
Synthesis of Methyl 4,6-Di-O-ethyl-α-d-glucopyranoside-Based Azacrown Ethers and Their Effects in Asymmetric Reactions




Abstract
:
1. Introduction
2. Results and Discussion
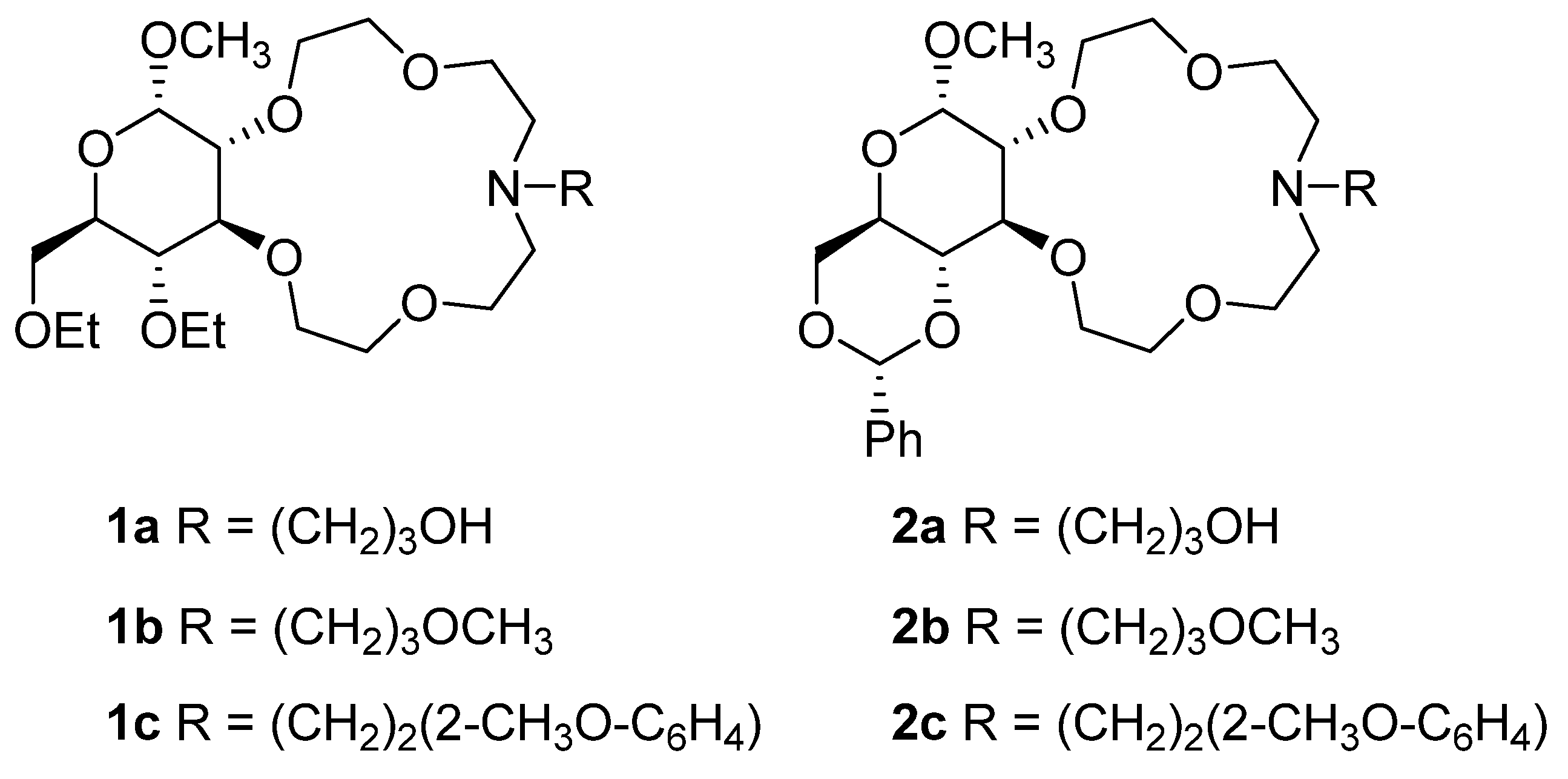
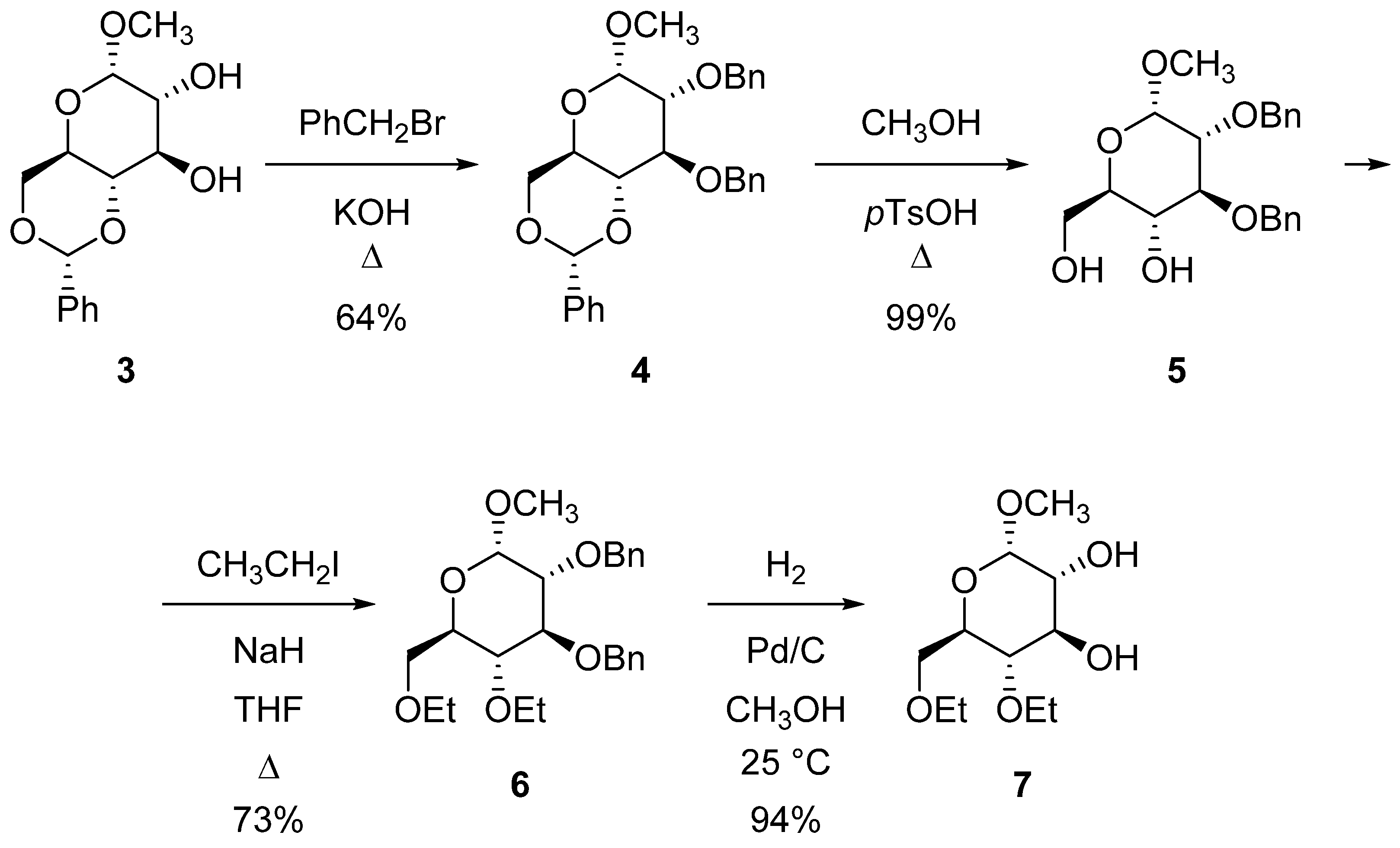
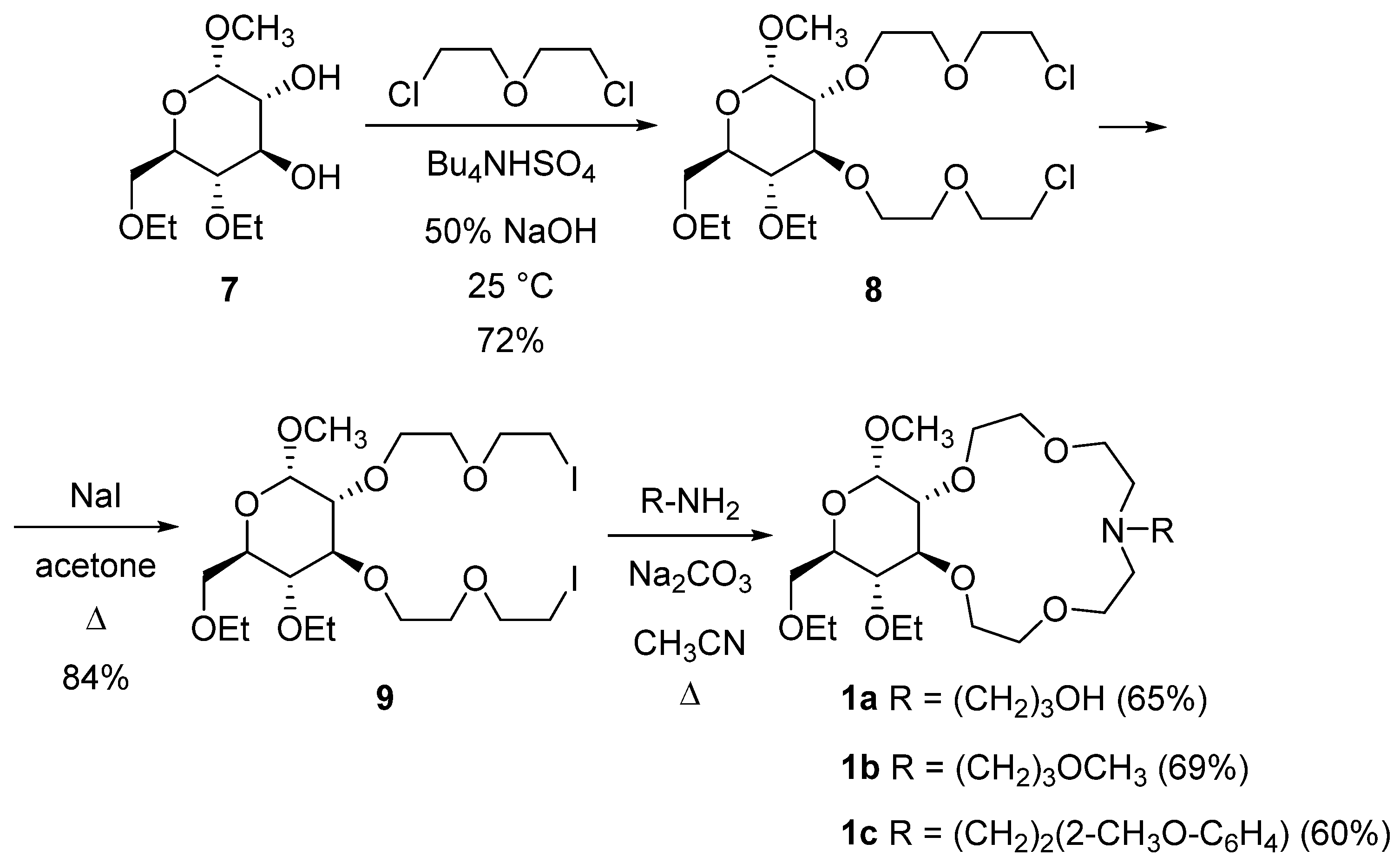
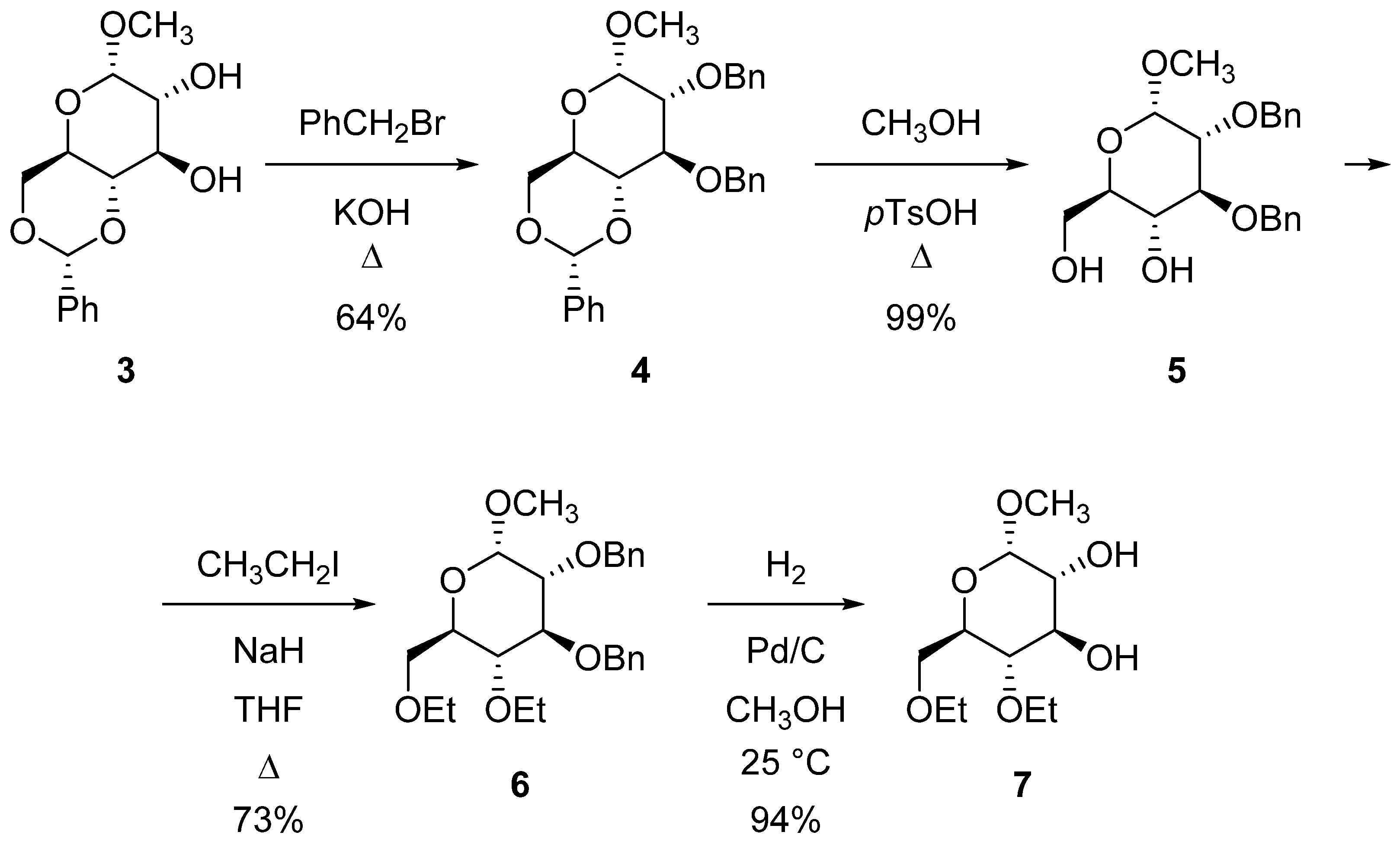
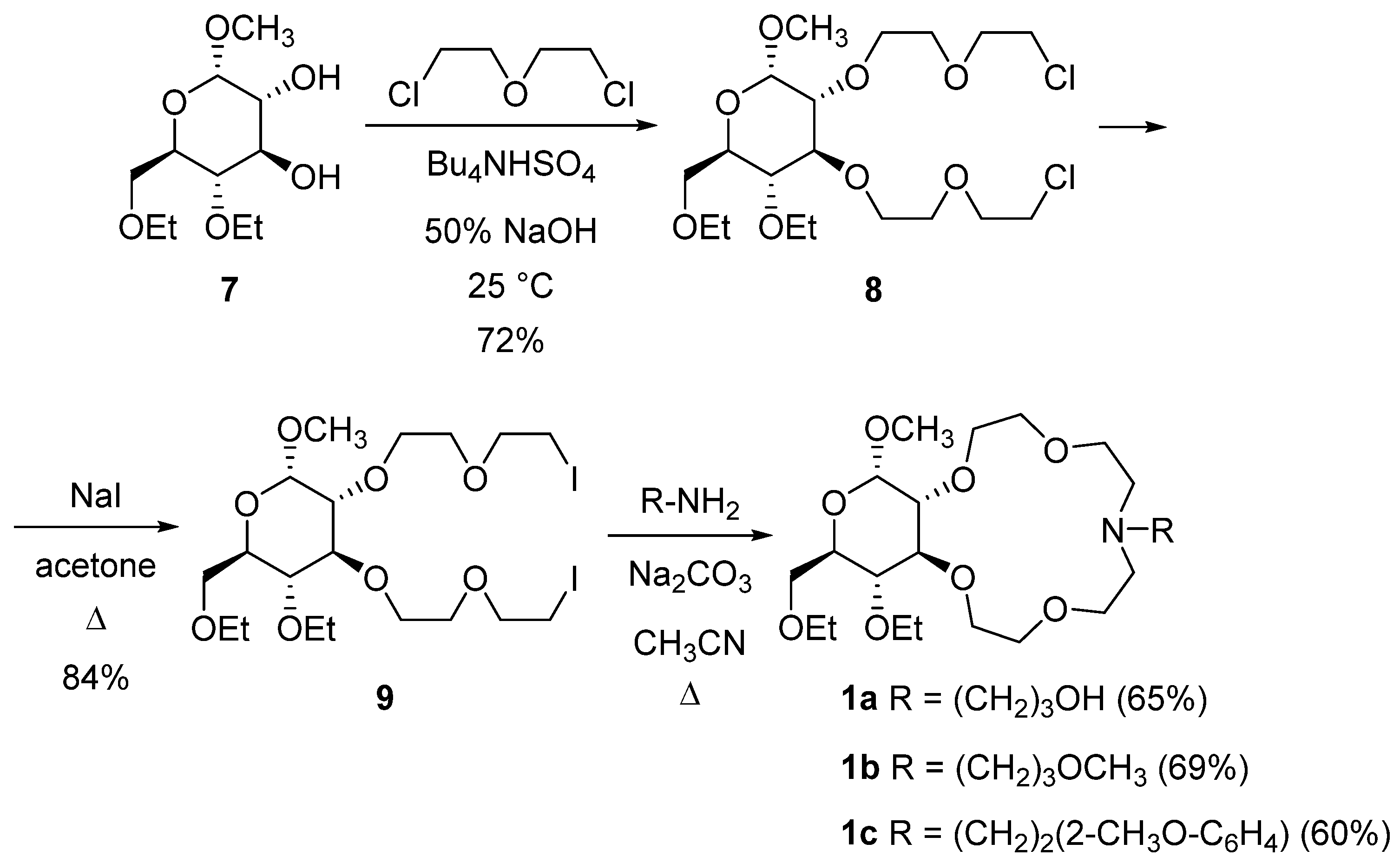
2.1. Synthesis of Azacrown Ethers
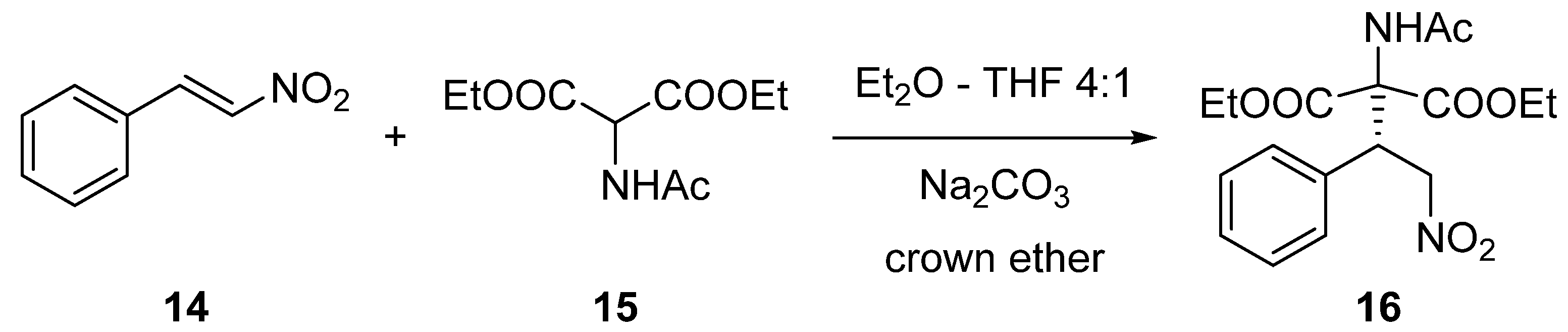
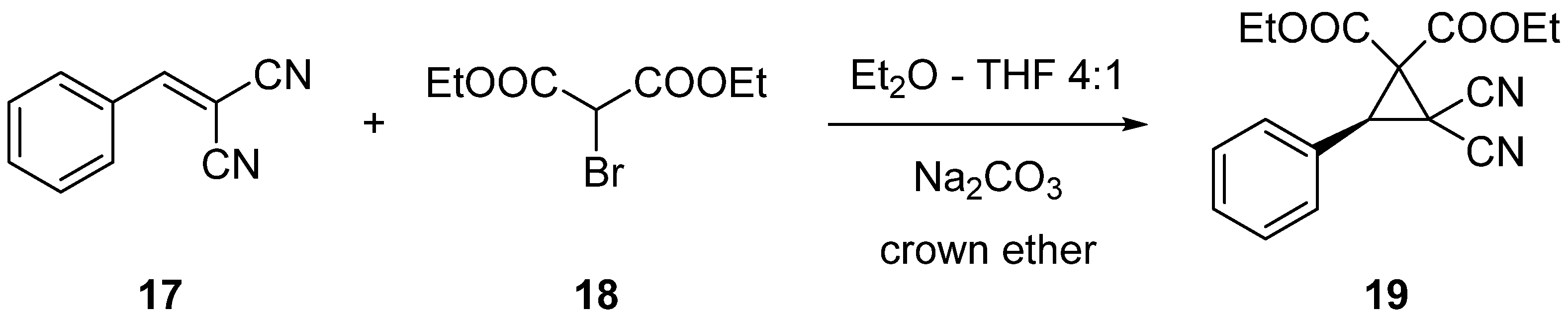
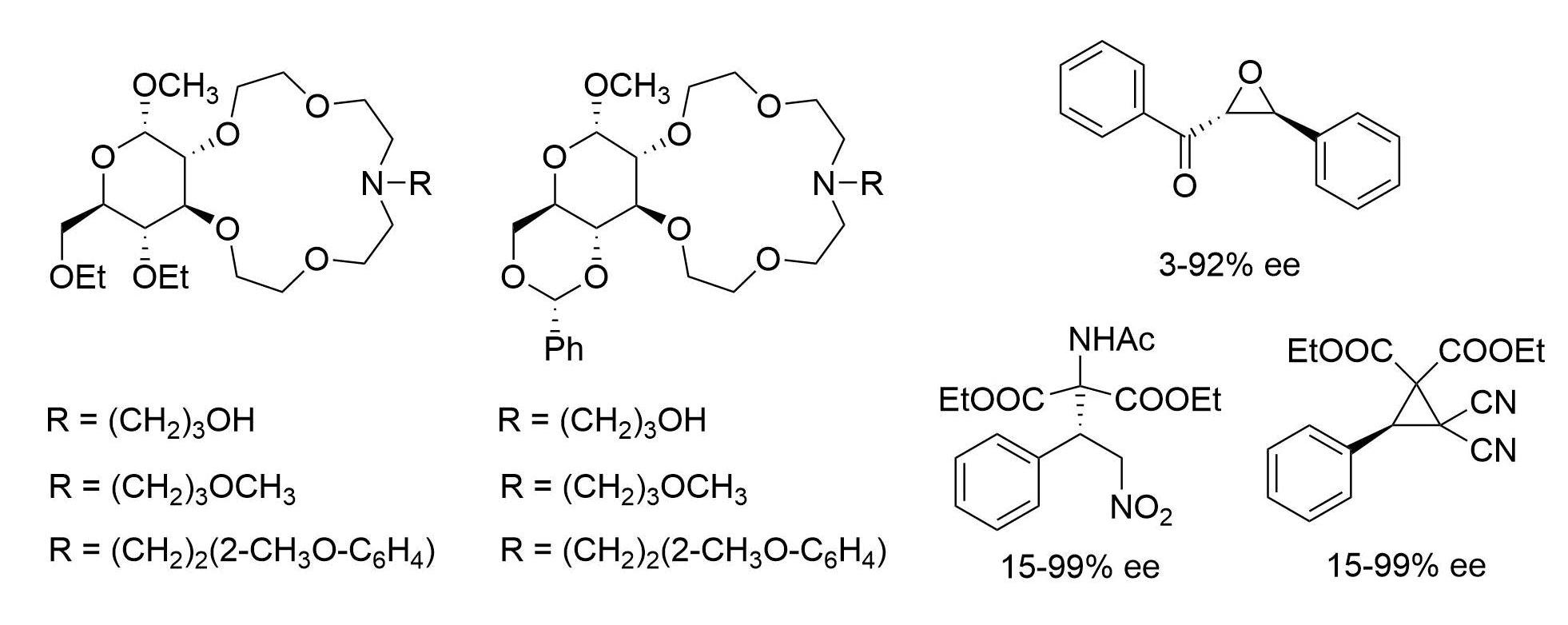
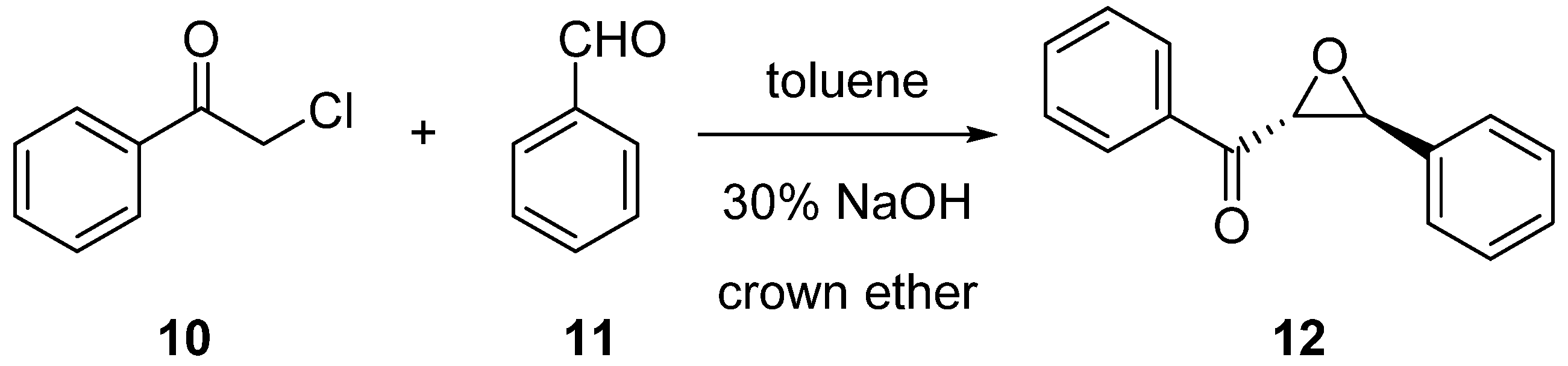
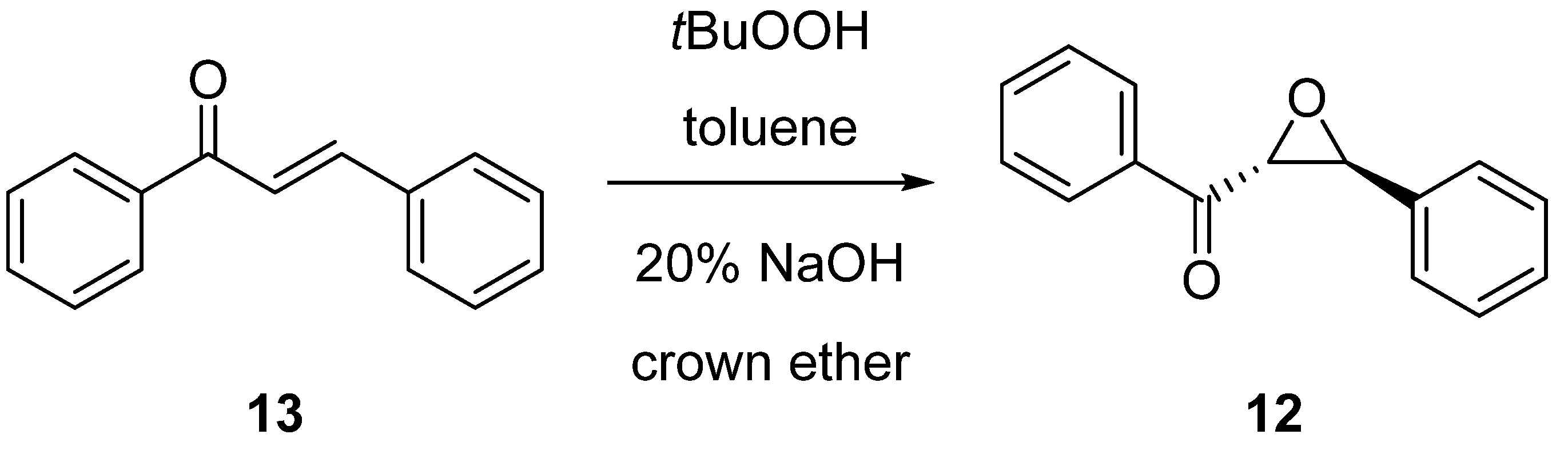
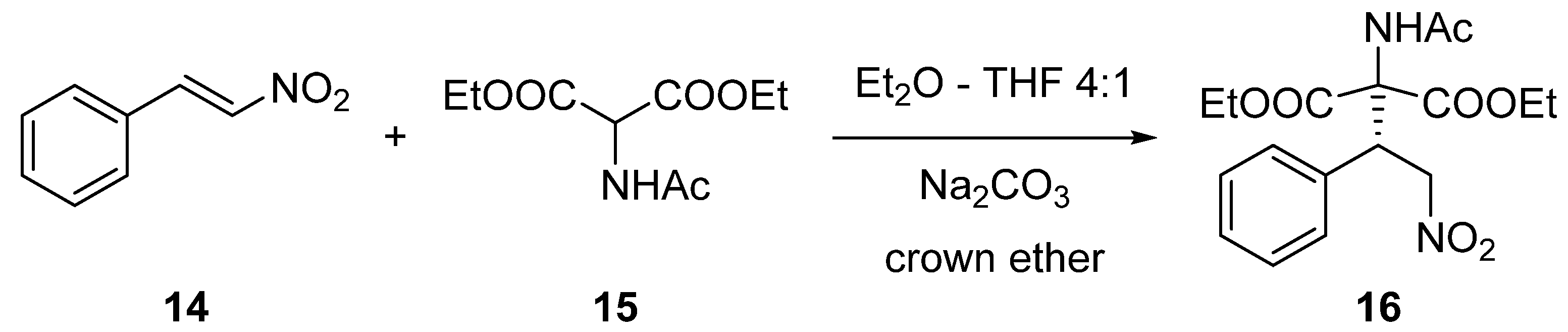
2.2. Enantioselective Reactions
3. Materials and Methods
3.1. General
3.2. Synthesis of Crown Ethers
3.2.1. Methyl-2,3-di-O-benzyl-4,6-di-O-ethyl-α-d-glucopyranoside (6)
3.2.2. Methyl-4,6-di-O-ethyl-α-d-glucopyranoside (7)
3.2.3. Methyl-4,6-di-O-ethyl-2,3-bis-O-[(2-chloroethoxy)ethyl]-α-d-glucopyranoside (8)
3.2.4. Methyl-4,6-di-O-ethyl-2,3-bis-O-[(2-iodoethoxy)ethyl]-α-d-glucopyranoside (9)
3.2.5. Methyl-4,6-di-O-ethyl-2,3-dideoxy-α-d-glucopyranosido[2,3-h]-N-[3-hydroxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1a)
3.2.6. Methyl-4,6-di-O-ethyl-2,3-dideoxy-α-d-glucopyranosido[2,3-h]-N-[3-methoxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1b)
3.2.7. Methyl-4,6-di-O-ethyl-2,3-dideoxy-α-d-glucopyranosido[2,3-h]-N-[2-(2-methoxyphenyl)ethyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1c)
3.3. Asymmetric Reactions
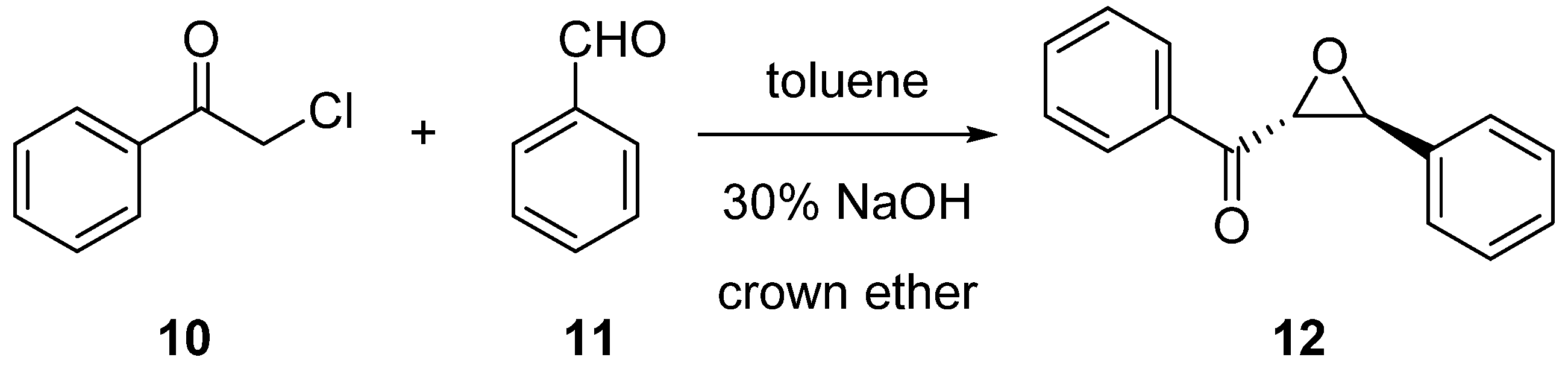
3.3.1. Synthesis of (2R,3S)-Phenyl(3-phenyloxirane-2-yl)methanone (12) via Darzens Condensation
