
NHC Polymeric Particles Obtained by Self-Assembly and Click Approach of Calix[4]Arene Amphiphiles as Support for Catalytically Active Pd Nanoclusters

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
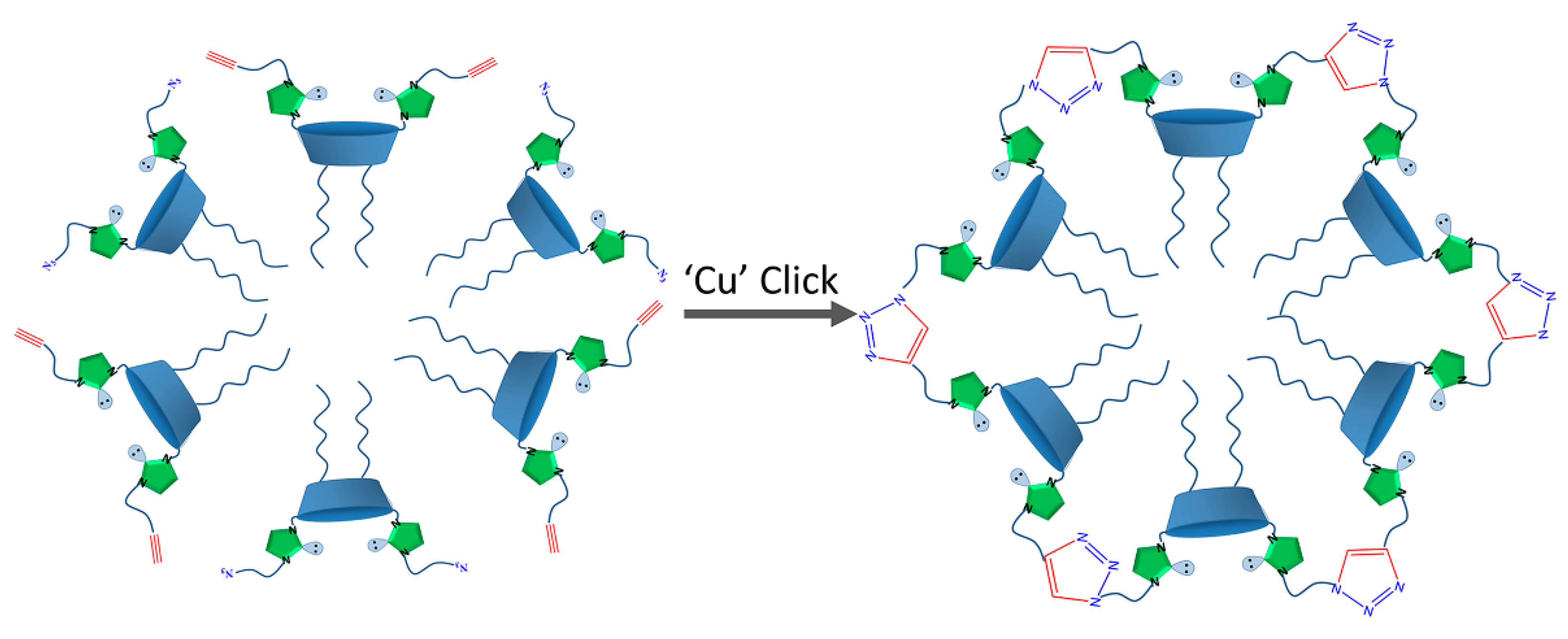
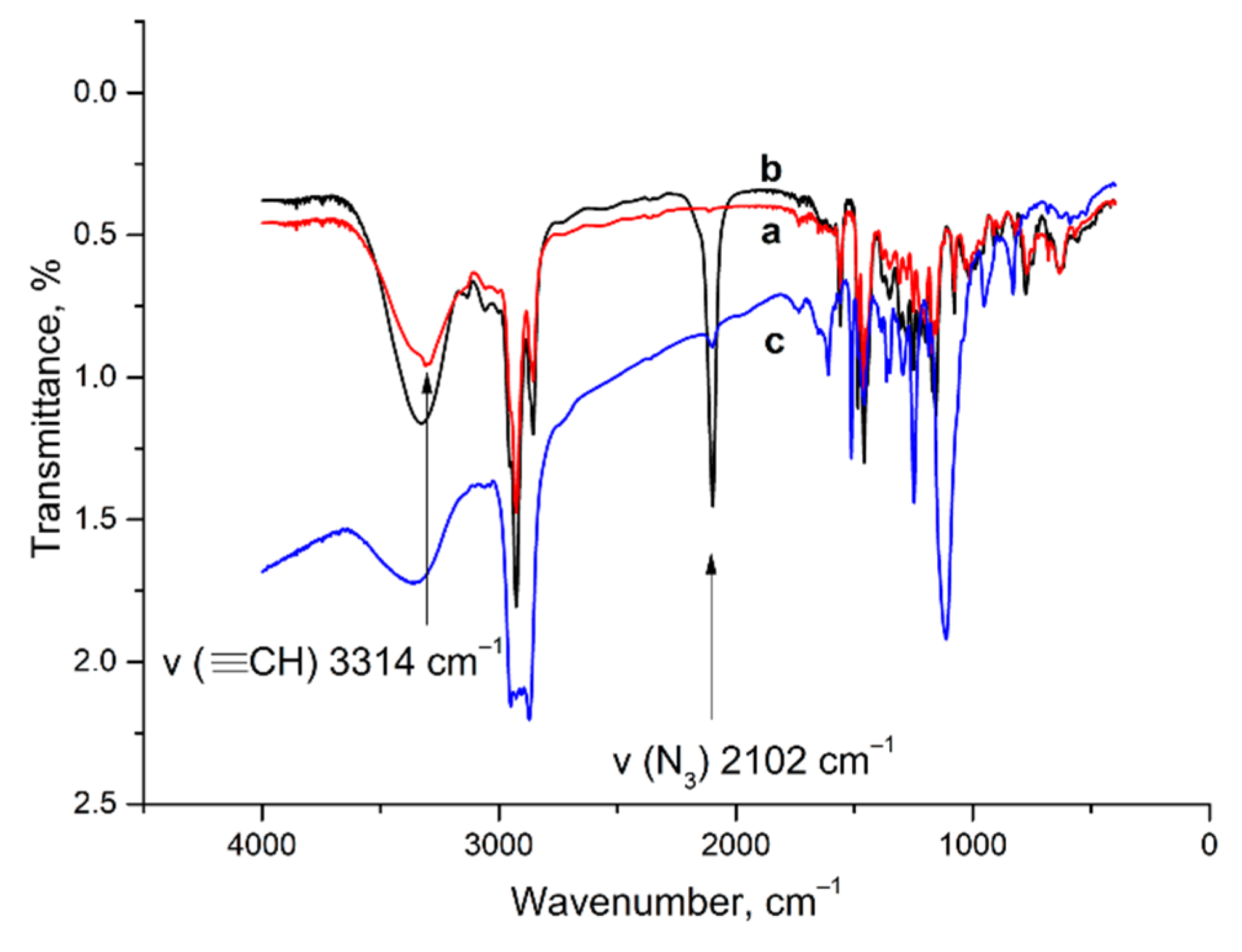
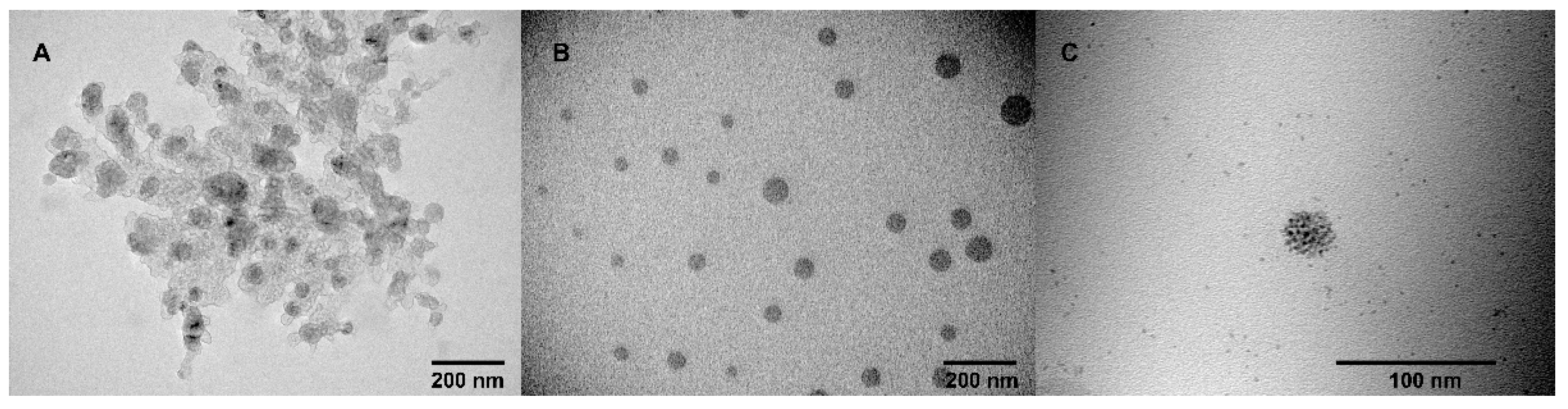
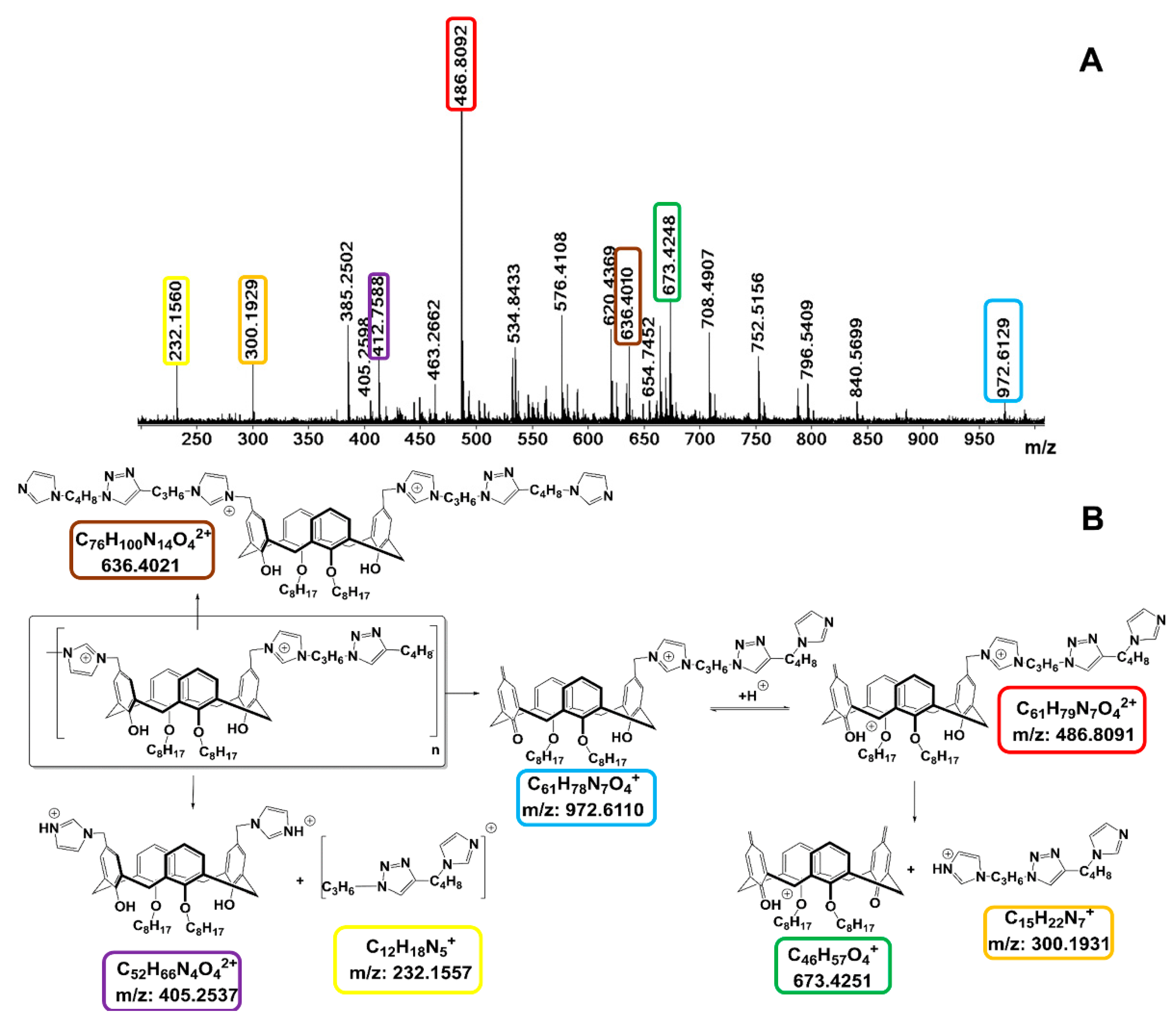
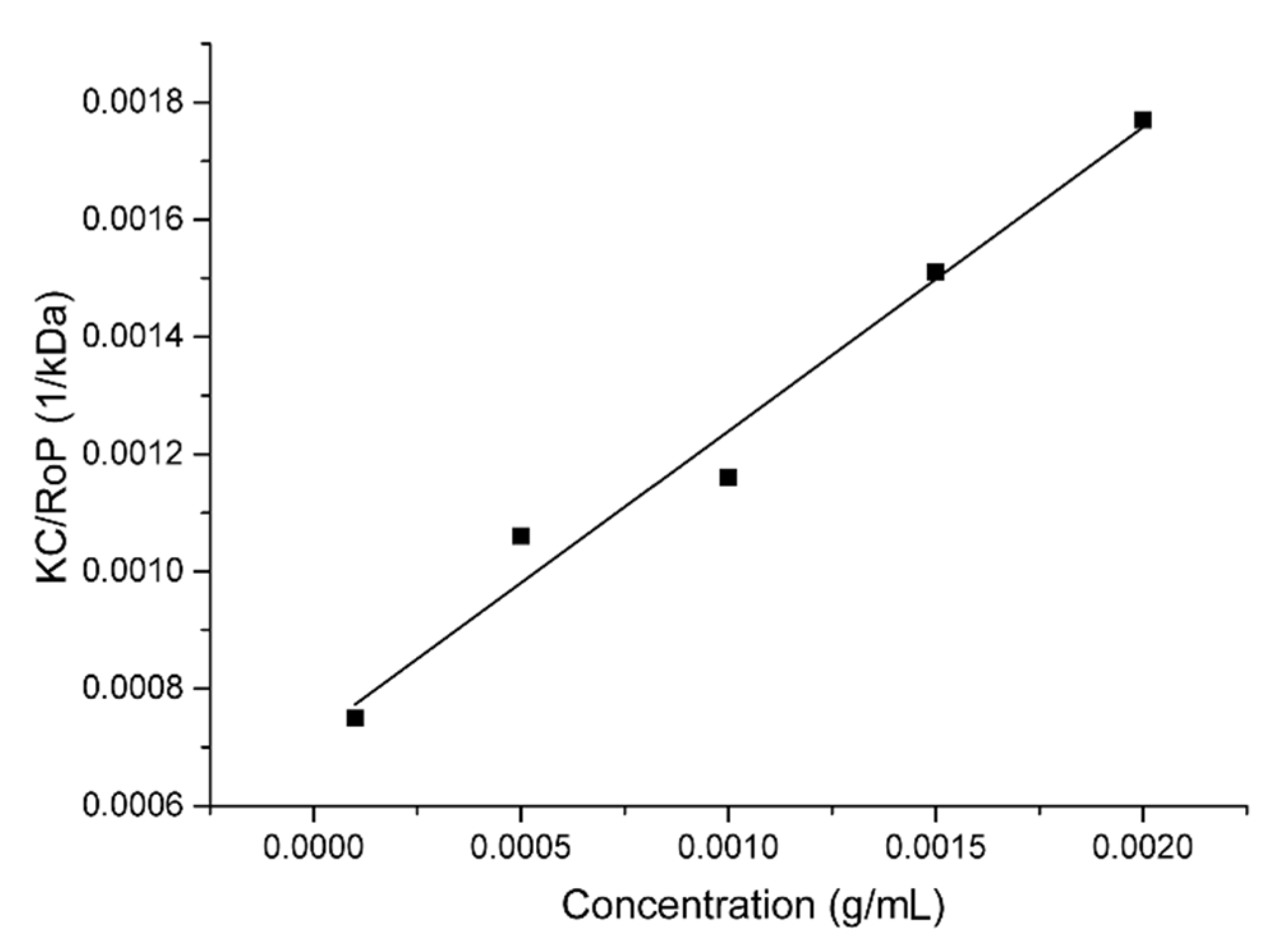
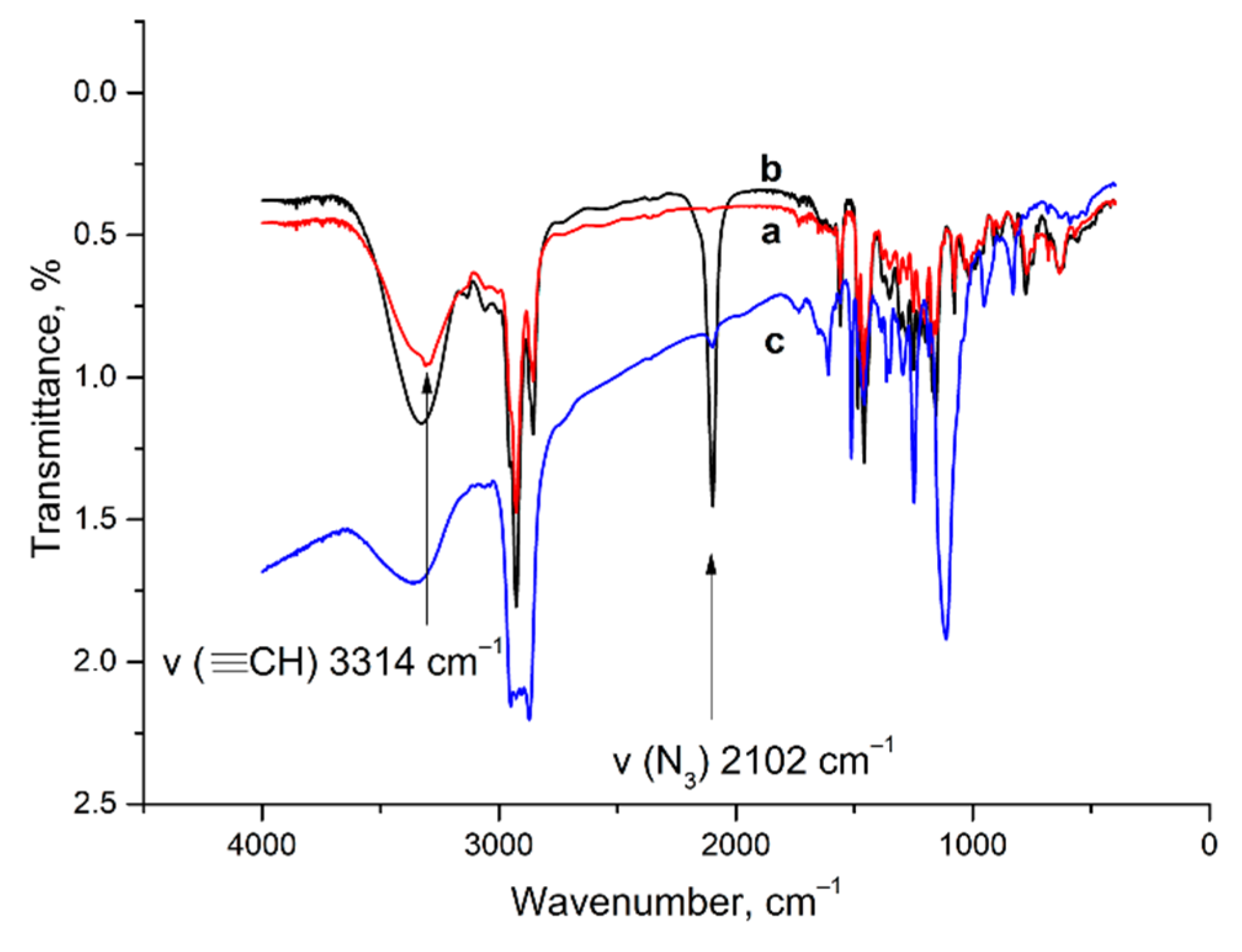
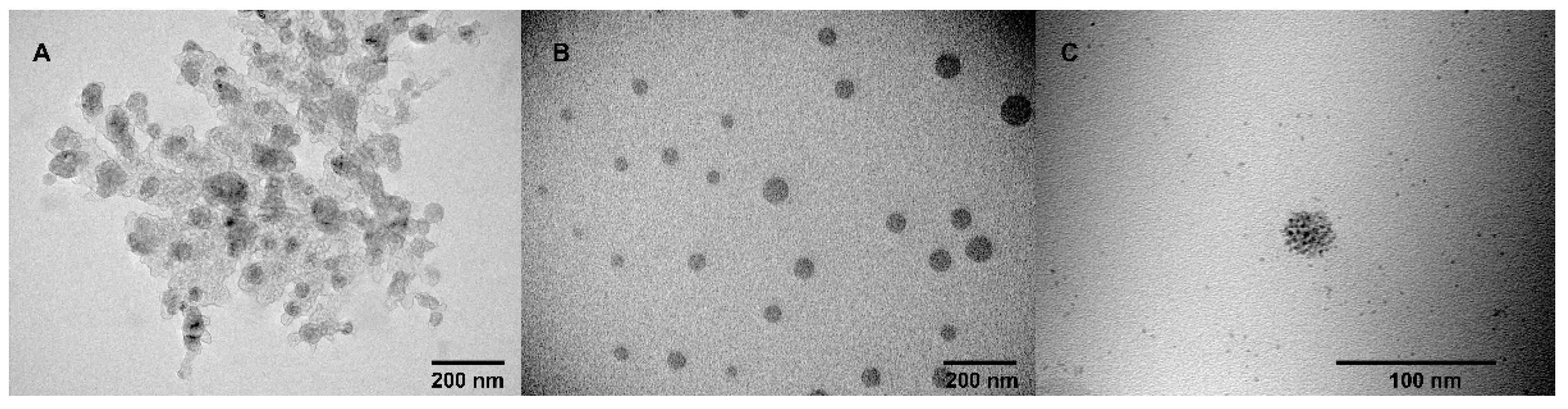
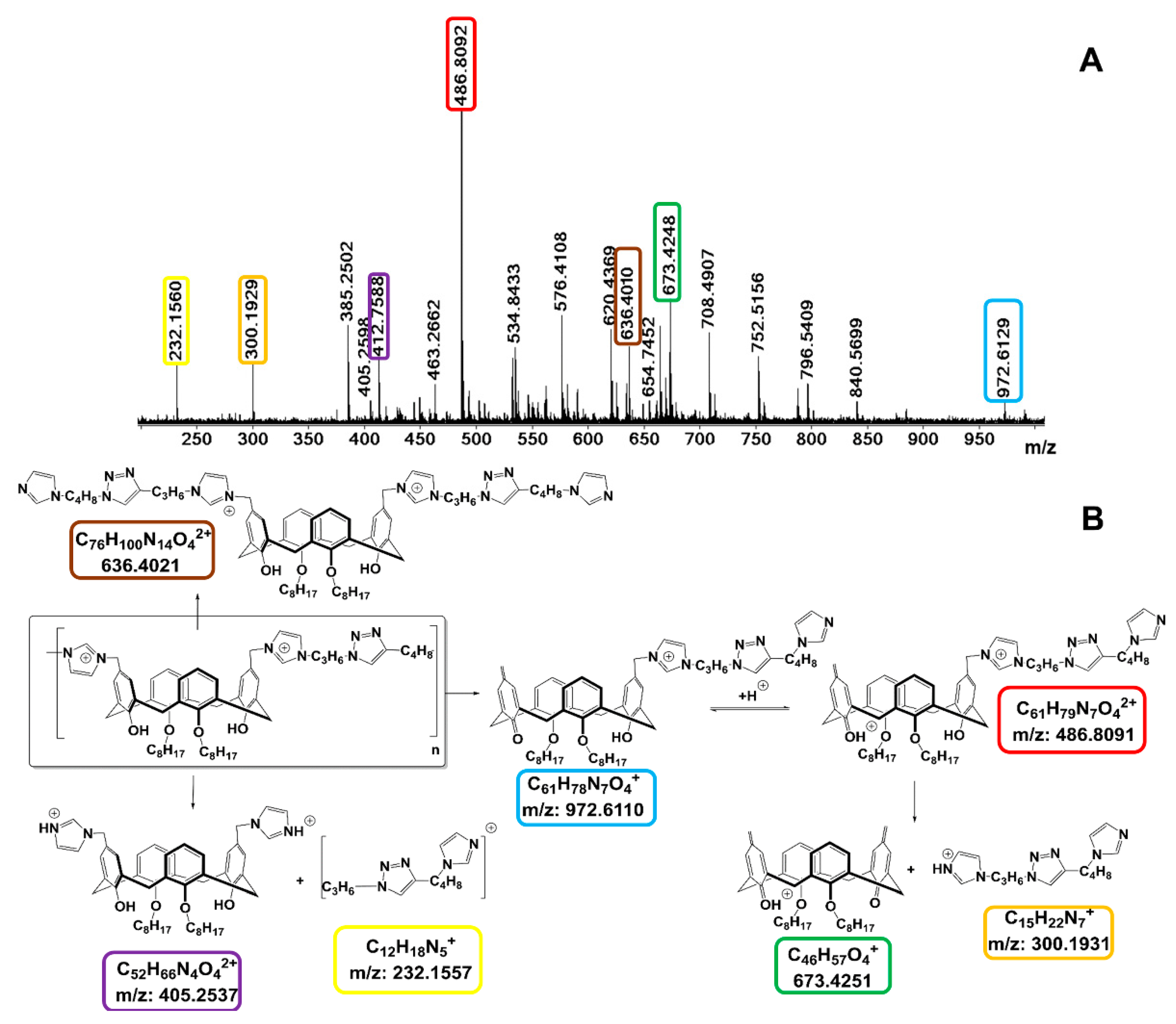
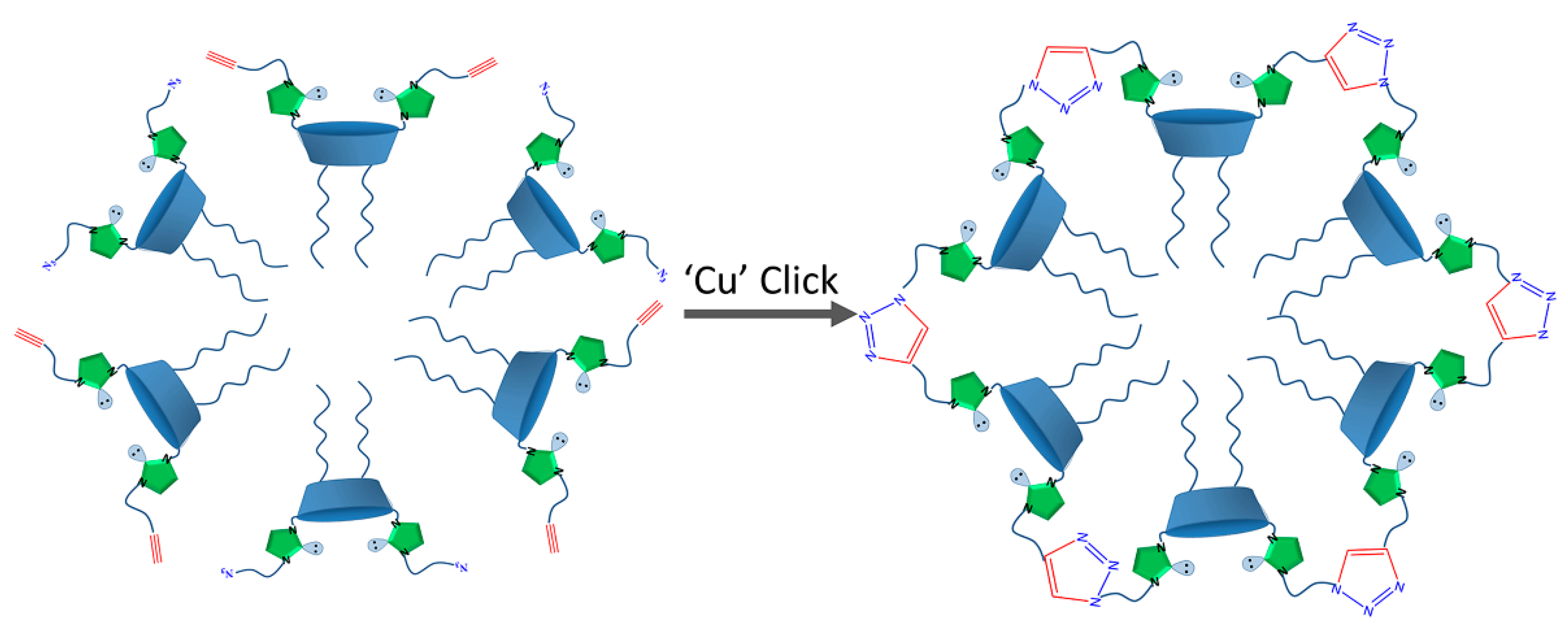
2.1. Aggregation and CuAAC Polymerization of the Core
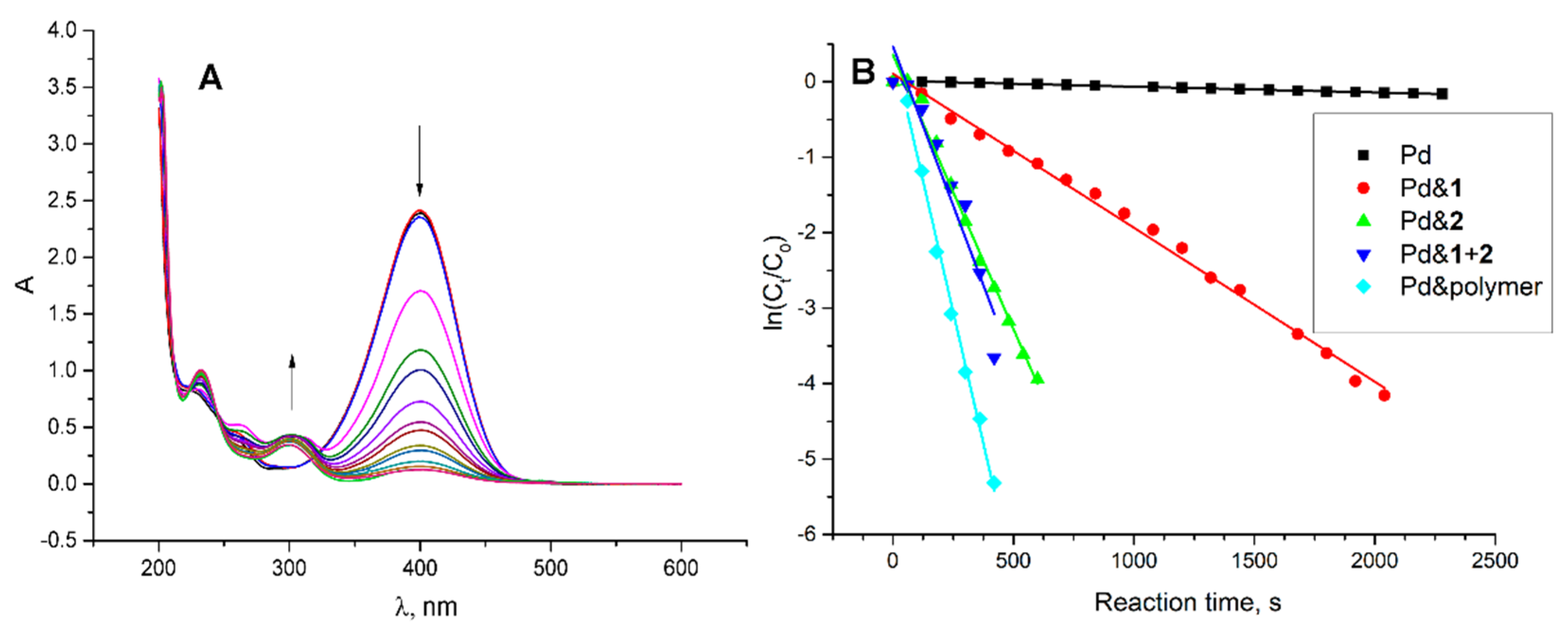
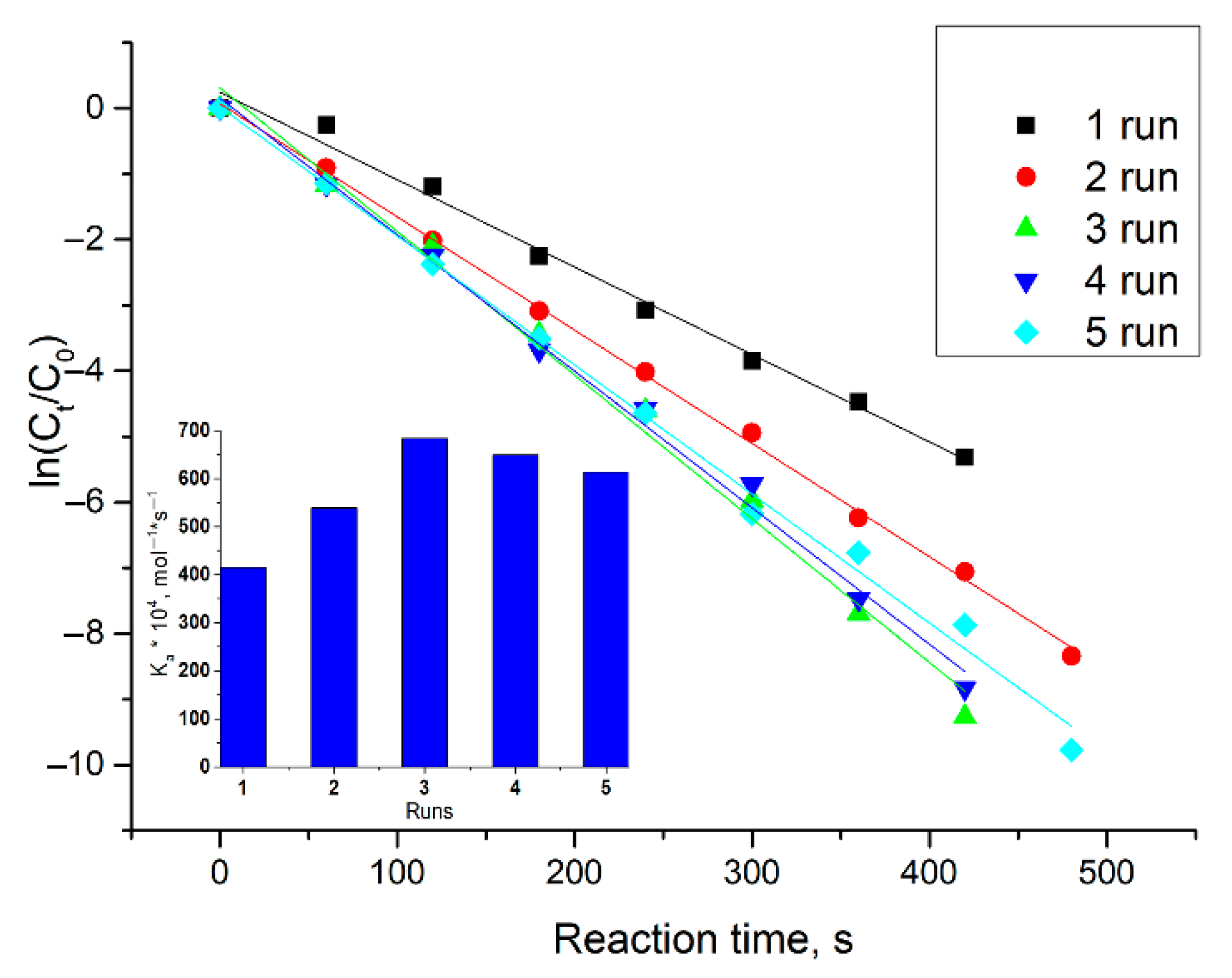
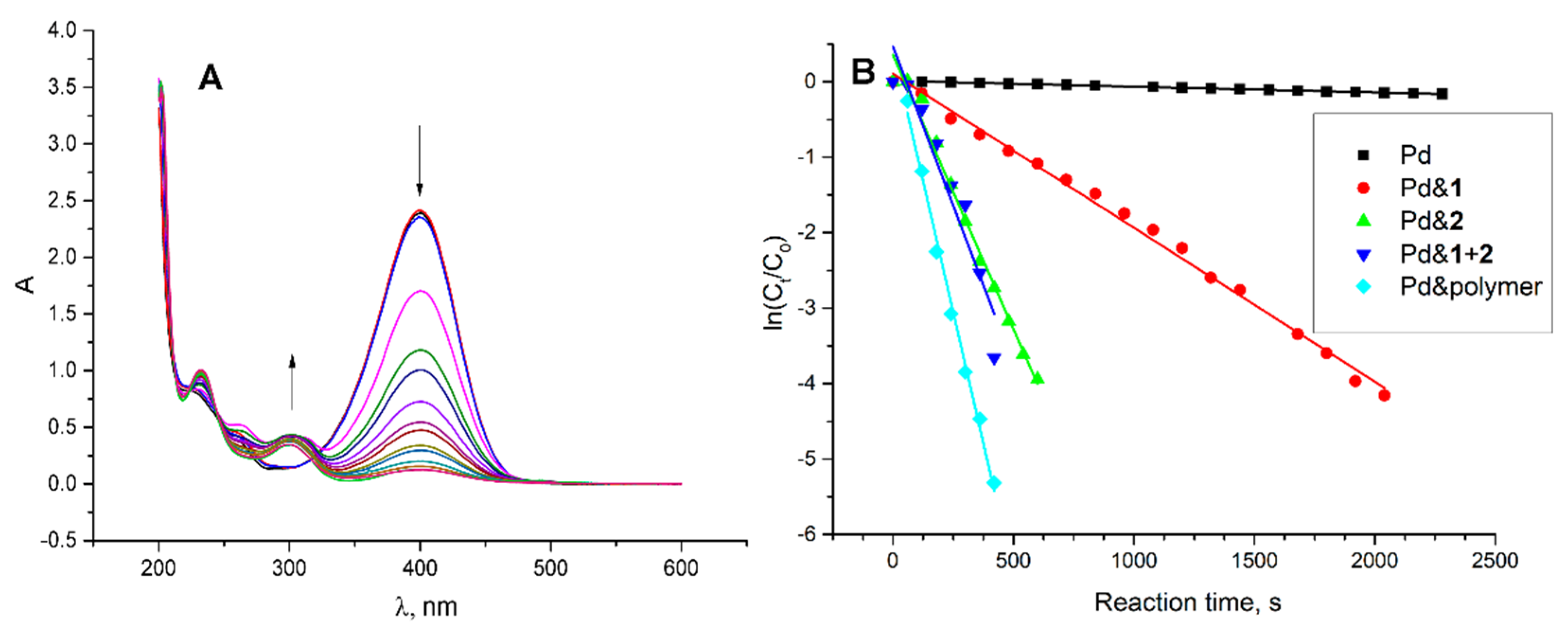
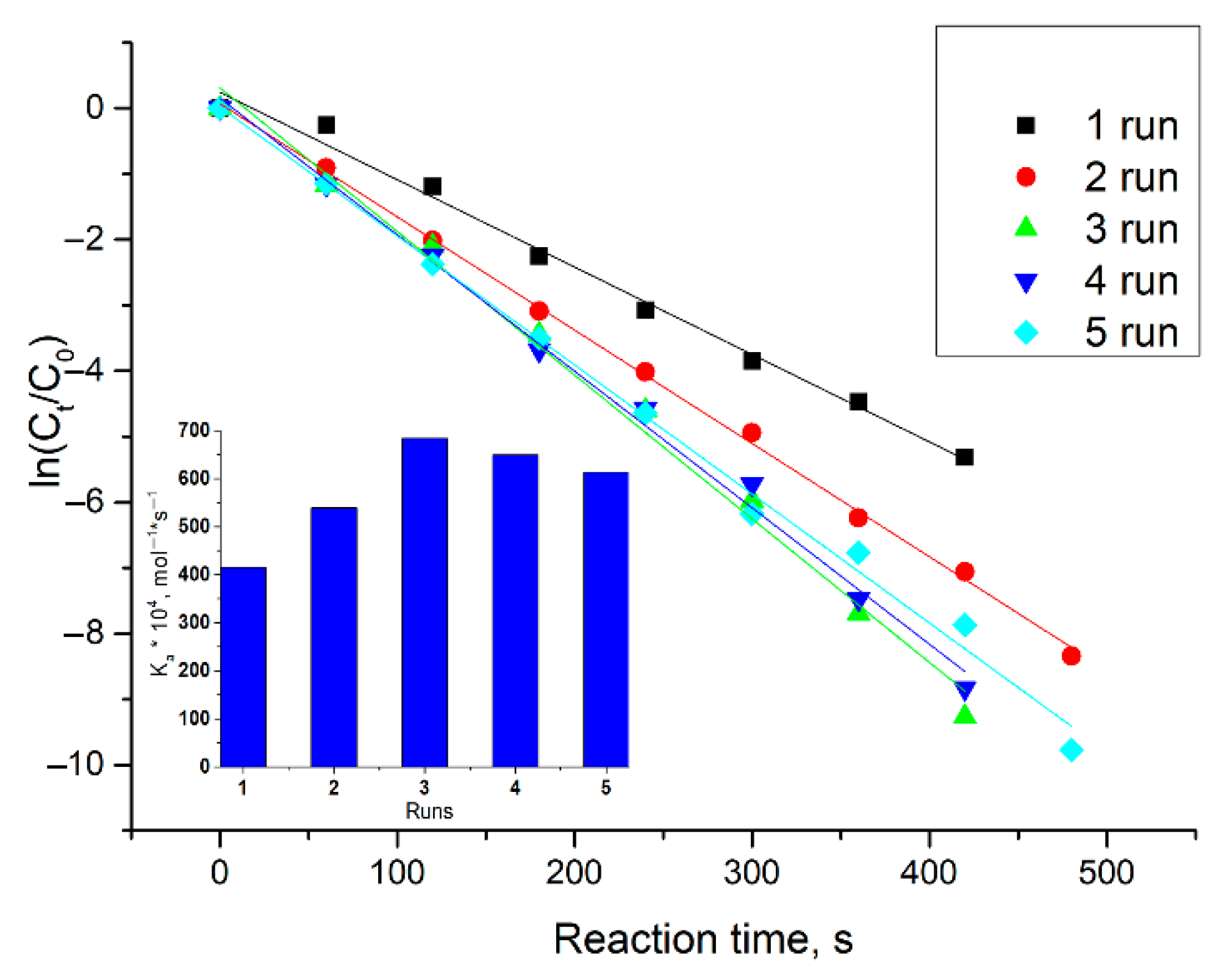
2.2. Decoration of the Core by Pd and Catalytic Activities of Received Nanoparticles in Model Reduction Reaction of p-Nitrophenol
3. Methods
3.1. Dynamic, Static, and Electrophoretic Light Scattering (DLS, SLS, and ELS)
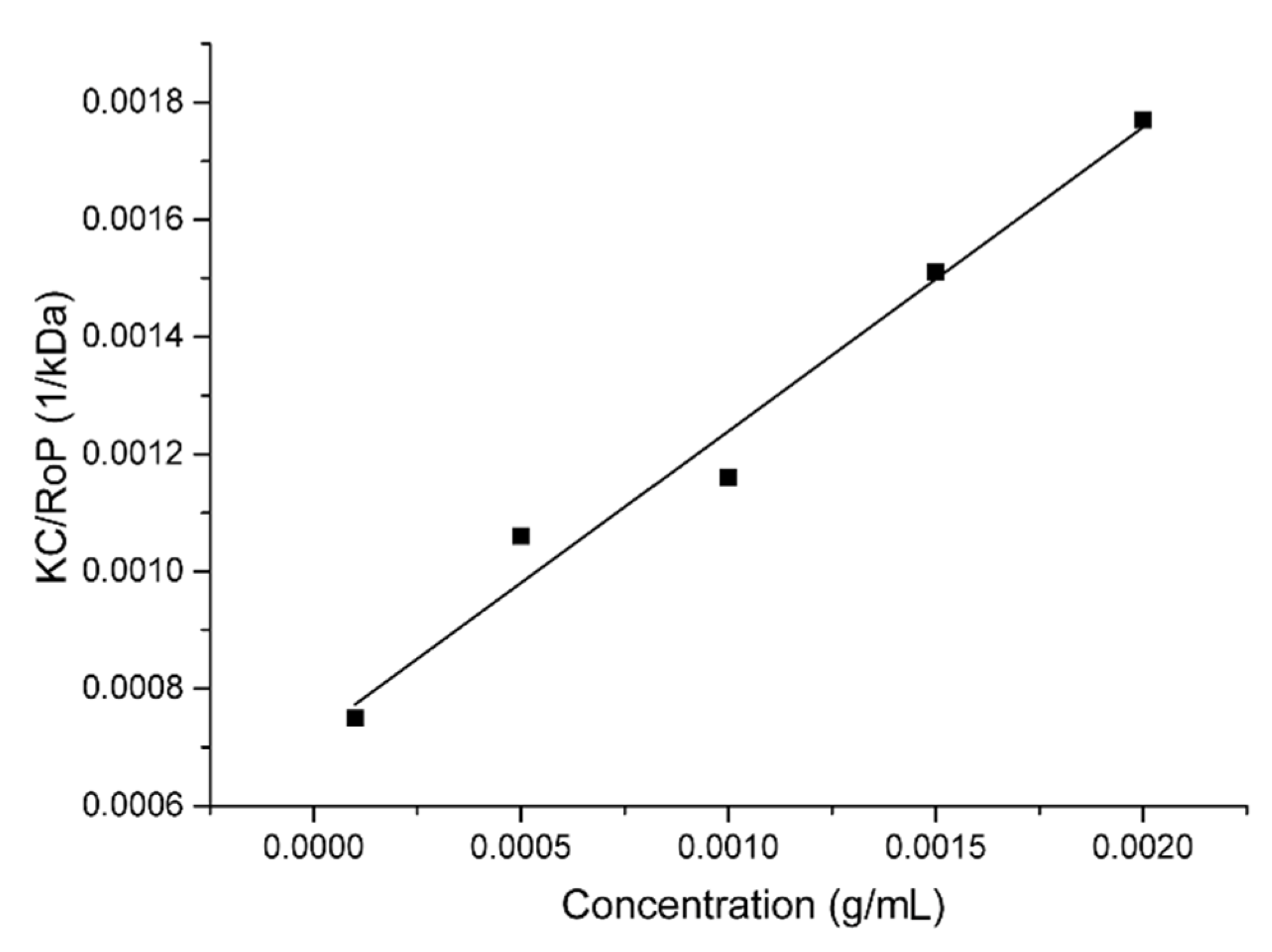
3.2. Critical Aggregation Concentration (CAC) Determination
3.3. Transition Electron Microscopy (TEM)
3.4. IR Spectra
3.5. HRESI Mass Spectrometry
3.6. Refractometry
3.7. Catalytic Reduction of PNP
4. Materials
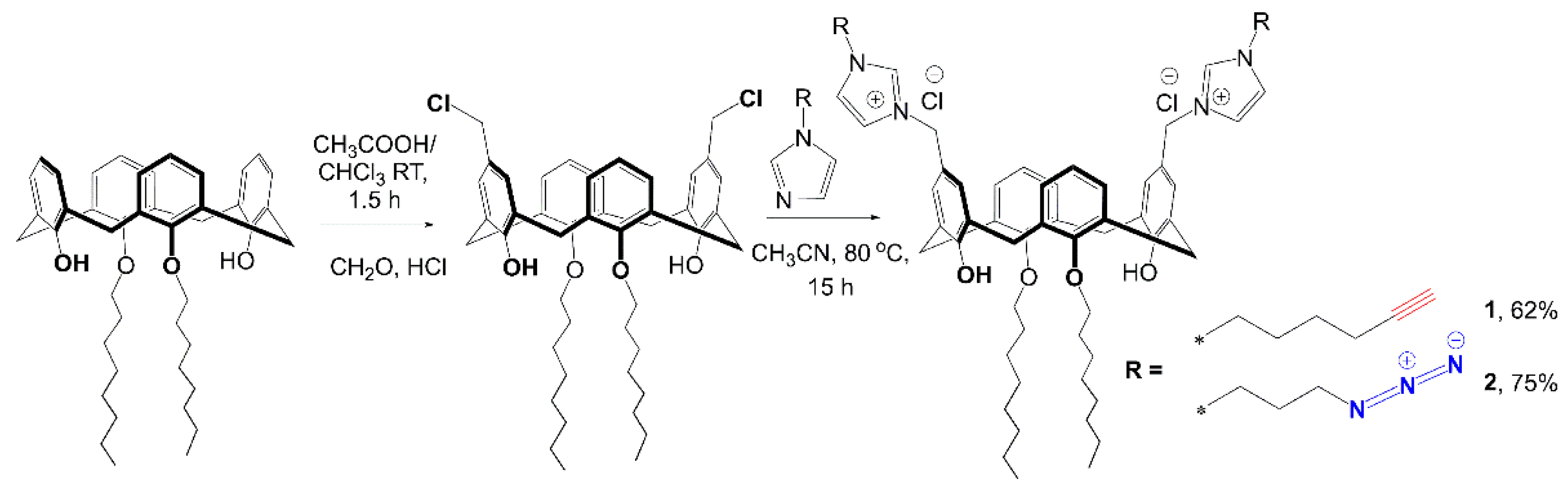
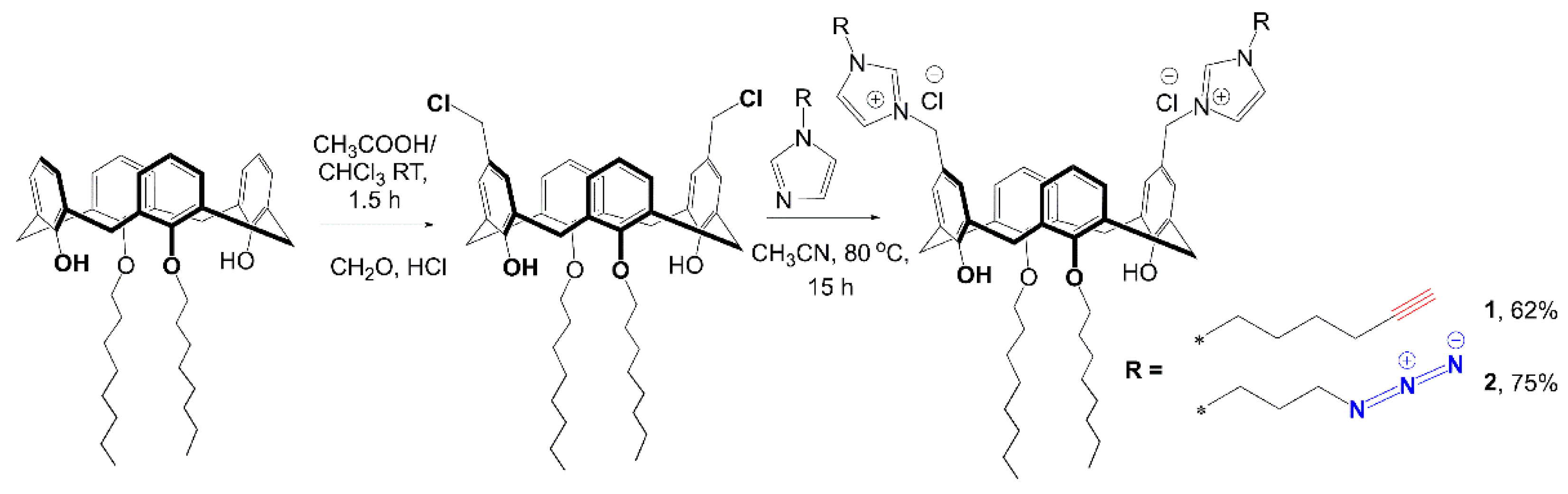
4.1. Synthesis of 11,23-bis[3-(1-(hex-5-yn-1-yl)-1H-imidazolium)methyl]-25,27-dihydroxy-26,28-dioctyloxycalix[4]arene dichloride 1
