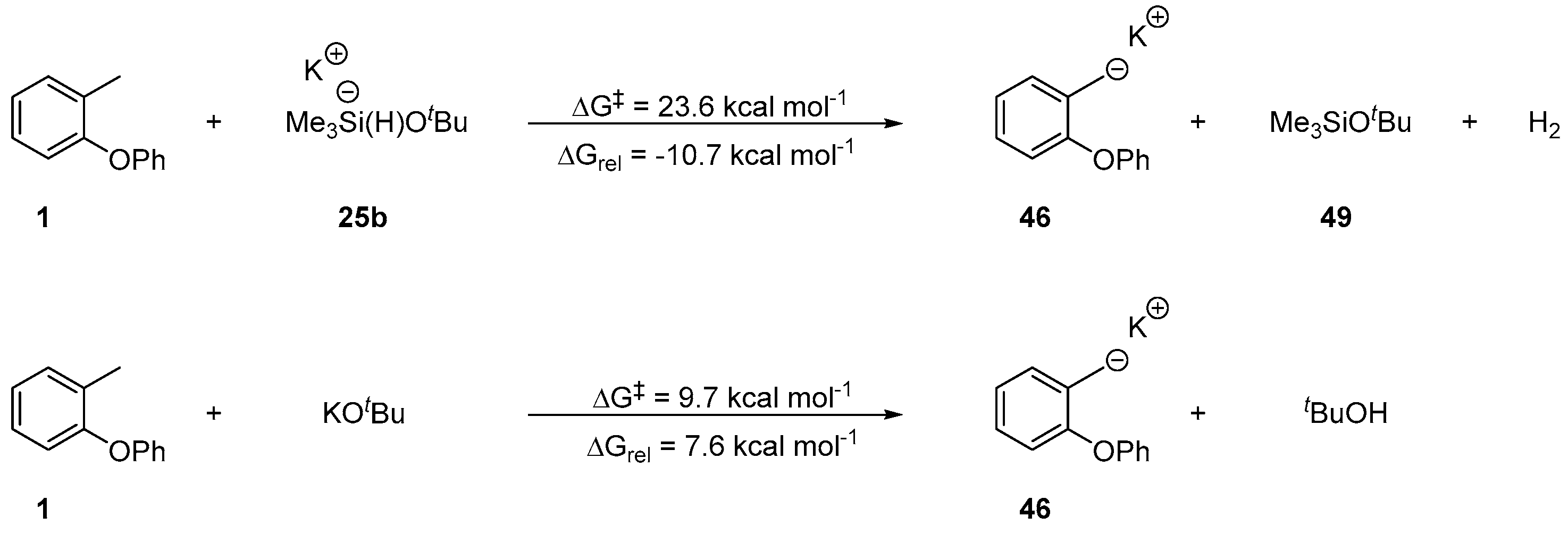
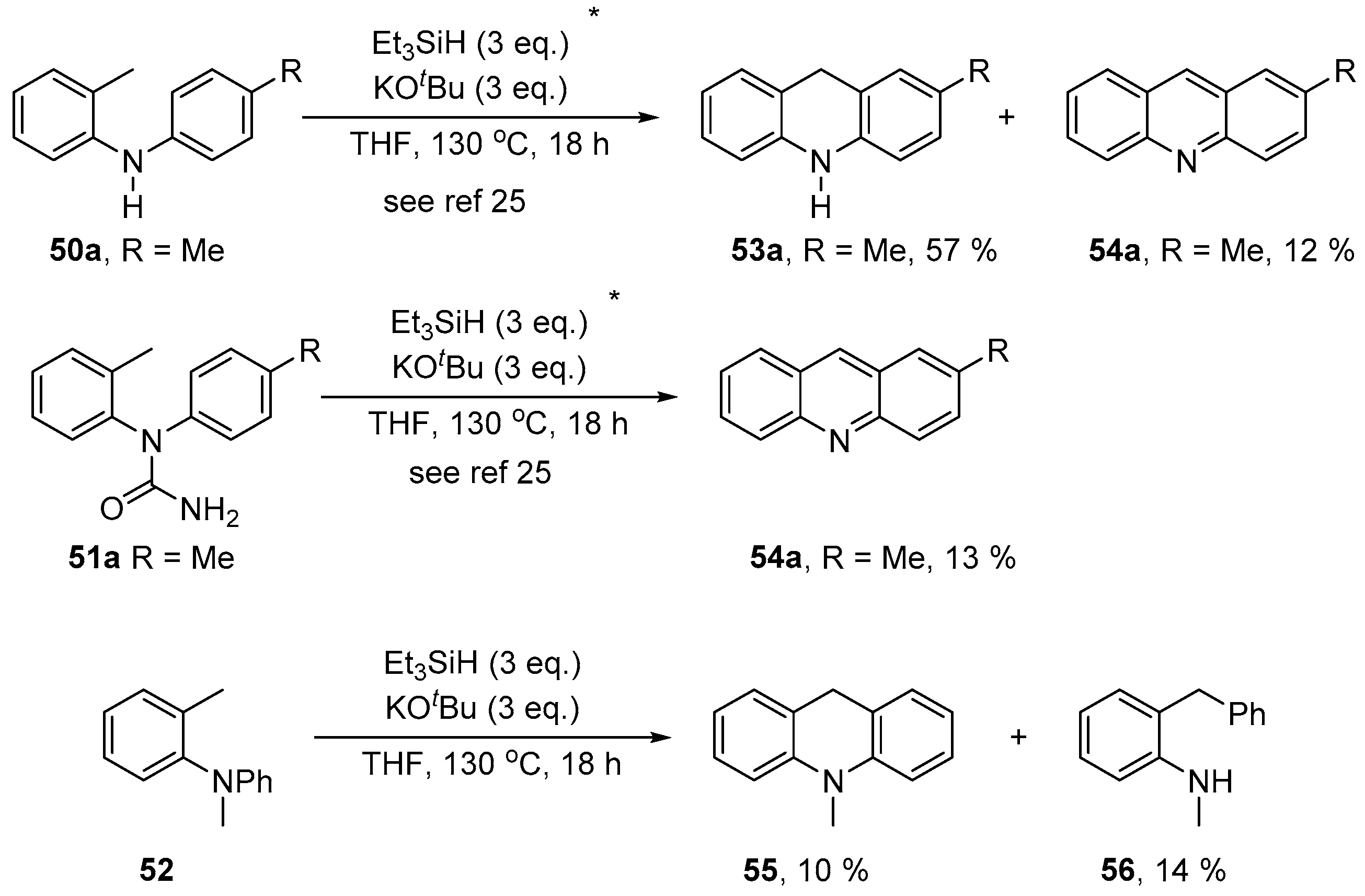
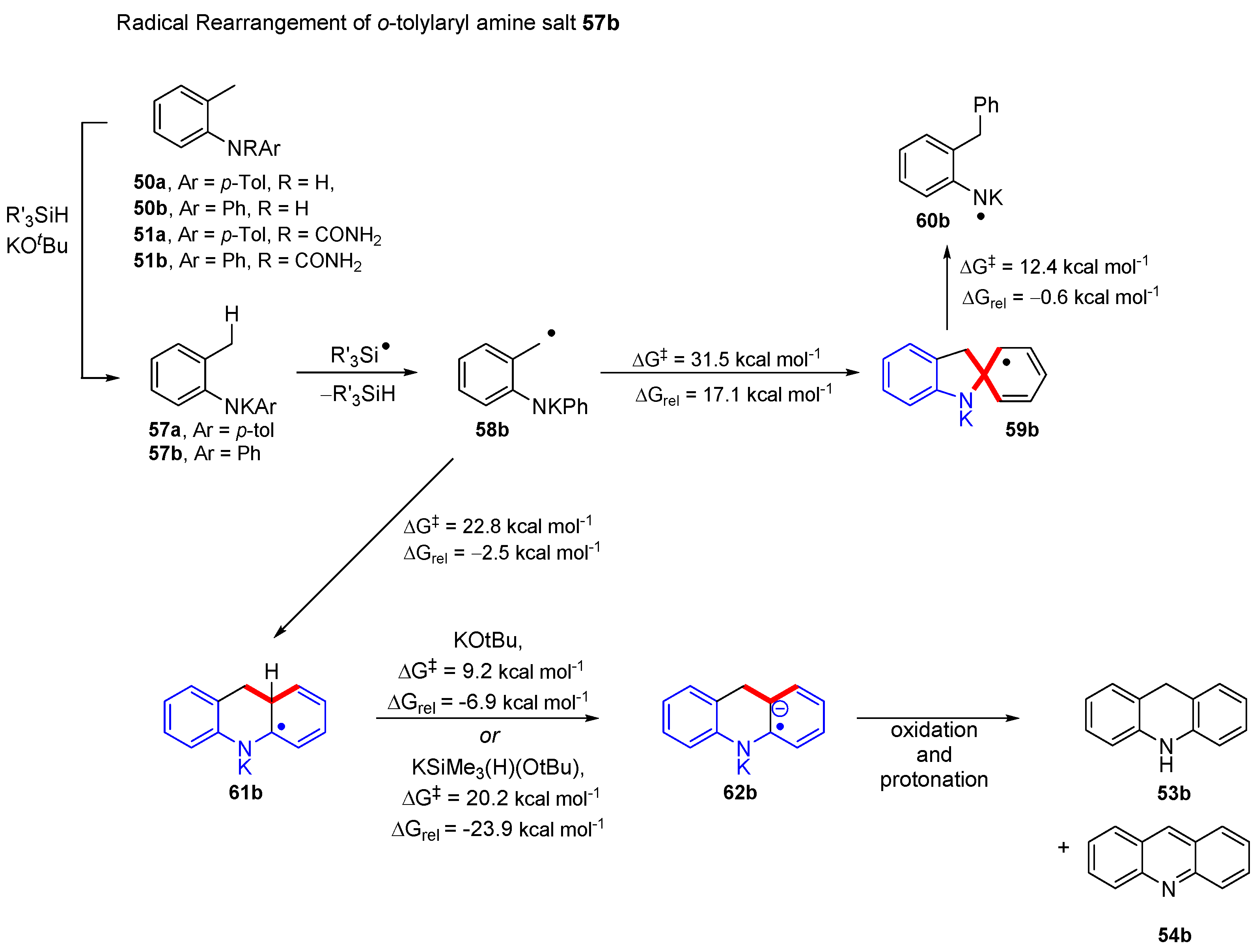
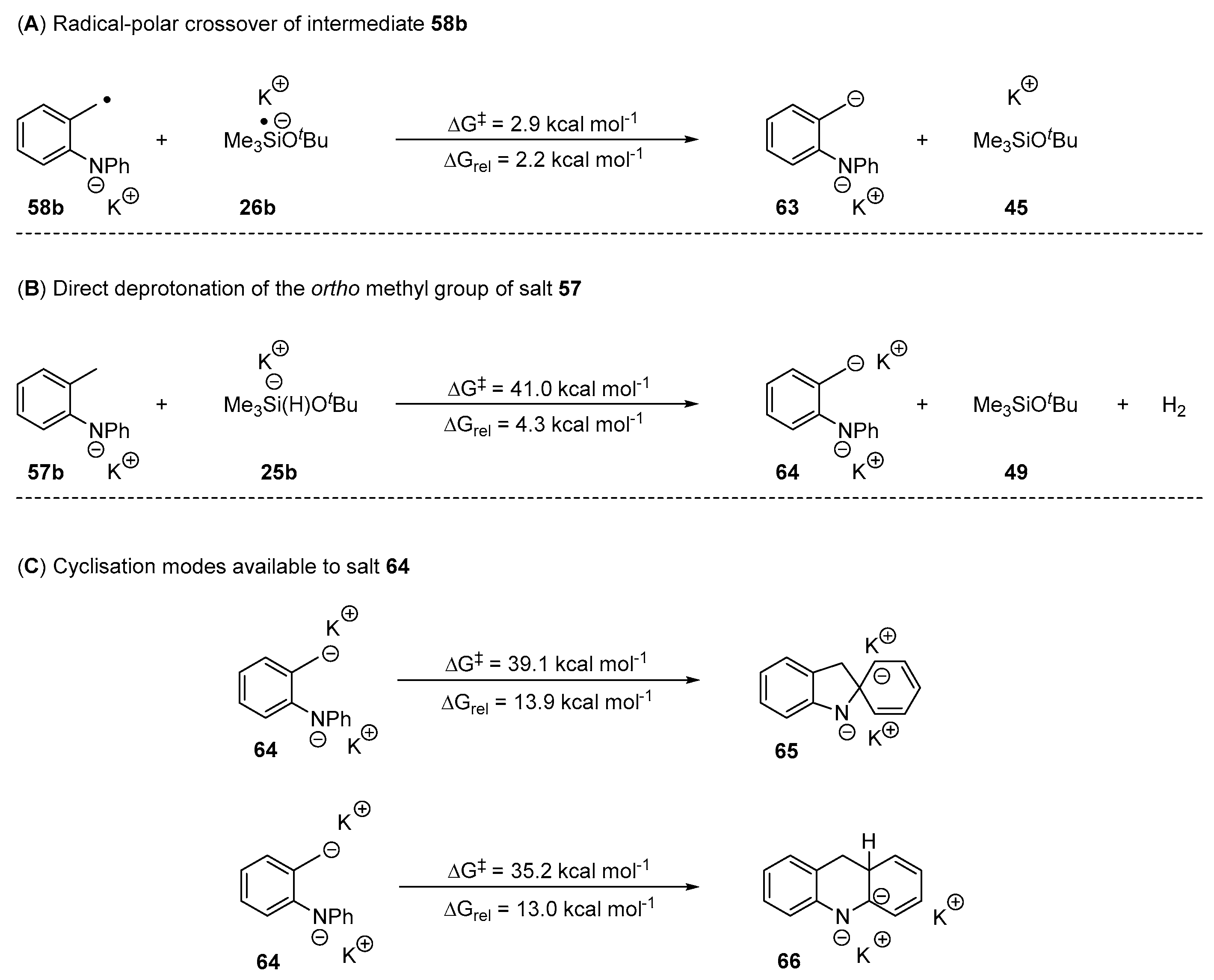
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Details
3.2. Substrate Synthesis
3.2.1. Preparation of 1-methyl-2-phenoxybenzene (1)

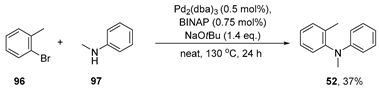
3.2.2. Preparation of N,2-dimethyl-N-phenylaniline (52)
3.3. Reactions of Substrates
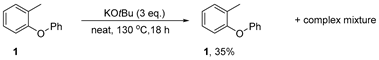
3.3.1. Reaction of 1-methyl-2-phenoxybenzene 1 with KOtBu—Neat
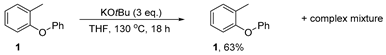
3.3.2. Reaction of 1-methyl-2-phenoxybenzene 1 with KOtBu—in THF
3.3.3. Reaction of N,2-dimethyl-N-phenylaniline 52 with KOtBu/Et3SiH—in THF
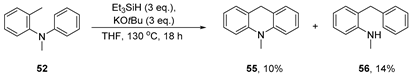
3.3.4. Reaction of N,2-dimethyl-N-phenylaniline 52 with KOtBu/Et3SiH—Neat
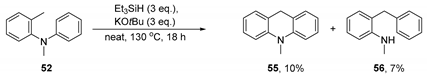
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wieland, H. Über Triphenylmethyl-peroxyd, Ein Beitrag zur Chemie der freien Radikale. Chem. Ber. 1911, 44, 2550. [Google Scholar] [CrossRef] [Green Version]
- Studer, A.; Bossart, M. Radical aryl migration reactions. Tetrahedron 2001, 57, 9649–9667. [Google Scholar] [CrossRef]
- Chen, Z.-M.; Zhang, X.-M.; Tu, Y.Q. Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 2015, 44, 5220–5245. [Google Scholar] [CrossRef]
- Warren, L.A.; Samuel, S. CLXXI.—Dehydro-2-naphtholsulphone. J. Chem. Soc. 1930, 1327–1331. [Google Scholar] [CrossRef]
- Warren, W.A.; Smiles, S. CXXII.—The Conversion of iso-β-Naphthol Sulphide into 2-Naphthol 1-Sulphide. J. Chem. Soc. 1931, 914–922. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Zahler, R.E. Aromatic nucleophilic substitution reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Truce, E.W.; Kreider, M.E.; Brand, W.W. The Smiles and Related Rearrangements of Aromatic Systems. Org. React. 1970, 18, 99–215. [Google Scholar]
- Makosza, M. Nucleophilic substitution in nitroarenes: A general corrected mechanism. ChemTexts 2019, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Holden, C.M.; Greaney, M.F. Modern Aspects of the Smiles Rearrangement. Chem. Eur. J. 2017, 23, 8992–9008. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Seayad, J.; Greaney, M.F. Truce–Smiles Rearrangements by Strain Release: Harnessing Primary Alkyl Radicals for Metal-Free Arylation. Angew. Chem. Int. Ed. 2021, 60, 22219–22223. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Duong, H.A.; Greaney, M.F. A visible light-mediated, decarboxylative, desulfonylative Smiles rearrangement for general arylethylamine syntheses. Chem. Commun. 2020, 56, 11493–11496. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Duong, H.A.; Greaney, M.F. Alkene Carboarylation through Catalyst-Free, Visible Light-Mediated Smiles Rearrangement. Chem. Eur. J. 2019, 25, 1927–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayles, R.; Johnson, M.C.; Maisey, R.F.; Turner, R.W. A Smiles Rearrangement Involving Non-Activated Aromatic Systems; the Facile Conversion of Phenols to Anilines. Synthesis 1977, 33–34. Available online: https://pascal-francis.inist.fr/vibad/index.php?action=getRecordDetail&idt=PASCAL7760286965 (accessed on 13 October 2021). [CrossRef]
- Coutts, I.G.C.; Southcott, M.R. The conversion of phenols to primary and secondary aromatic amines via a Smiles rearrangement. J. Chem. Soc. Perkin Trans. 1 1990, 767–771. [Google Scholar] [CrossRef]
- Ten Hoeve, W.; Kruse, C.G.; Luteyn, J.M.; Thiecke, J.R.G.; Wynberg, H. Direct Substitution of Aromatic Ethers by Lithium Amides. A New Aromatic Amination Reaction. J. Org. Chem. 1993, 58, 5101–5106. [Google Scholar] [CrossRef]
- Bonini, C.; Cristiani, G.; Funicello, M.; Viggiani, L. Facile entry to 4- and 5-hydroxybenzofuran and to their amino derivatives. Synth. Commun. 2006, 36, 1983–1990. [Google Scholar] [CrossRef]
- Snape, T.J. A truce on the Smiles rearrangement: Revisiting an old reaction–the Truce–Smiles rearrangement. Chem. Soc. Rev. 2008, 37, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Monos, T.M.; McAtee, R.C.; Stephenson, C.R.J. Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation. Science 2018, 361, 1369–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.A.; Dominey, A.P.; Williams, G.D.; Murphy, J.A. Visible light-mediated Smiles rearrangements and annulations of non-activated aromatics. Chem. Commun. 2020, 56, 11445–11448. [Google Scholar] [CrossRef] [PubMed]
- Loven, R.; Speckamp, W.N. A Novel 1,4 arylradical rearrangement. Tetrahedron Lett. 1972, 1567–1570. [Google Scholar] [CrossRef]
- Köhler, J.J.; Speckamp, W.N. Intramolecular Radical Reactions in α-halomethyl substituted piperidine sulfonamides. Tetrahedron Lett. 1977, 631–634. [Google Scholar] [CrossRef]
- Motherwell, W.B.; Pennell, A.M.K. A novel route to biaryls via intramolecular free radical ipso substitution reactions. J. Chem. Soc. Chem. Commun. 1991, 877–879. [Google Scholar] [CrossRef]