3.3.2. Synthesis of (2R,3S)-Phenyl(3-phenyloxirane-2-yl)methanone (12) via Epoxidation of trans-Chalcone
3.3.3. Synthesis of (S)-Diethyl 2-Acetamido-2-(2-nitro-1-phenylethyl)malonate (16) via Michael Addition
3.3.4. Synthesis of (R)-Diethyl 2,2-Dicyano-3-phenylcyclopropane-1,1-dicarboxylate (19) via MIRC Reaction
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- List, B.; Lerner, A.R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels–Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis, 1st ed.; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Macmillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, K.; Gawronski, J. Cinchona Alkaloids and Their Derivatives: Versatile Catalysts and Ligands in Asymmetric Synthesis. Synthesis 2001, 961–998. [Google Scholar]
- McCooey, S.H.; Connon, S.J. Urea- and Thiourea-Substituted Cinchona Alkaloid Derivatives as Highly Efficient Bifunctional Organocatalysts for the Asymmetric Addition of Malonate to Nitroalkenes: Inversion of Configuration at C9 Dramatically Improves Catalyst Performance. Angew. Chem. Int. Ed. 2005, 44, 6367–6370. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly Enantioselective Conjugate Addition of Nitromethane to Chalcones Using Bifunctional Cinchona Organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.; Schubert, G.; Soós, A.T.; Pápai, I. Theoretical Studies on the Bifunctionality of Chiral Thiourea-Based Organocatalysts: Competing Routes to C–C Bond Formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Carlone, A.; Cavalli, A.; Locatelli, M.; Mazzanti, A.; Ricci, P.; Sambri, L.; Melchiorre, P. Organocatalytic Asymmetric Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides. Angew. Chem. Int. Ed. 2006, 45, 4966–4970. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [Green Version]