4.2. Cross-Linking of 11,23-Bis[3(1-(hex-5-yn-1-yl)-1H-imidazolium]-25,27-dihydroxy-26,28-dioctyloxycalix[4]arene dichloride 1, 11,23-Bis[3(1-(3-azidopropyl))-1H-imidazolium]-25,27-dihydroxy-26,28-dioctyloxycalix[4]arene dichloride 2
4.3. Synthesis of Pd Nanoparticles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with poly(N-heterocyclic carbene) ligands: Structural features and catalytic applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef] [PubMed]
- Dez-Gonzalez, S.; Marion, N.; Nolan, S.P. N-heterocyclic carbenes in late transition metal catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef] [PubMed]
- Nahra, F.; Nelson, D.J.; Nolan, S.P. Design concept for N-heterocyclic carbene ligands. Trends Chem. 2020, 2, 1096–1113. [Google Scholar] [CrossRef]
- Hock, S.; Schaper, L.; Herrmann, W.; Kîhn, F. Group 7 transition metal complexes with N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 5073–5089. [Google Scholar] [CrossRef]
- Baczewska, P.; Śniady, K.; Kośnik, W.; Michalak, M. Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts 2021, 11, 972. [Google Scholar] [CrossRef]
- Koy, M.; Bellotti, P.; Das, M.; Glorius, F. N-Heterocyclic carbenes as tunable ligands for catalytic metal surfaces. Nat. Catal. 2021, 4, 352–363. [Google Scholar] [CrossRef]
- De, S.; Udvardy, A.; Czégéni, C.E.; Joó, F. Poly-N-heterocyclic carbene complexes with applications in aqueous media. Coord. Chem. Rev. 2019, 400, 213038–213069. [Google Scholar] [CrossRef]