- Da Mata, M.L.E.N.; Motherwell, W.B.; Ujjainwalla, F. Steric and electronic effects in the synthesis of biaryls and their heterocyclic congeners using intramolecular free radical [1,5] ipso substitution reactions. Tetrahedron Lett. 1997, 38, 137–140. [Google Scholar] [CrossRef]
- Kong, W.; Casimiro, M.; Merino, E.; Nevado, C. Copper-catalyzed one-Pot trifluoromethylation/aryl migration/desulfonylation and C(sp2)-N bond formation of conjugated tosyl amides. J. Am. Chem. Soc. 2013, 135, 14480–14483. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Merino, E.; Nevado, C. Arylphosphonylation and arylazidation of activated alkenes. Angew. Chem. Int. Ed. 2014, 53, 5078–5082. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Kong, W.; Fernández-Sánchez, L.; Merino, E.; Nevado, C. Cyclization cascades via N-amidyl radicals toward highly functionalized heterocyclic scaffolds. J. Am. Chem. Soc. 2015, 137, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.J.; Albright, H.; Sevrin, M.J.; Cole, K.P.; Stephenson, C.R.J. A visible-light-mediated radical Smiles rearrangement and its application to the synthesis of a difluoro-substituted spirocyclic ORL-1 antagonist. Angew. Chem. Int. Ed. 2015, 54, 14898–14902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khartabil, H.; Doudet, L.; Allart-Simon, I.; Ponce-Vargas, M.; Gérard, S.; Hénon, E. Mechanistic insights into Smiles rearrangement. Focus on π–π stacking interactions along the radical cascade. Org. Biomol. Chem. 2020, 18, 6840–6848. [Google Scholar] [CrossRef]
- Arokianathar, J.N.; Kolodziejczak, K.; Bugden, F.E.; Clark, K.F.; Tuttle, T.; Murphy, J.A. Benzylic C−H Functionalisation by [Et3SiH+KOtBu] leads to Radical Rearrangements in o-tolyl Aryl Ethers, Amines and Sulfides. Adv. Synth. Catal. 2020, 362, 2260–2267. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, A.; Toutov, A.A.; Swisher, N.A.; Grubbs, R.H. Lewis-base silane activation: From reductive cleavage of aryl ethers to selective ortho-silylation. Chem. Sci. 2013, 4, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Toutov, A.A.; Salata, M.; Fedorov, A.; Yang, Y.F.; Liang, Y.; Cariou, R.; Betz, K.N.; Couzijn, E.P.A.; Shabaker, J.W.; Houk, K.N.; et al. A potassium tert-butoxide and hydrosilane system for ultra-deep desulfurization of fuels. Nat. Energy 2017, 2, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.B.; Schuman, D.P.; Yang, Y.F.; Toutov, A.A.; Liang, Y.; Klare, H.F.T.; Nesnas, N.; Oestreich, M.; Blackmond, D.G.; Virgil, S.C.; et