
Chiral Aminoalcohols and Squaric Acid Amides as Ligands for Asymmetric Borane Reduction of Ketones: Insight to In Situ Formed Catalytic System by DOSY and Multinuclear NMR Experiments


Abstract
:
1. Introduction
2. Results and Discussion
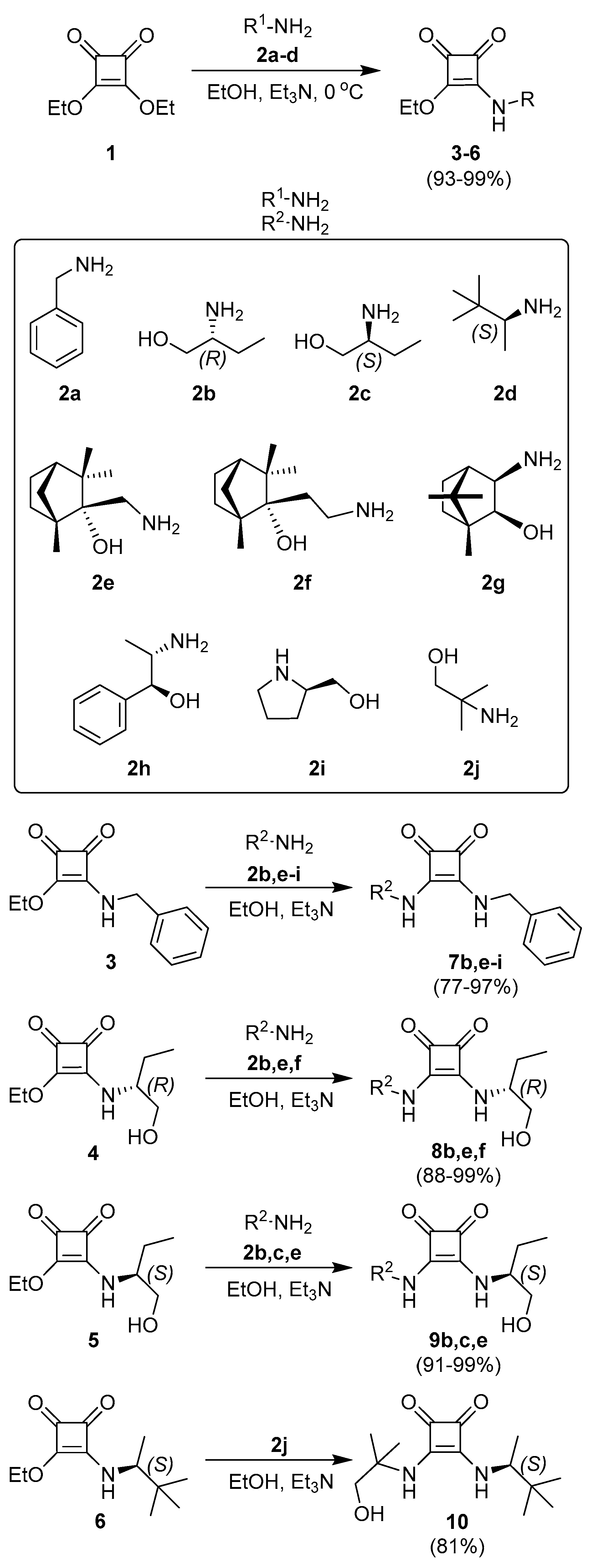
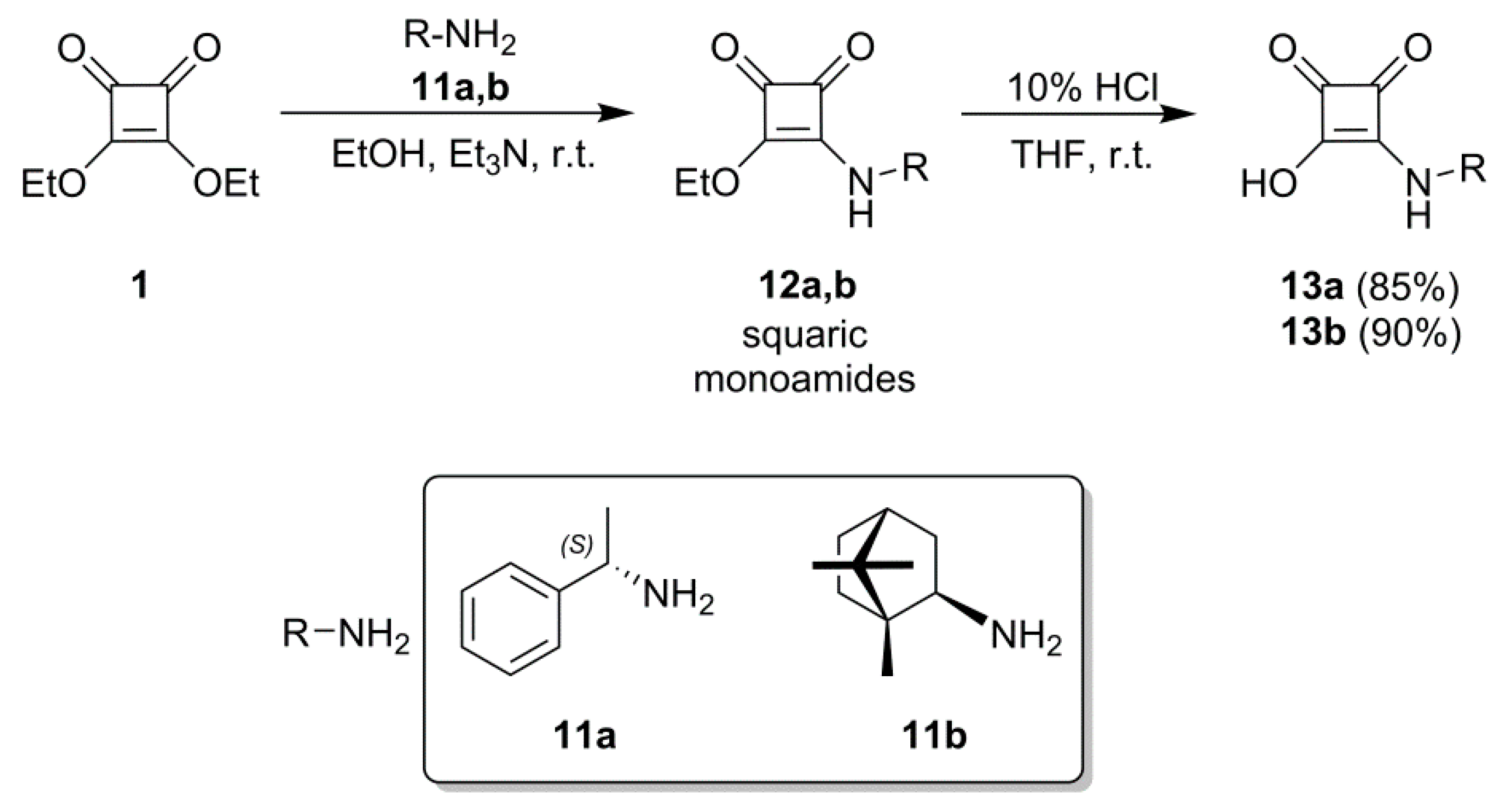
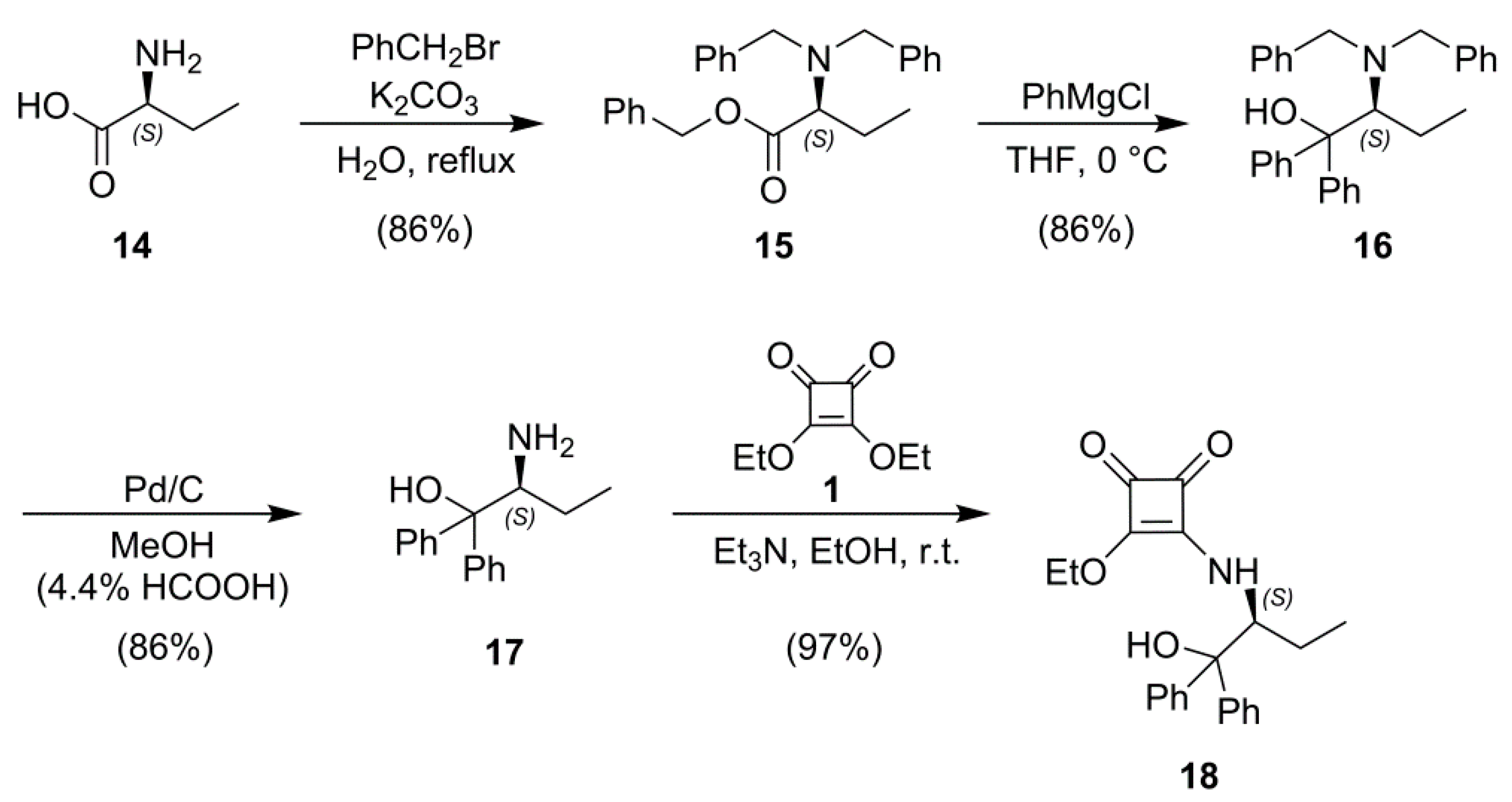
2.1. Synthesis
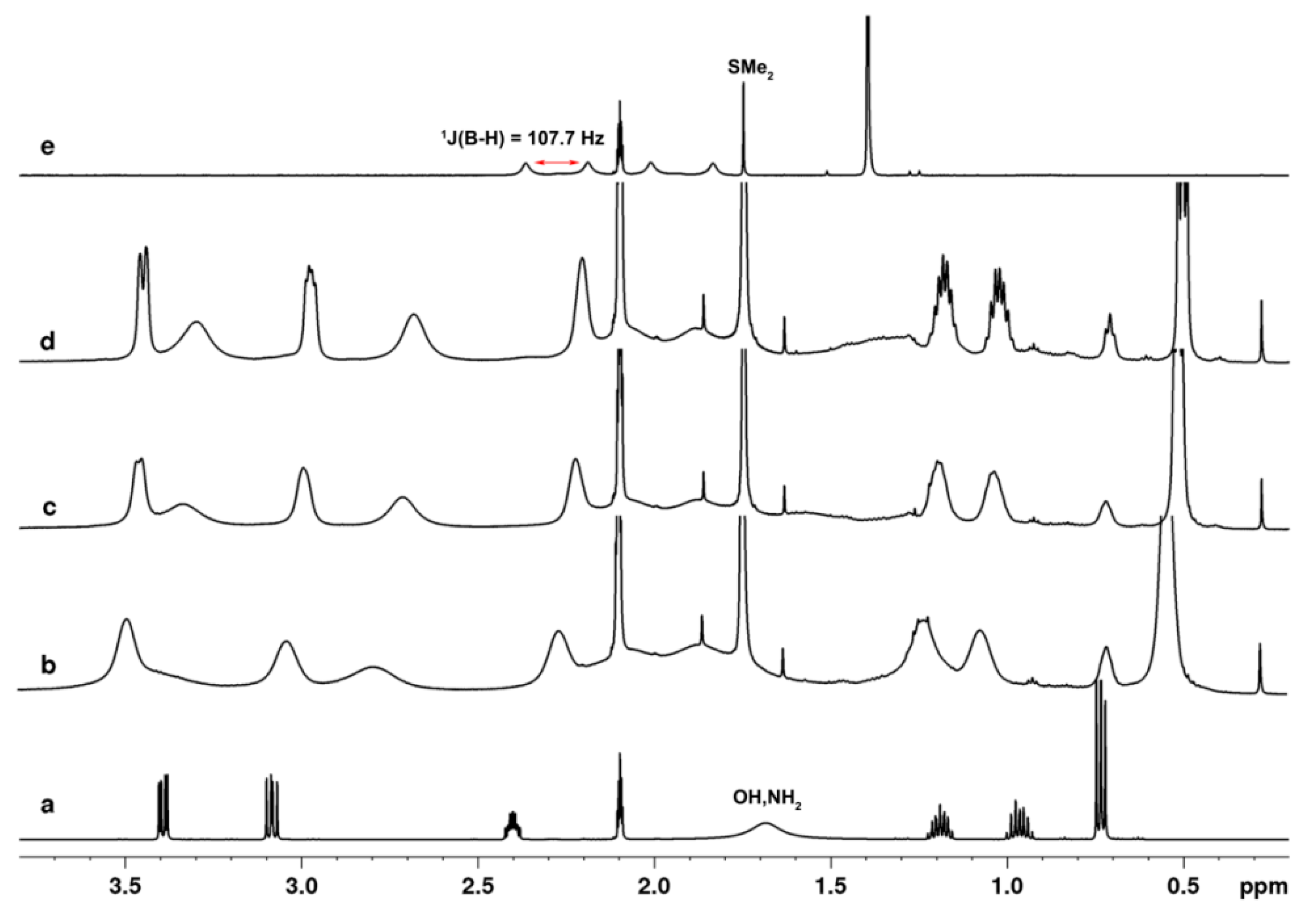
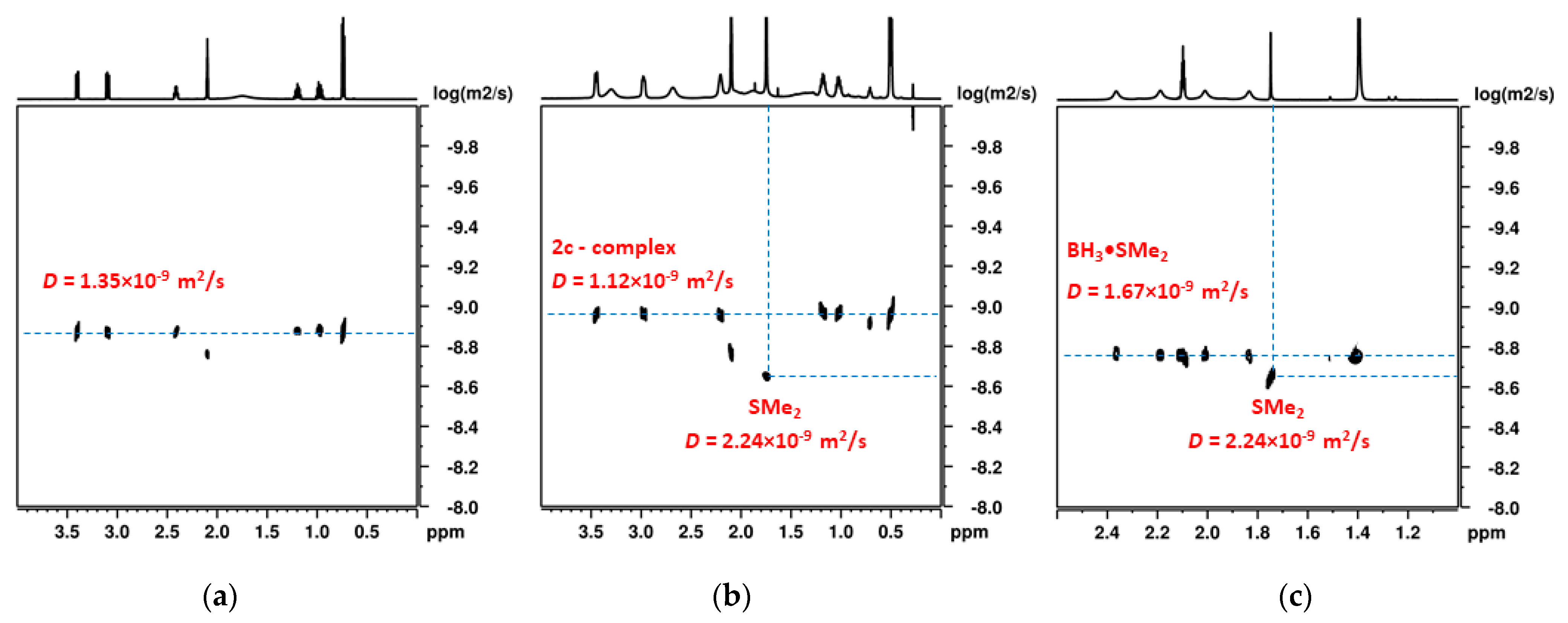
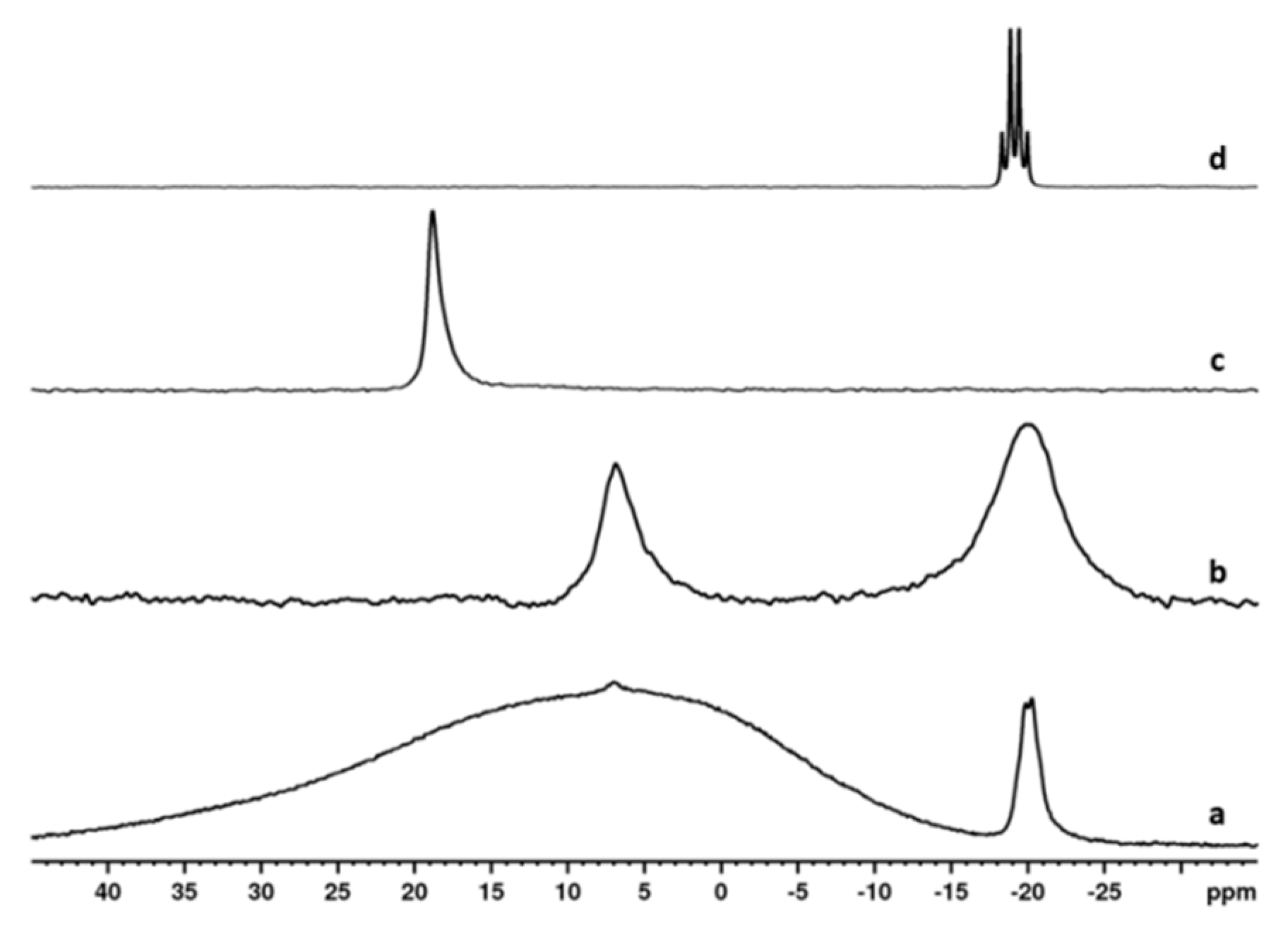
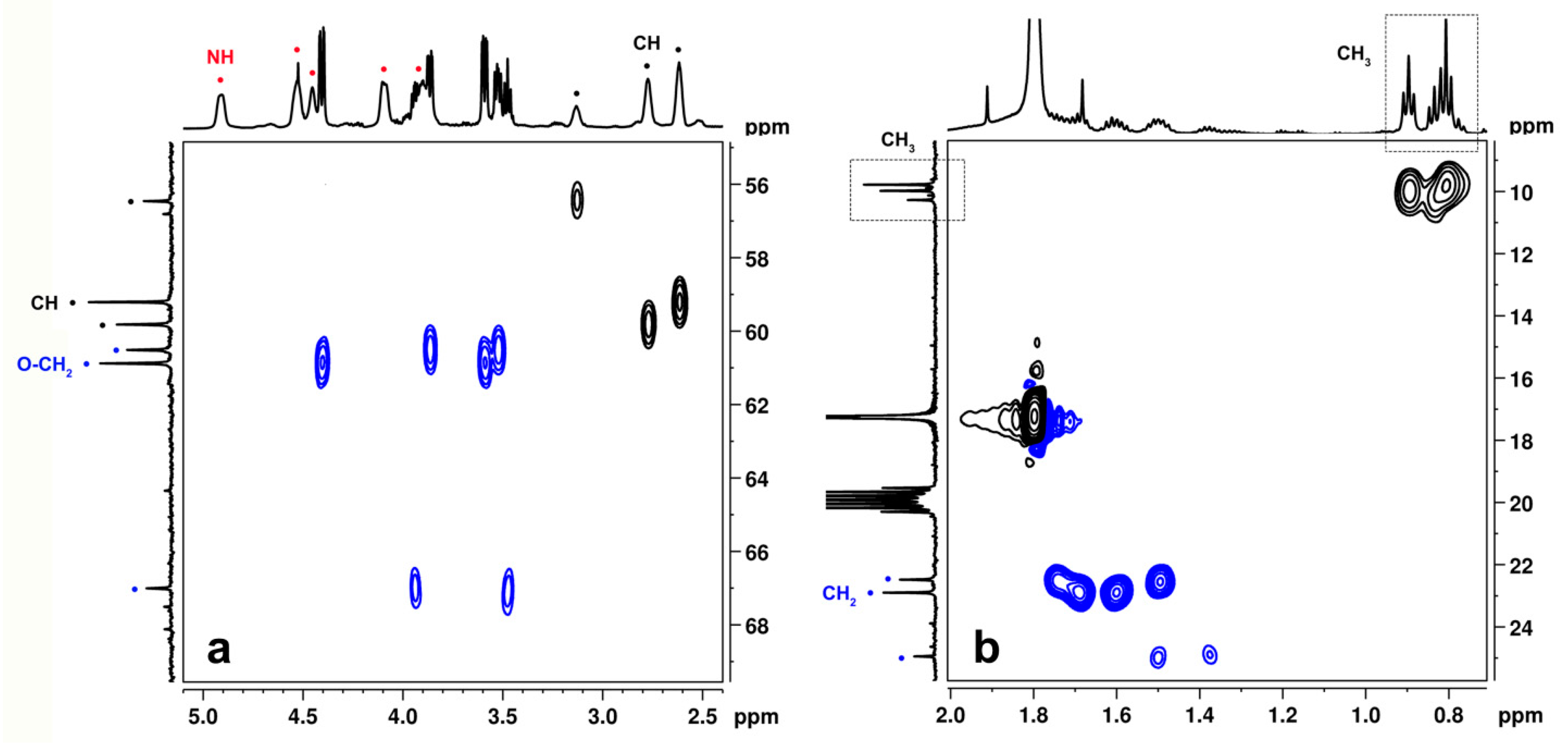
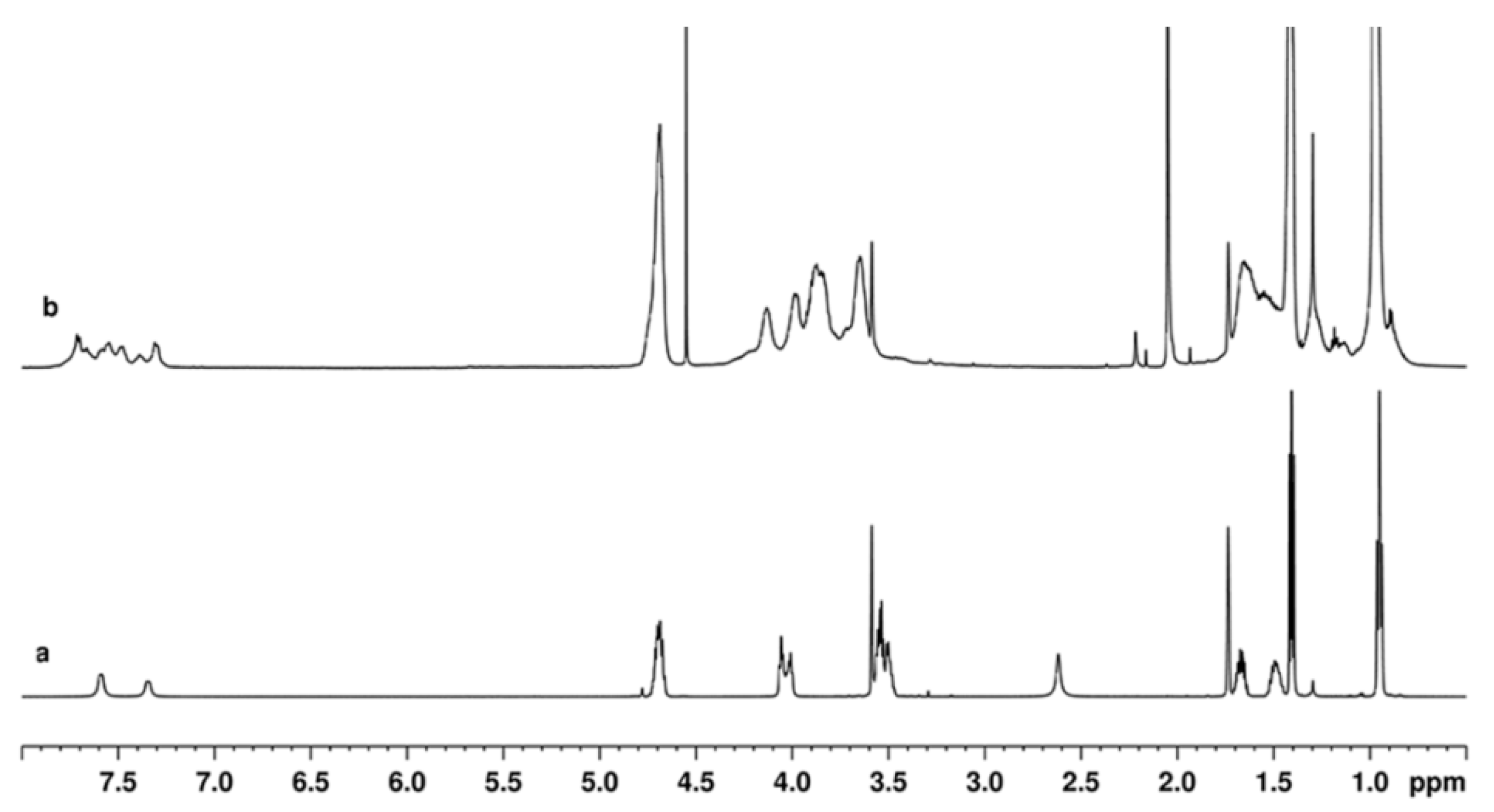
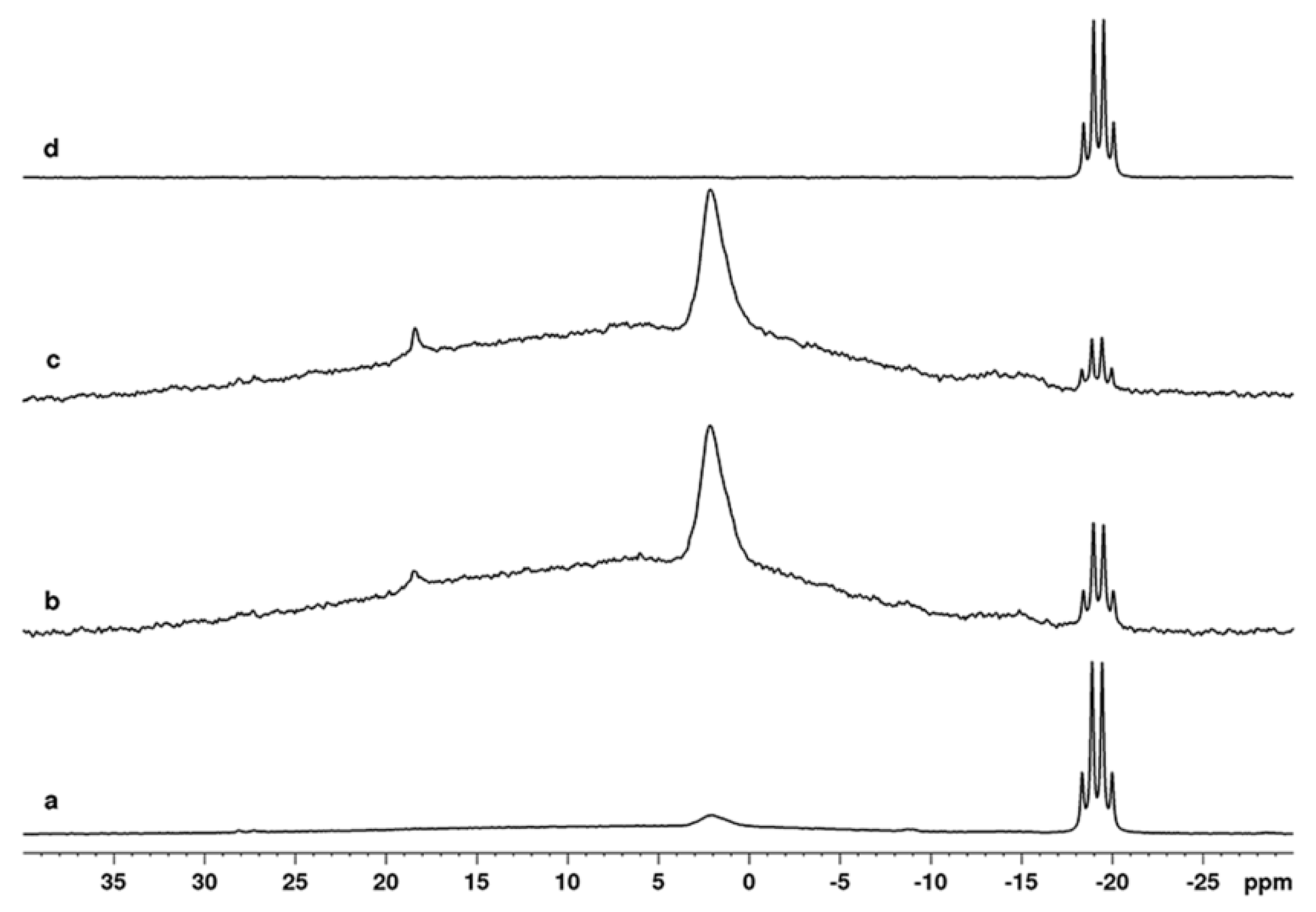
2.2. NMR Study of the Catalytic Systems
3. Materials and Methods
3.1. Chemistry
3.2. NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Abdul, R.; Taneja, S.C. Synthesis of Single-enantiomer Bioactive Molecules: A Brief Overview. Chirality 2014, 26, 63–78. [Google Scholar] [CrossRef]
- Lin, G.; Li, Y.; Chan, A.S.C. Asymmetric Catalytic Hydrogenation and Other Reduction Reactions. In Principles and Applications of Asymmetric Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; pp. 331–396. [Google Scholar] [CrossRef]
- Haftchenary, S.; Nelson, S.D.; Furst, L.; Dandapani, S.; Ferrara, S.J.; Bošković, Ž.V.; Figueroa Lazú, S.; Guerrero, A.M.; Serrano, J.C.; Crews, D.K.; et al. Efficient Routes to a Diverse Array of Amino Alcohol-Derived Chiral Fragments. ACS Comb. Sci. 2016, 18, 569–574. [Google Scholar] [CrossRef]
- Heravi, M.; Lashaki, T.B.; Fattahi, B.; Zadsirjan, V. Application of asymmetric Sharpless aminohydroxylation in total synthesis of natural products and some synthetic complex bio-active molecules. RSC Adv. 2018, 8, 6634–6659. [Google Scholar] [CrossRef] [Green Version]
- Listunov, D.; Maraval, V.; Chauvin, R.; Genisson, Y. Chiral alkynylcarbinols from marine sponges: Asymmetric synthesis and biological relevance. Nat. Prod. Rep. 2015, 32, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.K.S.; Mandal, M.; Schweizer, S.; Petasis, N.A.; Olah, G.A. A Facile Stereocontrolled Synthesis of anti-α-(Trifluoromethyl)-β-amino Alcohols. Org. Lett. 2000, 2, 3173–3176. [Google Scholar] [CrossRef]
- Sun, D.-Y.; Han, G.-Y.; Gong, J.-X.; Nay, B.; Li, X.-W.; Guo, Y.-W. Asymmetric Total Synthesis of Distaminolyne A and Revision of Its Absolute Configuration. Org. Lett. 2017, 19, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Tolomelli, A.; Ammazzalorso, A.; Bruno, I.; Amoroso, R. A Review of Strategies for the Development of Alkyl Prolines in Drug Discovery. Curr. Bioact. Compd. 2016, 12, 146–160. [Google Scholar] [CrossRef]