- Biffis, A.; Baron, M.; Tubaro, C. Poly-NHC Complexes of Transition Metals: Recent Applications and New Trends. Adv. Organomet. Chem. 2015, 63, 203–288. [Google Scholar]
- Wang, W.; Cui, L.; Sun, P.; Shi, L.; Yue, C.; Li, F. Reusable N-Heterocyclic Carbene Complex Catalysts and Beyond: A Perspective on Recycling Strategies. Chem. Rev. 2018, 118, 9843–9929. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, T.; Yang, H. Encapsulation of Hoveyda-Grubbs and Catalyst within Yolk-Shell Structured Silica for Olefin Metathesis. ACS Catal. 2015, 5, 2225–2231. [Google Scholar] [CrossRef]
- Lang, C.H.; Pahnke, K.; Kiefer, C.; Goldmann, A.S.; Roesky, P.W.; Barner-Kowollik, C. Consecutive modular ligation as an access route to palladium containing polymers. Polym. Chem. 2013, 4, 5456–5462. [Google Scholar] [CrossRef]
- Lambert, R.; Wirotius, A.L.; Vignolle, J.; Taton, D. C–C couplings in water by micellar catalysis at low loadings from a recyclable polymer-supported Pd(II)–NHC nanocatalyst. Polym. Chem. 2019, 10, 460. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, L.; Lv, H.; Zhang, G.; Xia, C.; Hahn, F.E.; Li, F. Modular “Click” Preparation of Bifunctional Polymeric Heterometallic Catalysts. Angew. Chem. Int. Ed. 2016, 55, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Burilov, V.; Garipova, R.; Mironova, D.; Sultanova, E.; Bogdanov, I.; Ocherednyuk, E.; Evtugyn, V.; Osin, Y.; Rizvanov, I.; Solovieva, S.; et al. New poly-imidazolium–triazole particles by CuAAC cross-linking of calix[4]arene bis-azide/alkyne amphiphiles—A prospective support for Pd in the Mizoroki–Heck reaction. RSC Adv. 2021, 11, 584–591. [Google Scholar] [CrossRef]