- Kótai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Pápai, I.; Soós, T. On the Mechanism of Bifunctional Squaramide-Catalyzed Organocatalytic Michael Addition: A Protonated Catalyst as an Oxyanion Hole. Chem. Eur. J. 2014, 20, 5631–5639. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Mika, L.T.; Csámpai, A.; Holczbauer, T.; Kardos, G.; Soós, T. Mechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction. RSC Adv. 2015, 5, 95079–95086. [Google Scholar] [CrossRef] [Green Version]
- Grayson, M.N. Mechanism and Origins of Stereoselectivity in the Cinchona Thiourea- and Squaramide-Catalyzed Asymmetric Michael Addition of Nitroalkanes to Enones. J. Org. Chem. 2017, 82, 4396–4401. [Google Scholar] [CrossRef] [PubMed]
- Boratynski, P.J.; Zielinska-Błajet, M.; Skarżewski, J. Chapter Two—Cinchona Alkaloids—Derivatives and Applications. In The Alkaloids, 1st ed.; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 29–145. [Google Scholar]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 3rd ed.; VCH: New York, NY, USA, 1993. [Google Scholar]
- Starks, C.M.; Liotta, C.L.; Halpern, M.E. Phase Transfer Catalysis: Fundamentals, Applications, and Industrial Perspectives; Chapman & Hall: New York, NY, USA, 1994. [Google Scholar]