- Cho, B.T. Recent development and improvement for boron hydride-based catalytic asymmetric reduction of unsymmetrical ketones. Chem. Soc. Rev. 2009, 38, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Bakshi, R.K.; Shibata, S. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 1987, 109, 5551–5553. [Google Scholar] [CrossRef]
- Hirao, A.; Itsuno, S.; Nakahama, S.; Yamazaki, N. Asymmetric reduction of aromatic ketones with chiral alkoxy-amine-borane complexes. Chem. Commun. 1981, 315–317. [Google Scholar] [CrossRef]
- Corey, E.J.; Helal, C.J. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Singh, V.K. Practical and Useful Methods for the Enantioselective Reduction of Unsymmetrical Ketones. Synthesis 1992, 1992, 605–617. [Google Scholar] [CrossRef]
- Mathre, D.J.; Shinkai, I.; Zaidlewicz, M.; Krzemiński, M.; Łączkowski, K.; Patti, A.; Pedotti, S. Tetrahydro-1-methyl-3,3-diphenyl-1H, 3H-pyrrolo[1,2-c][1,3,2]oxazaborole. In Encyclopedia of Reagents for Organic Synthesis; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Brunel, J.M.; Maffei, M.; Buono, G. Enantioselective reduction of ketones with borane, catalyzed by (S)-(−)-proline or (S)-(+)-prolinol. Tetrahedron Asymmetry 1993, 4, 2255–2260. [Google Scholar] [CrossRef]
- Xu, J.; Wei, T.; Lin, S.-S.; Zhang, Q. Rationale on the Abnormal Effect of Temperature on the Enantioselectivity in the Asymmetric Borane Reduction of Ketones Catalyzed by L-Prolinol. Helv. Chim. Acta 2005, 88, 180–186. [Google Scholar] [CrossRef]
- Quallich, G.J.; Woodall, T.M. In Situ Oxazaborolidines, Practical Enantioselective Hydride Reagents. Synlett 1993, 12, 929–930. [Google Scholar] [CrossRef]
- Masui, M.; Shioiri, T. A practical method for preparation of optically pure oxazaborolidines from α-Pinene. Tetrahedron 1995, 51, 8363–8370. [Google Scholar] [CrossRef]
- Corey, E.J.; Link, J.O. A new process for the generation of 1,3,2-oxazaborolidines, catalysts for enantioselective synthesis. Tetrahedron Lett. 1992, 33, 4141–4144. [Google Scholar] [CrossRef]
- Wu, X.F.; Min, C.; Nyamzundui, E.; Zhou, H.B.; Dong, C. A novel C 3-symmetric prolinol-squaramide catalyst for the asymmetric reduction of ketones by borane. Tetrahedron Asymmetry 2011, 22, 1640–1643. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, H.B.; Lü, S.M.; Luo, M.M.; Xie, R.G.; Choi, M.C.K.; Zhou, Z.Y.; Chan, A.S.C.; Yang, T.K. Chiral squaric prolinols: A new type of ligand for the asymmetric reduction of prochiral ketones by borane. Tetrahedron Asymmetry 2001, 12, 1907–1912. [Google Scholar] [CrossRef]