- Kaya, Z.; Bentouhami, E.; Pelzer, K.; Armspach, D. Cavity-shaped ligands for asymmetric metal catalysis. Coord. Chem. Rev. 2021, 445, 214066. [Google Scholar] [CrossRef]
- Boldrini, C.L.; Manfredi, N.; Montini, T.; Baldini, L.; Abbotto, A.; Fornasiero, P. Calix[4]arene-based molecular photosensitizers for sustainable hydrogen production and other solar applications. Curr. Opin. Green Sustain. Chem. 2021, 32, 100534–100541. [Google Scholar] [CrossRef]
- Antipin, I.S.; Alfimov, M.V.; Arslanov, V.V.; Burilov, V.A.; Vatsadze, S.Z.; Voloshin, Y.Z.; Volcho, K.P.; Gorbatchuk, V.V.; Gorbunova, Y.G.; Gromov, S.P.; et al. Functional supramolecular systems: Design and applications. Russ. Chem. Rev. 2021, 90, 895–1107. [Google Scholar] [CrossRef]
- Burilov, V.; Garipova, R.; Sultanova, E.; Mironova, D.; Grigoryev, I.; Solovieva, S.; Antipin, I. New Amphiphilic Imidazolium/Benzimidazolium Calix[4]arene Derivatives: Synthesis, Aggregation Behavior and Decoration of DPPC Vesicles for Suzuki Coupling in Aqueous Media. Nanomaterials 2020, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Burilov, V.A.; Gafiatullin, M.B.K.; Mironova, D.A.; Sultanova, E.D.; Evtugyn, V.G.; Osin, Y.N.; Islamov, D.R.; Usachev, K.S.; Solovieva, S.E.; Antipin, I.S. Amphiphilic PdII-NHC complexes on 1,3-alternate p-tert-butylthiacalix[4]arene platform: Synthesis and catalytic activities in coupling and hydrogenation reactions. Eur. J. Org. Chem. 2020, 2020, 2180–2189. [Google Scholar] [CrossRef]