al. Potassium tert-Butoxide-Catalyzed Dehydrogenative C-H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study. J. Am. Chem. Soc. 2017, 139, 6867–6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Yang, Y.F.; Jenkins, I.D.; Liang, Y.; Toutov, A.A.; Liu, W.B.; Schuman, D.P.; Grubbs, R.H.; Stoltz, B.M.; Krenske, E.H.; et al. Ionic and Neutral Mechanisms for C-H Bond Silylation of Aromatic Heterocycles Catalyzed by Potassium tert-Butoxide. J. Am. Chem. Soc. 2017, 139, 6880–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, P.; Hua, Y.; Bokka, A.; Thiamsiri, C.; Prasitwatcharakorn, W.; Karedath, A.; Chen, X.; Sardar, S.; Yum, K.; Leem, G.; et al. Catalytic hydrogen atom transfer from hydrosilanes to vinylarenes for hydrosilylation and polymerization. Nat. Catal. 2019, 2, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Young, A.; Rohrbach, S.; O’Connor, E.F.; Allison, M.; Wang, H.S.; Poole, D.L.; Tuttle, T.; Murphy, J.A. Electron-Transfer and Hydride-Transfer Pathways in the Stoltz–Grubbs Reducing System (KOtBu/Et3SiH). Angew. Chem. Int. Ed. 2017, 56, 13747–13751. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, F.; Rohrbach, S.; Tuttle, T.; Murphy, J.A. N-silylation of amines mediated by Et3SiH/KOtBu. Helv. Chim. Acta. 2019, 102, e1900235. [Google Scholar] [CrossRef]
- Smith, A.J.; Dimitrova, D.; Arokianathar, J.N.; Clark, K.F.; Poole, D.L.; Leach, S.G.; Murphy, J.A. Et3SiH + KOtBu provide multiple reactive intermediates that compete in the reactions and rearrangements of benzylnitriles and indolenines. Chem. Sci. 2020, 11, 12364–12370. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Dimitrova, D.; Arokianathar, J.N.; Kolodziejczak, K.; Young, A.; Allison, M.; Poole, D.L.; Leach, S.G.; Parkinson, J.A.; Tuttle, T.; et al. New reductive rearrangement of: N-arylindoles triggered by the Grubbs–Stoltz reagent Et3SiH/KOtBu. Chem. Sci. 2020, 11, 3719–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, K.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital stud-ies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Andersson, M.P.; Uvdal, P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis Set 6-311+G.(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Altshuller, A.P.; Rosenblum, L. Dielectric Properties of Some Alkylsilanes. J. Am. Chem. Soc. 1955, 77, 272–274. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: www.gaussian.com (accessed on 13 October 2021).