- Zhou, H.B.; Zhang, J.; Lü, S.M.; Xie, R.G.; Zhou, Z.Y.; Choi, M.C.K.; Chan, A.S.C.; Yang, T.K. Design, synthesis and structure of new chiral squaric acid monoaminoalcohols and diaminoalcohols and their use as catalysts in asymmetric reduction of ketones and diketones. Tetrahedron 2001, 57, 9325–9333. [Google Scholar] [CrossRef]
- Zou, H.-H.; Hu, J.; Zhang, J.; You, J.-S.; Ma, D.; Lü, D.; Xie, R.-G. Asymmetric reduction of prochiral ketones with borane using chiral squaric amino alcohols derived from camphor as catalysts. J. Mol. Catal. A Chem. 2005, 242, 57–61. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Trifunctional Squaramide Catalyst for Efficient Enantioselective Henry Reaction Activation. Adv. Synth. Catal. 2016, 358, 1801–1809. [Google Scholar] [CrossRef] [Green Version]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Guerola, M.; Escolano, M.; Alzuet-Piña, G.; Gómez-Bengoa, E.; Ramírez de Arellano, C.; Sánchez-Roselló, M.; del Pozo, C. Synthesis of substituted piperidines by enantioselective desymmetrizing intramolecular aza-Michael reactions. Org. Biomol. Chem. 2018, 16, 4650–4658. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-L.; Chang, C.-H. An asymmetric assembly of spirooxindole dihydropyranones through a direct enantioselective organocatalytic vinylogous aldol-cyclization cascade reaction of 3-alkylidene oxindoles with isatins. Chem. Commun. 2016, 52, 2322–2325. [Google Scholar] [CrossRef]
- Jin, X.; Min, Q.; Zheng, Y.; Wang, P.; Zhu, J.; Zhou, H.B. Synthesis and structural features of chiral cyclic squaramides and their application in asymmetric catalytic reaction. Arkivoc 2010, 11, 322–335. [Google Scholar] [CrossRef] [Green Version]
- Pace, W.H.; Mo, D.L.; Reidl, T.W.; Wink, D.J.; Anderson, L.L. Catalytic Asymmetric Synthesis of Dihydropyrido[1,2-a]indoles from Nitrones and Allenoates. Angew. Chem. Int. Ed. 2016, 55, 9183–9186. [Google Scholar] [CrossRef]
- Zhao, M.-X.; Dong, Z.-W.; Zhu, G.-Y.; Zhao, X.-L.; Shi, M. Diastereo- and enantioselective Mannich/cyclization cascade reaction of isocyanoacetates with cyclic sulfamide ketimines by cinchona alkaloid squaramide/AgOAc cooperative catalysis. Org. Biomol. Chem. 2018, 16, 4641–4649. [Google Scholar] [CrossRef]
- Cortadellas, O.; Galibert, A.M.; Soula, B.; Donnadieu, B.; Fabre, P.L. Palladium(II) complexes obtained from 3,4-bis(cyanamido)cyclobutane-1,2-dione dianion. Inorg. Chim. Acta 2004, 357, 746–754. [Google Scholar] [CrossRef]
- Seleem, H.S.; Ramadan, A.A.T.; Taha, A.; Samy, F. Unreported Coordination Behavior of a Squaric Bis (Hydrazone) Ligand. Res. J. Chem. Sci. 2011, 1, 109–116. [Google Scholar]