- O’ Donnell, M.I. Asymmetric Phase Transfer Reactions. In Catalytic Asymmetric Synthesis, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, NY, USA, 2000; pp. 727–745. [Google Scholar]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Marouka, K. Asymmetric Phase Transfer Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Kumaraguru, D.; Arockiam, J.B.; Paulpandian, S.; Rajendiran, B.; Siva, A. Highly enantioselective asymmetric Michael addition reactions with new chiral multisite phase-transfer catalysts. Synlett 2014, 25, 1685–1691. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A new generation of chiral phase-transfer catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef] [Green Version]
- Schettini, R.; De Riccardis, F.; Della Sala, G.; Izzo, I. Enantioselective Alkylation of Amino Acid Derivatives Promoted by Cyclic Peptoids under Phase-Transfer Conditions. J. Org. Chem. 2016, 81, 2494–2505. [Google Scholar] [CrossRef] [PubMed]
- Schörgenhumer, J.; Tiffner, M.; Waser, M. Chiral phase-transfer catalysis in the asymmetric α-heterofunctionalization of prochiral nucleophiles. Beilstein J. Org. Chem. 2017, 13, 1753–1769. [Google Scholar] [CrossRef] [Green Version]
- Schettini, R.; Sicignano, M.; De Riccardis, F.; Izzo, I.; Della Sala, G. Macrocyclic Hosts in Asymmetric Phase-Transfer Catalyzed Reactions. Synthesis 2018, 50, 4777–4795. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, G. Enantioselective Mannich reaction of γ-malonate-substituted α,β-unsaturated esters with N-Boc imines catalyzed by chiral bifunctional thiourea-phosphonium salts. Tetrahedron 2019, 75, 1697–1705. [Google Scholar] [CrossRef]