- Studer, A.; Curran, D.P. Organocatalysis and C-H activation meet radical- and electron-transfer reactions. Angew. Chem. Int. Ed. 2011, 50, 5018–5022. [Google Scholar] [CrossRef]
- Dub, P.A.; Henson, N.J.; Martin, R.L.; Gordon, J.C. Unravelling the mechanism of the asymmetric hydrogenation of acetophenone by [RuX2(diphosphine)(1,2-diamine)] catalysts. J. Am. Chem. Soc. 2014, 136, 3505–3521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sha, S.-C.; Bellomo, A.; Trongsiriwat, N.; Gao, F.; Tomson, N.C.; Walsh, P.J. Positional Selectivity in C-H Functionalizations of 2-Benzylfurans with Bimetallic Catalysts. J. Am. Chem. Soc. 2016, 138, 4260–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaga, A.; Peng, X.; Hirao, H.; Chiba, S. Diastereo-Divergent Synthesis of Saturated Azaheterocycles Enabled by tBuOK-Mediated Hydroamination of Alkenyl Hydrazones. Chem. Eur. J. 2015, 21, 19112–19118. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Tong, B.M.K.; Hirao, H.; Chiba, S. Inorganic-base-mediated hydroamination of alkenyl oximes for the synthesis of cyclic nitrones. Angew. Chem. Int. Ed. 2014, 53, 1959–1962. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Cation-π Interactions in Organic Synthesis. Chem. Rev. 2018, 118, 11353–11432. [Google Scholar] [CrossRef] [PubMed]
- Roth, H.G.; Roth, H.G.; Romero, N.A.; Nicewicz, D.A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Sim, B.A.; Griller, D.; Wayner, D.D.M. Reduction Potential for Substituted Benzyl Radicals: PKa values for the Corresponding Toluenes. J. Am. Chem. Soc. 1989, 111, 754–755. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of Inner Shell Marcus Terms for Amino Nitrogen Compounds by Molecular Orbital Calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Costil, R.; Dale, H.J.A.; Fey, N.; Whitcombe, G.; Matlock, J.V.; Clayden, J. Heavily Substituted Atropisomeric Diarylamines by Unactivated Smiles Rearrangement of N-Aryl Anthranilamides. Angew. Chem. Int. Ed. 2017, 56, 12533–12537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costil, R.; Lefebvre, Q.; Clayden, J. Medium-Sized-Ring Analogues of Dibenzodiazepines by a Conformationally Induced Smiles Ring Expansion. Angew. Chem. Int. Ed. 2017, 56, 14602–14606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, D.J.; Ward, J.W.; Clayden, J. Asymmetric α-arylation of amino acids. Nature 2018, 562, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mas-Roselló, J.; Okoh, M.; Clayden, J. Enantioselectively functionalised phenytoin derivatives by auxiliary-directed N to C aryl migration in lithiated α-amino nitriles. Chem. Commun. 2018, 54, 10985–10988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, R.; Clayden, J. Photocatalytic Difunctionalization of Vinyl Ureas by Radical Addition Polar Truce–Smiles Rearrangement Cascades. Angew. Chem. Int. Ed. 2020, 59, 11600–11606. [Google Scholar] [CrossRef]
- Millward, M.J.; Ellis, E.; Ward, J.W.; Clayden, J. Hydantoin-bridged medium ring scaffolds by migratory insertion of urea-tethered nitrile anions into aromatic C–N bonds. Chem. Sci. 2021, 12, 2091–2096. [Google Scholar] [CrossRef]
- Abrams, R.; Jesani, M.H.; Browning, A.; Clayden, J. Triarylmethanes and their Medium-Ring Analogues by Unactivated Truce–Smiles Rearrangement of Benzanilides. Angew. Chem. Int. Ed. 2021, 60, 11272–11277. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, S.; Smith, A.J.; Pang, J.H.; Poole, D.L.; Tuttle, T.; Chiba, S.; Murphy, J.A. Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem. Int. Ed. 2019, 58, 16368–16388. [Google Scholar] [CrossRef] [Green Version]
- Legnani, L.; Porta, A.; Caramella, P.; Toma, L.; Zanoni, G.; Vidari, G. Computational mechanistic study of the Julia-Kocieński reaction. J. Org. Chem. 2015, 80, 3092–3100. [Google Scholar] [CrossRef]
- Nawaz, F.; Mohanan, K.; Charles, L.; Rajzmann, M.; Bonne, D.; Chuzel, O.; Rodriguez, J.; Coquerel, Y. Temporary intramolecular generation of pyridine carbenes in metal-free three-component C–H bond functionalisation/aryl-transfer reactions. Chem. Eur. J. 2013, 19, 17578–17583. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, A.; García, J.J. Bond Activation with Low-Valent Nickel in Homogeneous Systems. Eur. J. Inorg. Chem. 2010, 2010, 4063–4074. [Google Scholar] [CrossRef]
- Estimation of Approximate Figures for Rate Constants for Unimolecular Reactions with Particular Energy Barriers. Using This Tool, We Calculate a Ball-Park Figure of 3 × 10−2 s−1 for a Unimolecular Reaction with a Barrier of 26 kcal mol−1 Carried Out at 130 °C. Available online: https://www.unige.ch/sciences/chiorg/lacour/correl (accessed on 13 October 2021).