- Zhang, X.; Zuo, Z.; Tang, J.; Wang, K.; Wang, C.; Chen, W.; Li, C.; Xu, W.; Xiong, X.; Yuntai, K.; et al. Design, synthesis and biological evaluation of novel estrogen-derived steroid metal complexes. Bioorg. Med. Chem. Lett. 2013, 23, 3793–3797. [Google Scholar] [CrossRef]
- Johnson, C.S. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Denkova, P.; Momekova, D.; Rangelov, S.; Lambov, N.; Willem, R. Investigation of sterically stabilized liposomes by diffusion ordered NMR spectroscopy. J. Control. Release 2010, 148, e47–e48. [Google Scholar] [CrossRef]
- Denkova, P.S.; Van Lokeren, L.; Willem, R. Mixed Micelles of Triton X-100, Sodium Dodecyl Dioxyethylene Sulfate, and Synperonic L61 Investigated by NOESY and Diffusion Ordered NMR Spectroscopy. J. Phys. Chem. B 2009, 113, 6703–6709. [Google Scholar] [CrossRef]
- Kowalczuk, A.; Stoyanova, E.; Mitova, V.; Shestakova, P.; Momekov, G.; Momekova, D.; Koseva, N. Star-shaped nano-conjugates of cisplatin with high drug payload. Int. J. Pharm. 2011, 404, 220–230. [Google Scholar] [CrossRef]
- Ulrich, K.; Sanders, M.; Grinberg, F.; Galvosas, P.; Vasenkov, S. Application of Pulsed Field Gradient NMR with High Gradient Strength for Studies of Self-Diffusion in Lipid Membranes on the Nanoscale. Langmuir 2008, 24, 7365–7370. [Google Scholar] [CrossRef]
- Valentini, M.; Vaccaro, A.; Rehor, A.; Napoli, A.; Hubbell, J.A.; Tirelli, N. Diffusion NMR Spectroscopy for the Characterization of the Size and Interactions of Colloidal Matter: The Case of Vesicles and Nanoparticles. J. Am. Chem. Soc. 2004, 126, 2142–2147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Beckham, H.W. Direct Synthesis of Cyclodextrin-Rotaxanated Poly(ethylene glycol)s and Their Self-Diffusion Behavior in Dilute Solution. Macromolecules 2003, 36, 9859–9865. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusions-NMR-Spektroskopie in der Supramolekularen und Kombinatorischen Chemie: Ein alter Parameter—Neue Erkenntnisse. Angew. Chem. 2004, 117, 524–560. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Ma, Z.; Hopson, R.; Wei, Y.; Budil, D.; Gulla, S.; Moulton, B. Postsynthetic modification of a coordination compound with a paddlewheel motif via click reaction: DOSY and ESR studies. Inorg. Chem. Commun. 2012, 15, 78–83. [Google Scholar] [CrossRef]
- Pastor, A.; Martínez-Viviente, E. NMR spectroscopy in coordination supramolecular chemistry: A unique and powerful methodology. Coord. Chem. Rev. 2008, 252, 2314–2345. [Google Scholar] [CrossRef]