- Burilov, V.A.; Garipova, R.I.; Solovieva, S.E.; Antipin, I.S. Synthesis of Bifunctional Derivatives of Calix[4]arene Bearing Azidoalkyl Fragments in Cone Stereoisomeric Form. Dokl. Chem. 2020, 490, 1–5. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Iqbal, M.; Watson, T. p-tert-butylcalix[4]arene. Org. Synth. 1990, 68, 234. [Google Scholar]
- Huang, Z.-T.; Wang, G.-Q.; Yang, L.-M.; Lou, Y.-X. The Selective Chloromethylation of 25,27-Dihydroxy-26,28-Dimethoxycalix[4]arene and Nucleophilic Substitution Therefrom. Synth. Commun. 1995, 25, 1109–1118. [Google Scholar] [CrossRef]
- Steckel, A.; Schlosser, G. An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation. Molecules 2019, 24, 611. [Google Scholar] [CrossRef] [Green Version]
- Niessen, W.M.A.; Correa, R.A. Fragmentation of even-electron ions. In Interpretation of MS-MS Mass Spectra of Drugs and Pesticides, 1st ed.; Desiderio, M., Loo, J.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 71–128. [Google Scholar]
- Pashynska, V.A.; Kosevich, M.V.; Van den Heuvel, H.; Claeys, M. The effect of cone voltage on electrospray mass spectra of the bisquaternary ammonium salt decamethoxinum. Rapid Commun. Mass Spectrom. 2006, 20, 755–763. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, M.; Liu, M.; Xiao, Y.; Li, Z.; Chen, J.; Sun, Y.; Zhao, J.; Fang, S.; Jia, D.; et al. Homogeneous Pd nanoparticles produced in direct reactions: Green synthesis, formation mechanism and catalysis properties. J. Mater. Chem. 2014, 2, 1369–1374. [Google Scholar] [CrossRef]