- Newcomb, M. Radical Kinetics and Clocks. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 1, Chapter 5; ISBN 9781119953678. [Google Scholar] [CrossRef]
- Crombie, L.; Wyvill, R.D. β-Halogeno ether synthesis of olefinic alcohols: Stereochemistry and conformation of 2-substituted 3-halogenotetrahydro-pyran and -furan precursors. J. Chem. Soc. Perkin Trans. 1985, 1, 1971–1981. [Google Scholar] [CrossRef]
- Murphy, J.A.; Khan, T.A.; Zhou, S.; Thomson, D.W.; Mahesh, M. Highly Efficient Reduction of Unactivated Aryl and Alkyl Iodides by a Ground-State Neutral Organic Electron Donor. Angew. Chem. Int. Ed. 2005, 44, 1356–1360. [Google Scholar] [CrossRef]
- Murphy, J.A.; Zhou, S.; Thomson, D.W.; Schoenebeck, F.; Mahesh, M.; Park, S.R.; Tuttle, T.; Berlouis, L.E.A. The Generation of Aryl Anions by Double Electron Transfer to Aryl Iodides from a Neutral Ground-State Organic Super-Electron Donor. Angew. Chem. Int. Ed. 2007, 46, 5178–5183. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An Improved Catalyst System for Aromatic Carbon−Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J Am Chem Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Weber, P.; Scherpf, T.; Rodstein, I.; Lichte, D.; Scharf, L.T.; Gooßen, L.J.; Gessner, V.H. A Highly Active Ylide-Functionalized Phosphine for Palladium-Catalyzed Aminations of Aryl Chlorides. Angew. Chem. Int. Ed. 2019, 58, 3203–3207. [Google Scholar] [CrossRef]