- Mahajan, D.P.; Godbole, H.M.; Singh, G.P.; Shenoy, G.G. Synthesis of novel phase transfer catalysts derived from proline-mandelic acid/tartaric acid: Their evaluation in enantioselective epoxidation and Darzens condensation. J. Chem. Sci. 2019, 131, 22. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Wu, J.-H.; Zhang, H.; Ren, X.; Tan, J.-P.; Zhu, L.; Zhang, H.-S.; Jiang, C.; Wang, T. Highly Enantioselective Synthesis of Fused Tri- and Tetrasubstituted Aziridines: Aza-Darzens Reaction of Cyclic Imines with α-Halogenated Ketones Catalyzed by Bifunctional Phosphonium Salt. Angew. Chem. Int. Ed. 2019, 58, 7425–7430. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, K. Design of high-performance chiral phase-transfer catalysts with privileged structures. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, H. Chiral Phase-Transfer Catalysts with Hydrogen Bond: A Powerful Tool in the Asymmetric Synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Majdecki, M.; Tyszka-Gumkowska, A.; Jurczak, J. Highly Enantioselective Epoxidation of α,β-unsaturated ketones using amide-based cinchona alkaloids as hybrid phase-transfer catalysts. Org. Lett. 2020, 22, 8687–8691. [Google Scholar] [CrossRef]
- Lu, D.; Liu, X.; Wu, J.-H.; Zhang, S.; Tan, J.-P.; Yu, X.; Wang, T. Asymmetric Construction of Bispiro-Cyclopropane-Pyrazolones via a [2+1] Cyclization Reaction by Dipeptide-Based Phosphonium Salt Catalysis. Adv. Synth. Catal. 2020, 362, 1966–1971. [Google Scholar] [CrossRef]
- Tian, Z.; Meng, X.; Luo, Y.; Cao, S.; Zhao, G. A novel quaternary ammonium salts derived from α-amino acids with large steric hindrance group and its application in asymmetric Mannich reaction. Tetrahedron 2020, 76, 131484. [Google Scholar] [CrossRef]
- Genov, G.R.; Douthwaite, J.L.; Lahdenperä, A.S.K.; Gibson, D.C.; Phipps, R.J. Enantioselective remote C–H activation directed by a chiral cation. Science 2020, 367, 1246–1251. [Google Scholar] [CrossRef] [Green Version]