- Fernández, I.; Martínez-Viviente, E.; Breher, F.; Pregosin Paul, S. 7Li, 31P, and 1H Pulsed Gradient Spin-Echo (PGSE) Diffusion NMR Spectroscopy and Ion Pairing: On the Temperature Dependence of the Ion Pairing in Li(CPh3), Fluorenyllithium, and Li[N(SiMe3)2] amongst Other Salts. Chem. Eur. J. 2005, 11, 1495–1506. [Google Scholar] [CrossRef]
- Martínez-Viviente, E.; Rüegger, H.; Pregosin, P.S.; López-Serrano, J. 31P and 35Cl PGSE Diffusion Studies on Phosphine Ligands and Selected Organometallic Complexes. Solvent Dependence of Ion Pairing. Organometallics 2002, 21, 5841–5846. [Google Scholar] [CrossRef]
- Nama, D.; Anil Kumar, P.G.; Pregosin Paul, S. 195Pt, 1H and 31P PGSE diffusion studies on platinum complexes. Magn. Reson. Chem. 2004, 43, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Jasperse, C.P.; Sibi, M.P. Characterization of Brønsted Acid–Base Complexes by 19F DOSY. Org. Lett. 2015, 17, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Keresztes, I.; Hopson, R.; Williard, P.G. Characterization of Reactive Intermediates by Multinuclear Diffusion-Ordered NMR Spectroscopy (DOSY). Acc. Chem. Res. 2009, 42, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong Thi Kim, N.; Shestakova, P.; Mihaylov Tzvetan, T.; Absillis, G.; Pierloot, K.; Parac-Vogt Tatjana, N. Multinuclear Diffusion NMR Spectroscopy and DFT Modeling: A Powerful Combination for Unraveling the Mechanism of Phosphoester Bond Hydrolysis Catalyzed by Metal-Substituted Polyoxometalates. Chem. Eur. J. 2015, 21, 4428–4439. [Google Scholar] [CrossRef]
- Luong, T.K.N.; Shestakova, P.; Absillis, G.; Parac-Vogt, T.N. Detailed Mechanism of Phosphoanhydride Bond Hydrolysis Promoted by a Binuclear ZrIV-Substituted Keggin Polyoxometalate Elucidated by a Combination of 31P, 31P DOSY, and 31P EXSY NMR Spectroscopy. Inorg. Chem. 2016, 55, 4864–4873. [Google Scholar] [CrossRef]
- McArthur, D.; Butts, C.P.; Lindsay, D.M. A dialkylborenium ion via reaction of N-heterocyclic carbene-organoboranes with Bronsted acids-synthesis and DOSY NMR studies. Chem. Commun. 2011, 47, 6650–6652. [Google Scholar] [CrossRef] [PubMed]
- Nakakoshi, M.; Ueda, M.; Sakurai, S.; Miyata, O.; Sugiura, M.; Naito, T. Structure elucidation of the intermediate in triethylborane-mediated radical addition of oxime ethers with 2D- and 3D-DOSY NMR. Magn. Reson. Chem. 2006, 44, 807–812. [Google Scholar] [CrossRef]
- Qiao, Y.; Ge, W.; Jia, L.; Hou, X.; Wang, Y.; Pedersen, C.M. Glycosylation intermediates studied using low temperature 1H- and 19F-DOSY NMR: New insight into the activation of trichloroacetimidates. Chem. Commun. 2016, 52, 11418–11421. [Google Scholar] [CrossRef] [Green Version]