- Stopka, T.; Marzo, L.; Zurro, M.; Janich, S.; Würthwein, E.U.; Daniliuc, C.G.; Alemán, J.; Mancheño, O.G. Oxidative C-H Bond Functionalization and Ring Expansion with TMSCHN2: A Copper(I)-Catalyzed Approach to Dibenzoxepines and Dibenzoazepines. Angew. Chem. Int. Ed. 2015, 54, 5049–5053. [Google Scholar] [CrossRef] [PubMed]
- Creencia, E.C.; Taguchi, K.; Horaguchi, T. Thermal reactions of N-alkyl-2-benzylaniline and N-alkyl-N′-phenyl-o-phenylenediamine: An unusual route to 2-phenylindole and 2-phenylbenzimidazole. J. Heterocycl. Chem. 2008, 45, 837–843. [Google Scholar] [CrossRef]
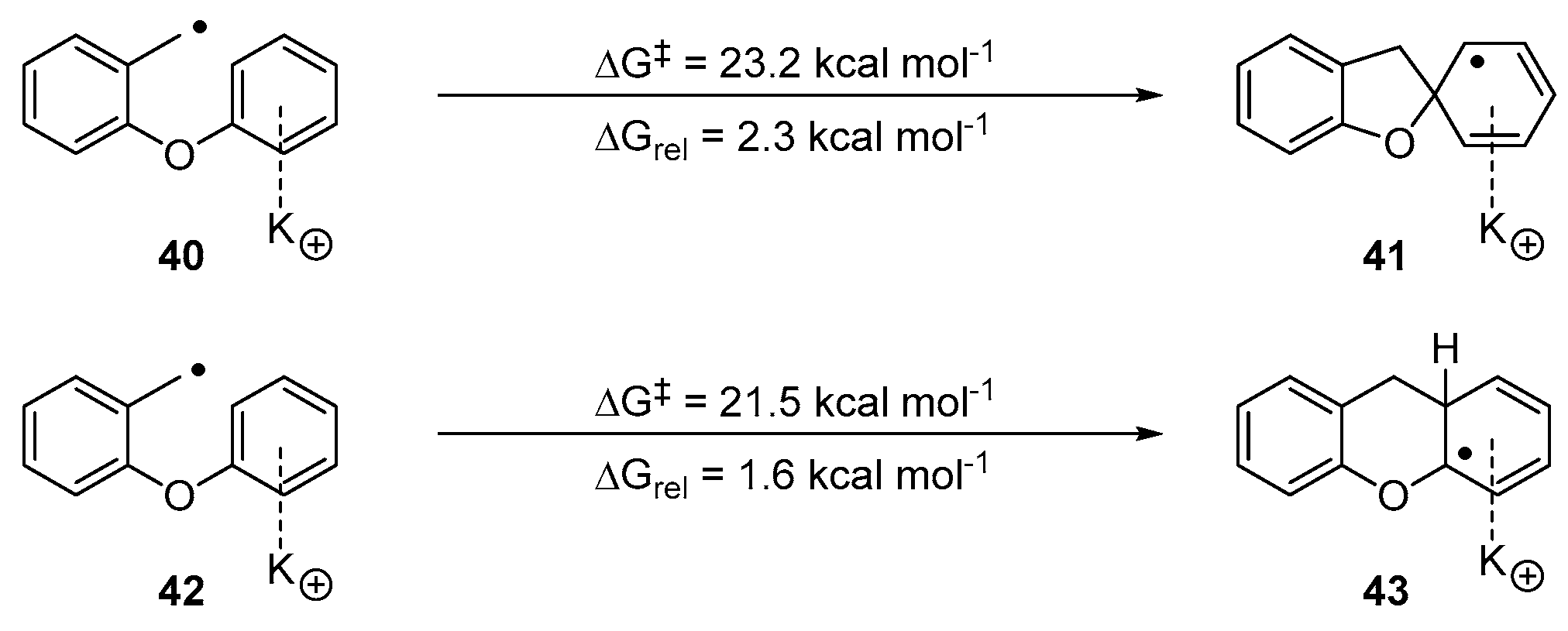
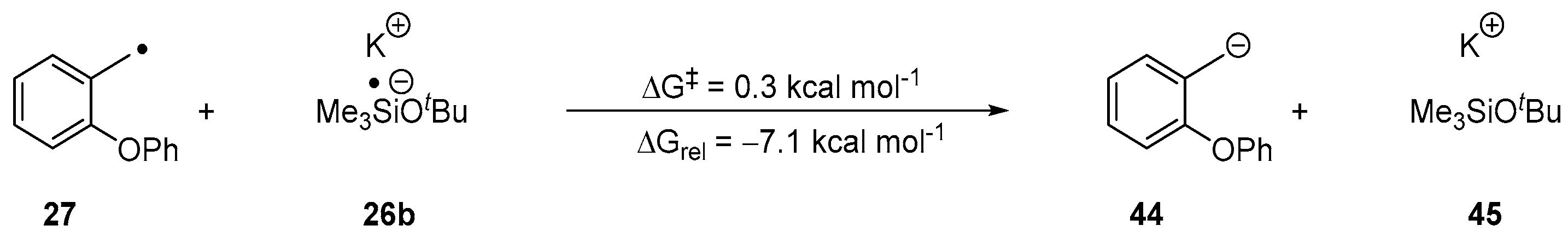
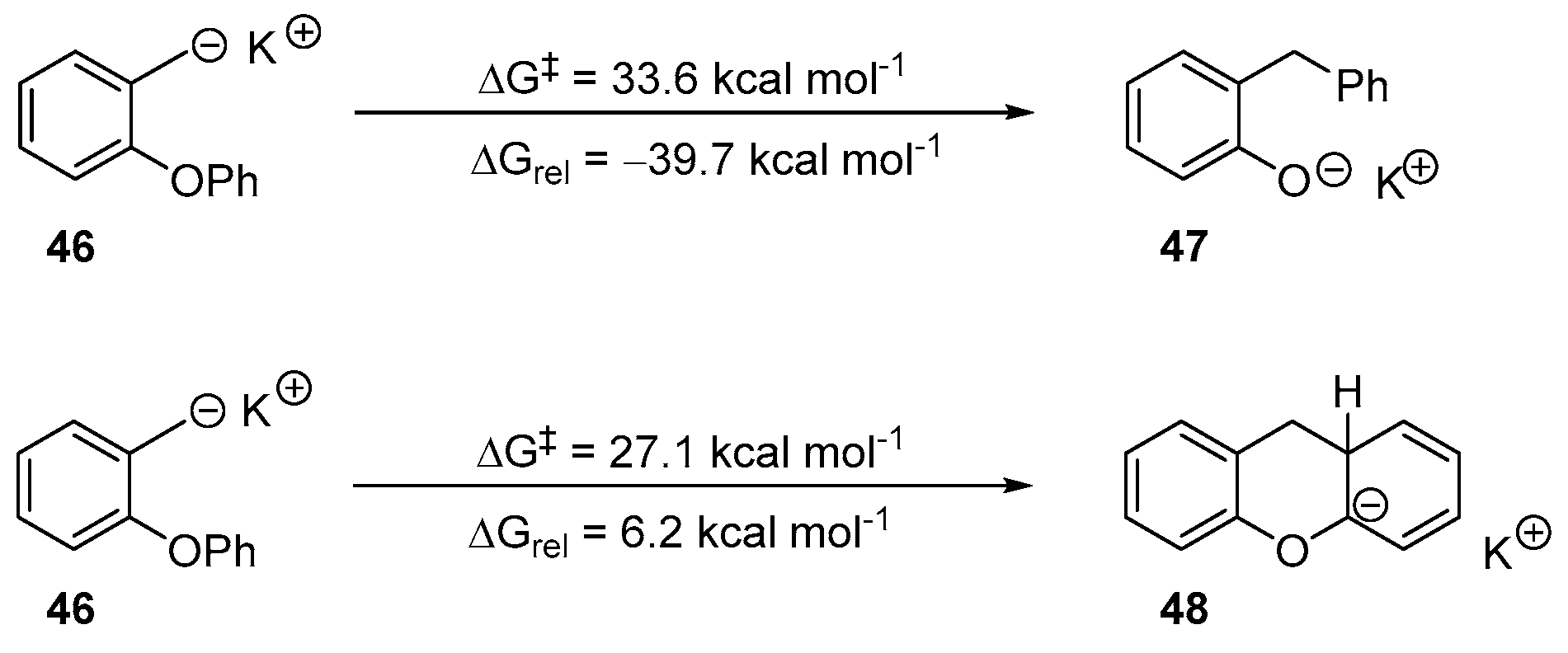


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
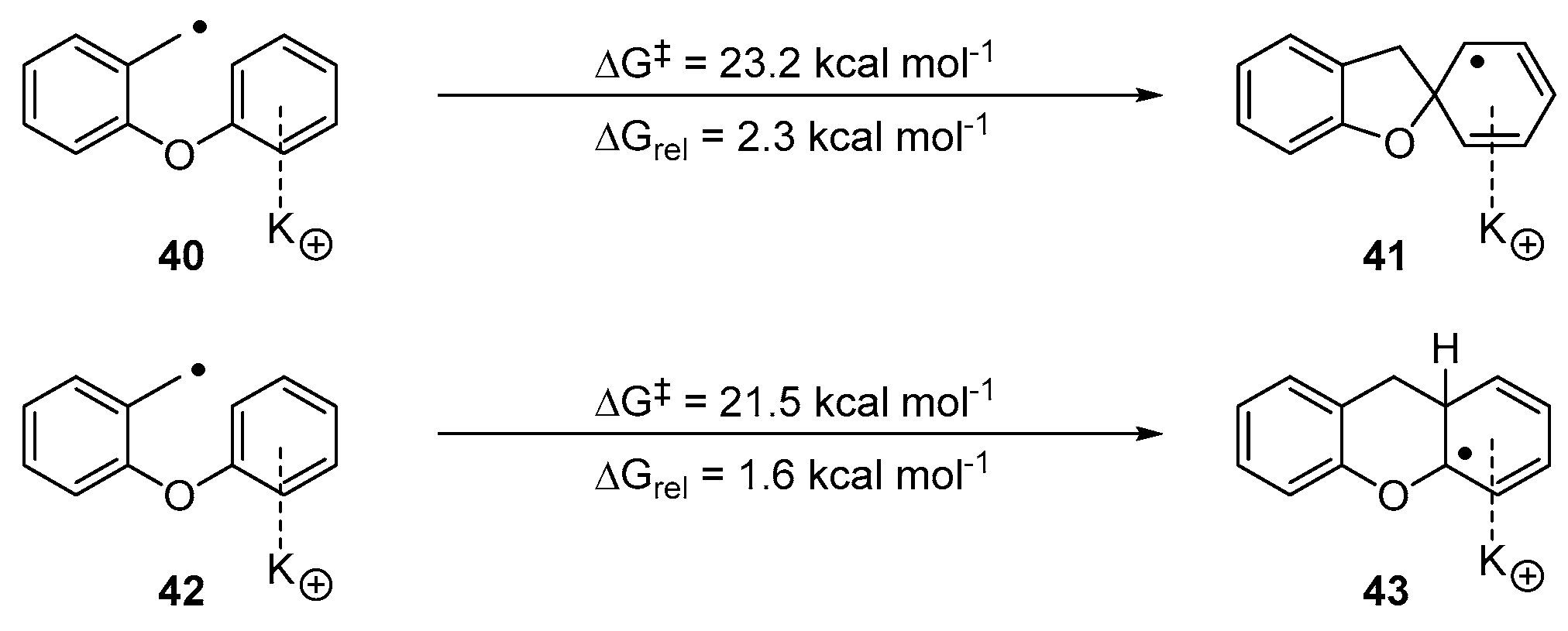{kind=link}
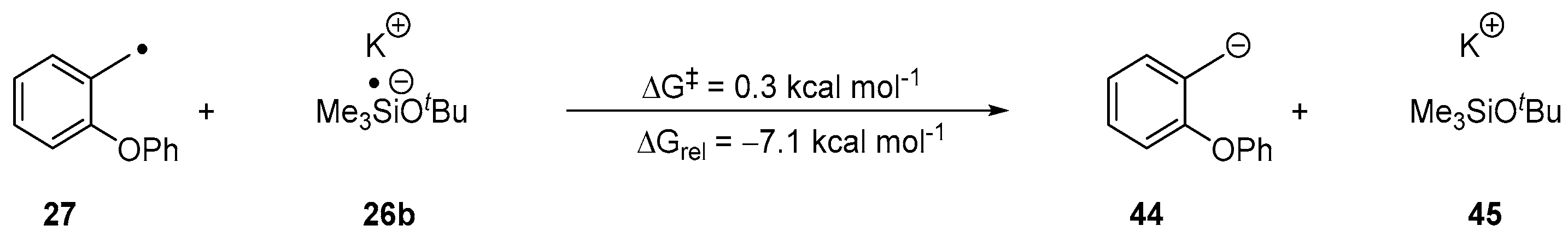{kind=link}
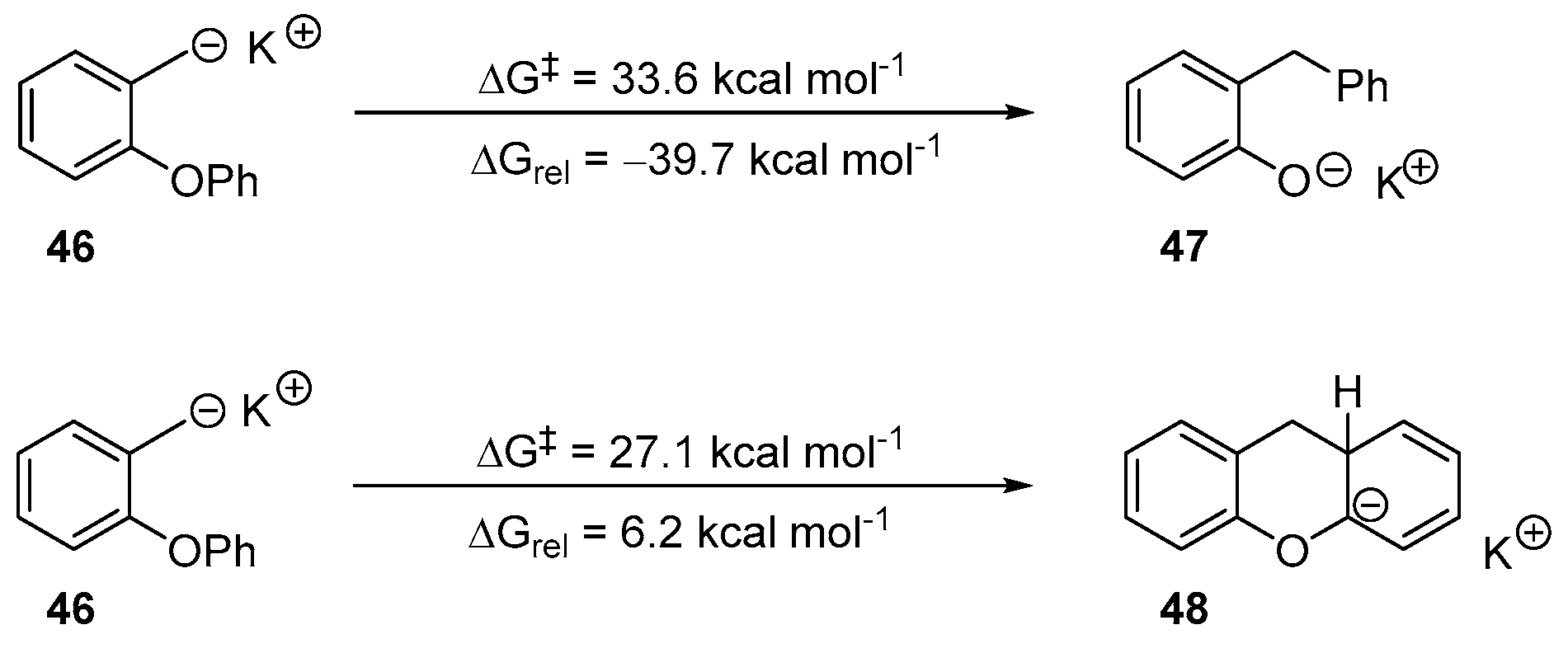{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Entry | R | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) |
| 1 | SiMe3 | 17.8 | 1.8 |
| 2 | Me | 18.2 | 0.4 |
 | |||
| Entry | R | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) |
| 1 | SiMe3 | 2.0 | −7.3 |
| 2 | Me | 3.8 | −8.6 |
 | |||
| Entry | R | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) |
| 1 | SiMe3 | 28.2 | −7.1 |
| 2 | Me | 25.3 | −7.8 |

| Entry | Anion | 5-exo-trig | 6-aryl | ||
| - | - | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) |
| 1 | 73 | 26.5 | 3.0 | 27.7 | 8.6 |