- Sabah, K.J.; Zahid, N.I.; Hashim, R. Synthesis of new chiral macrocycles-based glycolipids and its application in asymmetric Michael addition. Res. Chem. Intermed. 2021, 47, 2653–2667. [Google Scholar] [CrossRef]
- Cram, D.J.; Sogah, G.D.Y. Chiral Crown Complexes Catalyze Michael Addition Reactions to Give Adducts in High Optical Yields. J. Chem. Soc. Chem. Commun. 1981, 13, 625–628. [Google Scholar] [CrossRef]
- Stoddart, J.F. Synthetic chiral receptor molecules from natural products. In Progress in Macrocyclic Chemistry; Izatt, R.M., Christensen, J.J., Eds.; Wiley-Interscience: New York, NY, USA, 1981; Volume 2, pp. 173–250. [Google Scholar]
- Stoddart, J.F. Chiral crown ethers. In Topics in Stereochemistry; Eliel, E.L., Wielen, S.H., Eds.; Wiley: New York, NY, USA, 1988; Volume 17, pp. 207–288. [Google Scholar]
- Jarosz, S.; Listkowski, A. Sugar derived crown ethers and their analogs: Synthesis and properties. Curr. Org. Chem. 2006, 10, 643–662. [Google Scholar] [CrossRef]
- Miethchen, R.; Fehring, V. Chirale Kronenether mit integriertem, 1,4-verbrückten D-Glucopyranose-Baustein. Synthesis 1998, 1, 94–98. [Google Scholar] [CrossRef]
- Bakó, P.; Tôke, L. Novel monoaza- and diazacrown ethers incorporating sugar units and their extraction ability towards picrate salts. J. Incl. Phenom. Macrocycl. Chem. 1995, 23, 195–201. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Thurston, J.E.; Sek, D.C.; Joly, J.-P. Utility of crown ethers derived from methyl β-D-galactopyranoside and their lathanide couples as chiral NMR discriminating agents. Tetrahedron Asymmetry 2001, 12, 1125–1130. [Google Scholar] [CrossRef]