- Schober, K.; Hartmann, E.; Zhang, H.; Gschwind Ruth, M. 1H DOSY Spectra of Ligands for Highly Enantioselective Reactions—A Fast and Simple NMR Method to Optimize Catalytic Reaction Conditions. Angew. Chem. Int. Ed. 2010, 49, 2794–2797. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, V.; Dobrikov, G.; Genov, M. Chiral β- and γ-aminoalcohols derived from (+)-camphor and (-)-fenchone as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2001, 12, 1323–1329. [Google Scholar] [CrossRef]
- Chen, Y.K.; Jeon, S.J.; Walsh, P.J.; Nugent, W.A. (2S)-(-)-3-exo-(morpholino)isoborneol[(-)-MIB]. Org. Synth. 2005, 82, 87–92. [Google Scholar] [CrossRef]
- Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E.; Yin, J. Parallel Kinetic Resolution of Acyclic γ-Amino-α,β-unsaturated Esters: Application to the Asymmetric Synthesis of 4-Aminopyrrolidin-2-ones. Org. Lett. 2012, 14, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Santhi, V.; Rao, J.M. Asymmetric reduction of prochiral ketones using in situ generated oxazaborolidines derived from amino alcohols of (1R)-camphor as catalysts. Tetrahedron Asymmetry 2000, 11, 3553–3560. [Google Scholar] [CrossRef]
- Nevalainen, V. Quantum chemical modeling of chiral catalysis. Part 6. On the relative stability of dimers of chiral oxazaborolidines used in the catalytic enantioselective reduction of ketones. Tetrahedron Asymmetry 1992, 3, 933–945. [Google Scholar] [CrossRef]
- Nevalainen, V. Quantum chemical modeling of chiral catalysis. Part 3. On the role of a Lewis basic solvent in the mechanism of catalytic enantioselective reduction of carbonyl compounds by chiral oxazaborolidines. Tetrahedron Asymmetry 1991, 2, 827–842. [Google Scholar] [CrossRef]
- Kettouche, H.S. A DFT study on the reaction mechanism of enantioselective reduction of ketones with borane catalyzed by a B-methoxy-oxazaborolidine catalyst derived from (–)-β-pinene. J. Mol. Model. 2020, 26, 27. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Atherton, J.C.C. Asymmetric reduction using N-methyl and N-benzyl oxazaborolidines based upon cis-1-amino-2-indanol: A preliminary mechanistic study. Tetrahedron Asymmetry 2000, 11, 4543–4548. [Google Scholar] [CrossRef]
- Gilmore, N.J.; Jones, S. Evaluating the role of solvent and borane on the enantioselectivity of the oxazaborolidine reduction of prochiral ketones using catalysts derived from cis-(1R,2S)-1-amino-indan-2-ol. Tetrahedron Asymmetry 2003, 14, 2115–2118. [Google Scholar] [CrossRef]