| 2 | 74 | 26.5 | 1.1 | 31.0 | 8.5 |
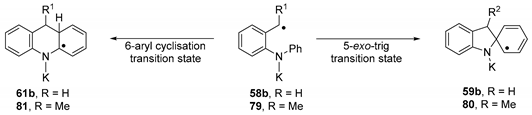
| Entry | Radical | 5-exo-trig | 6-aryl | ||
| - | - | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) | ΔG‡ (kcal mol−1) | ΔGrel (kcal mol−1) |
| 1 | 58b | 31.5 | 17.1 | 22.8 | −2.5 |
| 2 | 79 | 27.9 | 16.6 | 20.2 | −0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolodziejczak, K.; Stewart, A.J.; Tuttle, T.; Murphy, J.A. Radical and Ionic Mechanisms in Rearrangements of o-Tolyl Aryl Ethers and Amines Initiated by the Grubbs–Stoltz Reagent, Et3SiH/KOtBu. Molecules 2021, 26, 6879. https://doi.org/10.3390/molecules26226879
Kolodziejczak K, Stewart AJ, Tuttle T, Murphy JA. Radical and Ionic Mechanisms in Rearrangements of o-Tolyl Aryl Ethers and Amines Initiated by the Grubbs–Stoltz Reagent, Et3SiH/KOtBu. Molecules. 2021; 26(22):6879. https://doi.org/10.3390/molecules26226879
Chicago/Turabian StyleKolodziejczak, Krystian, Alexander J. Stewart, Tell Tuttle, and John A. Murphy. 2021. "Radical and Ionic Mechanisms in Rearrangements of o-Tolyl Aryl Ethers and Amines Initiated by the Grubbs–Stoltz Reagent, Et3SiH/KOtBu" Molecules 26, no. 22: 6879. https://doi.org/10.3390/molecules26226879
APA StyleKolodziejczak, K., Stewart, A. J., Tuttle, T., & Murphy, J. A. (2021). Radical and Ionic Mechanisms in Rearrangements of o-Tolyl Aryl Ethers and Amines Initiated by the Grubbs–Stoltz Reagent, Et3SiH/KOtBu. Molecules, 26(22), 6879. https://doi.org/10.3390/molecules26226879