- Herve’s, P.; Pe´rez-Lorenzo, M.; Liz-Marza´n, L.M.; Dzubiella, J.; Lu, Y.; Ballauff, M. Catalysis by metallic nanoparticles in aqueous solution: Model reactions. Chem. Soc. Rev. 2012, 41, 5577–5587. [Google Scholar] [CrossRef]
- Illam, P.M.; Donthireddy, S.N.R.; Chakrabartty, S.; Rit, A. Heteroditopic Ru(II)− and Ir(III)−NHC Complexes with Pendant 1,2,3-Triazole/Triazolylidene Groups: Stereoelectronic Impact on Transfer Hydrogenation of Unsaturated Compounds. Organometallics 2019, 38, 2610–2623. [Google Scholar] [CrossRef]
- Yu, Y.; Jung, E.; Kim, H.-J.; Cho, A.; Kim, J.; Yu, T.; Lee, J. Protein Particles Decorated with Pd Nanoparticles for the Catalytic Reduction of p-Nitrophenol to p-Aminophenol. ACS Appl. Nano Mater. 2020, 3, 10487–10496. [Google Scholar] [CrossRef]
- Veisi, H.; Joshani, Z.; Karmakar, B.; Tamoradi, T.; Heravi, M.M.; Javad, G. Ultrasound assisted synthesis of Pd NPs decorated chitosan-starch functionalized Fe3O4 nanocomposite catalyst towards Suzuki-Miyaura coupling and reduction of 4-nitrophenol. Int. J. Biol. Macromol. 2021, 172, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Ayad, A.I.; Luart, D.; Dris, A.O.; Guénin, E. Kinetic Analysis of 4-Nitrophenol Reduction by “Water-Soluble” Palladium Nanoparticles. Nanomaterials 2020, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, G. Revisiting Catalytic Model Reaction p-Nitrophenol/NaBH4 by Metallic Nanoparticles Coated onto Polymeric Spheres. Nanoscale 2013, 5, 11919–11927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals; Elsevier: New York, NY, USA, 2009. [Google Scholar]