- Stepanenko, V.; Ortiz-Marciales, M.; Barnes, C.E.; Garcia, C. Studies on the synthesis of borazines from borane and 1,2-aminoalcohols. Tetrahedron Lett. 2006, 47, 7603–7606. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Tang, M.; Zhao, J.; Sun, L.; Zhang, W.; Zhao, C.; Zhang, S.; Wang, H. A DFT study of the enantioselective reduction of prochiral ketones promoted by pinene-derived amino alcohols. Tetrahedron Asymmetry 2009, 20, 1020–1026. [Google Scholar] [CrossRef]
- Rotger, M.C.; Piña, M.N.; Frontera, A.; Martorell, G.; Ballester, P.; Deyà, P.M.; Costa, A. Conformational Preferences and Self-Template Macrocyclization of Squaramide-Based Foldable Modules. J. Org. Chem. 2004, 69, 2302–2308. [Google Scholar] [CrossRef]
- Jaganyi, D.; Mzinyati, A. An 11B NMR spectroscopy investigation of the mechanism of the reduction of nitriles by BH3·SMe2. Polyhedron 2006, 25, 2730–2736. [Google Scholar] [CrossRef]
- Krzemiński, M.P.; Ćwiklińska, M. Chiral terpene auxiliaries II. Spiroborate esters derived from α-pinene—New catalysts for asymmetric borane reduction of prochiral ketones. Tetrahedron Lett. 2011, 52, 3919–3921. [Google Scholar] [CrossRef]
- Király, P. Background-free solution boron NMR spectroscopy. Magn. Reson. Chem. 2012, 50, 620–626. [Google Scholar] [CrossRef]
- Brown, J.M.; Lloyd-Jones, G.C.; Layzell, T.P. Reversible dimerisation of ephedrine-derived oxazaborolidines. Tetrahedron Asymmetry 1993, 4, 2151–2154. [Google Scholar] [CrossRef]
- Corey, E.J.; Link, J.O. A new chiral catalyst for the enantioselective synthesis of secondary alcohols and deuterated primary alcohols by carbonyl reduction. Tetrahedron Lett. 1989, 30, 6275–6278. [Google Scholar] [CrossRef]
- Kawanami, Y.; Yanagita, R.C. Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols. Molecules 2018, 23, 2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siedle, A.R. 11B NMR Spectroscopy. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Academic Press: Cambridge, MA, USA, 1988; Volume 20, pp. 205–314. [Google Scholar]
- Wrackmeyer, B. Nuclear Magnetic Resonance Spectroscopy of Boron. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Academic Press: Cambridge, MA, USA, 1988; Volume 20, pp. 61–203. [Google Scholar]
- Bosiak, M.J.; Krzemiński, M.P.; Jaisankar, P.; Zaidlewicz, M. Asymmetric synthesis of N-1-(heteroaryl)ethyl-N-hydroxyureas. Tetrahedron Asymmetry 2008, 19, 956–963. [Google Scholar] [CrossRef]
- Carman, R.M.; Greenfield, K.L. The endo- and exo-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-amines (Bornan-2-amines) and their acetamides. Aust. J. Chem. 1984, 37, 1785–1790. [Google Scholar] [CrossRef]
- Ipaktschi, J. Reduktion von Oximen mit Natriumboranat in Gegenwart von Übergangsmetallverbindungen. Chem. Ber. 1984, 117, 856–858. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Squaric Acid Amides | Free Aminoalcohols | ||||||