- Miethchen, R.; Faltin, F.; Fehring, V. Chiral Crown Ethers Based on Galactopyranosides. Synthesis 2002, 2002, 1851–1856. [Google Scholar] [CrossRef]
- Pietraszkiewicz, M.; Salanski, P.; Jurczak, J. Synthesis of novel chiral [2.2.1]cryptands incorporating sugars. Tetrahedron 1984, 40, 2971–2973. [Google Scholar] [CrossRef]
- Bakó, P.; Mako, A.; Keglevich, G.; Kubinyi, M.; Pál, K. Synthesis of d-mannose-based azacrown ethers and their application in enantioselective reactions. Tetrahedron Asymmetry 2005, 16, 1861–1871. [Google Scholar] [CrossRef]
- Mako, A.; Menyhárd, D.K.; Bakó, P.; Keglevich, G.; Tôke, L. Theoretical study of the asymmetric phase-transfer mediated epoxidation of chalcone catalyzed by chiral crown ethers derived from monosaccharides. J. Mol. Struct. 2008, 892, 336–342. [Google Scholar] [CrossRef]
- Van Maarschalkerwaart, D.A.H.; Willard, N.P.; Pandit, U.K. Synthesis of carbohydrate containing crown ethers and their application as catalysts in asymmetric Michael additions. Tetrahedron 1992, 48, 8825–8840. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Reddy, V.G.; Krishna, P.R. Synthesis of new chiral 18-crown-6 ethers from D-xylose. Tetrahedron Asymmetry 1999, 10, 3777–3784. [Google Scholar] [CrossRef]
- Szabó, T.; Rapi, Z.; Keglevich, G.; Szöllősy, Á.; Drahos, L.; Bakó, P. Synthesis of L-arabinose-based crown ethers and their applications as enantioselective phase transfer catalysts. Arkivoc 2012, 2012, 36–48. [Google Scholar] [CrossRef]