- Deschamps, J.; Balog, M.; Boury, B.; Ben Yahia, M.; Filhol, J.-S.; van der Lee, A.; Choueiry, A.A.; Barisien, T.; Legrand, L.; Schott, M.; et al. Tuning Topochemical Polymerization of Diacetylenes: A Joint Synthetic, Structural, Photophysical, and Theoretical Study of a Series of Analogues of a Known Reactive Monomer, 1,6-Bis(diphenylamino)-2,4-hexadiyne (THD). Chem. Mater. 2010, 22, 3961. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | CAC, µM | d, nm | PDI | ζ. mV |
|---|---|---|---|---|
| 1 | 24 | 122 ± 5 | 0.465 ± 0.045 | +54 ± 11 |
| 2 | 130 | 98 ± 9 | 0.432 ± 0.124 | +97 ± 1 |
| 1 + 2 | 40 | 167 ± 22 | 0.424 ± 0.043 | +70 ± 10 |
| polymer | 236 ± 34 | 0.401 ± 0.043 | +66 ± 10 |
| System | d, nm | PDI | ζ. mV |
|---|---|---|---|
| Pd0 | 230 ± 21 | 0.967 ± 0.058 | +2 ± 0.6 |
| Pd & 1 | 61 ± 1 | 0.322 ± 0.001 | +66 ± 2 |
| Pd & 2 | 93 ± 17 | 0.261 ± 0.096 | +58 ± 7 |
| Pd & 1 + 2 | 65 ± 2 | 0.438 ± 0.073 | +62 ± 9 |
| Pd & polymer | 198 ± 7 | 0.382 ± 0.073 | +60 ± 6 |
| Catalytic System | RC, k, × 102 s−1 | SAA, Ka, × 104 mol−1s−1 |
|---|---|---|
| Pd0 | 0.012 ± 0.002 | 4 ± 0.6 |
| Pd and 1 | 0.198 ± 0.016 | 62 ± 5 |
| Pd and 2 | 0.771 ± 0.023 | 241 ± 7 |
| Pd and 1 + 2 | 0.950 ± 0.070 | 297 ± 22 |
| Pd and polymer 1 run | 1.328 ± 0.099 | 415 ± 31 |
| Pd and polymer 2 run | 1.724 ± 0.038 | 539 ± 12 |