| Entry | Ligand | Yield (%) | eea (Config.) | Entry | Ligand | Yield (%) | eea (Config.) |
| 1 | 4 | 99 | 53 (R) | 19 | 2b | 98 | 79 (R) |
| 2 | 5 | 99 | 31 (S) | ||||
| 3 | 7b | 72 | 39 (R) | ||||
| 4 | 8b | 99 | 42 (R) | 20 | 2c | 97 | 83 (S) |
| 5 | 9b | 86 | 0 | ||||
| 6 | 9c | 99 | 19 (S) | ||||
| 7 | 7f | 92 | 0 | 21 | 2f | 99 | 9 (S) |
| 8 | 8f | 99 | 56 (R) | ||||
| 9 | 7e | 95 | 10 (R) | 22 | 2e | 99 | 49 (R) |
| 10 | 8e | 99 | 49 (R) | ||||
| 11 | 9e | 99 | 35 (S) | ||||
| 12 | 7h | 80 | 13 (S) | 23 | 2h | 80 | 61 (S) |
| 13 | 7i | 99 | 37 (S) | 24 | 2i b | 98 | 44 (R) |
| 14 | 7g | 99 | 56 (R) | 25 | 2gc | 94 | 79 (S) |
| 15 | 10 | 98 | 0 | 26 | 6 | 96 | 0 |
| 16 | 13a | 99 | 0 | - | - | - | - |
| 17 | 13b | 93 | 0 | - | - | - | - |
| 18 | 18 | 92 | 26 (S) | 27 | 17 | 99 | 94 (S) |
| Entry | Ligand (Equiv.) a | Temperature (°C) | Solvent | Time (min) | Yield (%) | eec (Config.) |
|---|---|---|---|---|---|---|
| 1 | 2c (0.1) | 50 | THF | 20 | 96 | 83 (S) |
| 2 | 2c (0.1) b | 50 | THF | 5 | 91 | 86 (S) |
| 3 | 2c (0.1) | 25 | THF | 30 | 92 | 73 (S) |
| 4 | 2c (0.1) | 0 | THF | 240 | 99 | 22 (S) |
| 5 | 2c (0.1) | 50 | Toluene | 20 | 97 | 70 (S) |
| 6 | 2c (0.1) | 100 | Toluene | 5 | 91 | 80 (S) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolova, Y.; Dobrikov, G.M.; Petkova, Z.; Shestakova, P. Chiral Aminoalcohols and Squaric Acid Amides as Ligands for Asymmetric Borane Reduction of Ketones: Insight to In Situ Formed Catalytic System by DOSY and Multinuclear NMR Experiments. Molecules 2021, 26, 6865. https://doi.org/10.3390/molecules26226865
Nikolova Y, Dobrikov GM, Petkova Z, Shestakova P. Chiral Aminoalcohols and Squaric Acid Amides as Ligands for Asymmetric Borane Reduction of Ketones: Insight to In Situ Formed Catalytic System by DOSY and Multinuclear NMR Experiments. Molecules. 2021; 26(22):6865. https://doi.org/10.3390/molecules26226865
Chicago/Turabian StyleNikolova, Yana, Georgi M. Dobrikov, Zhanina Petkova, and Pavletta Shestakova. 2021. "Chiral Aminoalcohols and Squaric Acid Amides as Ligands for Asymmetric Borane Reduction of Ketones: Insight to In Situ Formed Catalytic System by DOSY and Multinuclear NMR Experiments" Molecules 26, no. 22: 6865. https://doi.org/10.3390/molecules26226865
APA StyleNikolova, Y., Dobrikov, G. M., Petkova, Z., & Shestakova, P. (2021). Chiral Aminoalcohols and Squaric Acid Amides as Ligands for Asymmetric Borane Reduction of Ketones: Insight to In Situ Formed Catalytic System by DOSY and Multinuclear NMR Experiments. Molecules, 26(22), 6865. https://doi.org/10.3390/molecules26226865