| Pd and polymer 3 run | 2.185 ± 0.035 | 683 ± 11 |
| Pd and polymer 4 run | 2.076 ± 0.016 | 649 ± 5 |
| Pd and polymer 5 run | 1.964 ± 0.048 | 614 ± 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burilov, V.; Mironova, D.; Sultanova, E.; Garipova, R.; Evtugyn, V.; Solovieva, S.; Antipin, I. NHC Polymeric Particles Obtained by Self-Assembly and Click Approach of Calix[4]Arene Amphiphiles as Support for Catalytically Active Pd Nanoclusters. Molecules 2021, 26, 6864. https://doi.org/10.3390/molecules26226864
Burilov V, Mironova D, Sultanova E, Garipova R, Evtugyn V, Solovieva S, Antipin I. NHC Polymeric Particles Obtained by Self-Assembly and Click Approach of Calix[4]Arene Amphiphiles as Support for Catalytically Active Pd Nanoclusters. Molecules. 2021; 26(22):6864. https://doi.org/10.3390/molecules26226864
Chicago/Turabian StyleBurilov, Vladimir, Diana Mironova, Elsa Sultanova, Ramila Garipova, Vladimir Evtugyn, Svetlana Solovieva, and Igor Antipin. 2021. "NHC Polymeric Particles Obtained by Self-Assembly and Click Approach of Calix[4]Arene Amphiphiles as Support for Catalytically Active Pd Nanoclusters" Molecules 26, no. 22: 6864. https://doi.org/10.3390/molecules26226864
APA StyleBurilov, V., Mironova, D., Sultanova, E., Garipova, R., Evtugyn, V., Solovieva, S., & Antipin, I. (2021). NHC Polymeric Particles Obtained by Self-Assembly and Click Approach of Calix[4]Arene Amphiphiles as Support for Catalytically Active Pd Nanoclusters. Molecules, 26(22), 6864. https://doi.org/10.3390/molecules26226864