- Joly, J.-P.; Nazhaoui, M.; Dumont, B. Synthesis and complexation behaviour of some crown ethers derived from D-hexopyranosides and D-hexopyranosides and D-mannitol towards racemic phenylglycine salts. Bull. Soc. Chim. Fr. 1994, 131, 369–380. [Google Scholar]
- Gryko, D.T.; Piatek, P.; Jurczak, J. An Efficient Method for Preparation of Chiral Macrocyclic Bisamides Starting from Diol Derivatives of D-Mannitol and L-Tartaric Acid. Synthesis 1999, 336–340. [Google Scholar] [CrossRef]
- Nemcsok, T.; Rapi, Z.; Keglevich, G.; Grün, A.; Bakó, P. Synthesis of D-mannitol-based crown ethers and their application as catalysts in asymmetric phase transfer reactions. Chirality 2018, 30, 407–419. [Google Scholar] [CrossRef]
- Rapi, Z.; Nemcsok, T.; Pálvölgyi, Á.; Keglevich, G.; Grün, A.; Bakó, P. Synthesis of L-threitol-based crown ethers and their application as enantioselective phase transfer catalyst in Michael additions. Chirality 2017, 29, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Bakó, P.; Keglevich, G.; Rapi, Z. Asymmetric phase transfer reactions catalyzed by chiral crown ethers derived from monosaccharides. Lett. Org. Chem. 2010, 7, 645–656. [Google Scholar] [CrossRef]
- Orbán, I.; Bakó, P.; Rapi, Z. Carbohydrate-Based Azacrown Ethers in Asymmetric Syntheses. Chemistry 2021, 3, 550–577. [Google Scholar] [CrossRef]
- Rapi, Z.; Bakó, P.; Drahos, L.; Keglevich, G. Side-Arm Effect of a Methyl α-D-Glucopyranoside Based Lariat Ether Catalysts in Asymmetric Syntheses. Heteroatom Chem. 2015, 26, 63–71. [Google Scholar] [CrossRef]
- Pálvölgyi, M.; Rapi, Z.; Ozohanics, O.; Toth, G.; Keglevich, G.; Bakó, P. Synthesis of alkyl α- and β-d-glucopyranoside-based chiral crown ethers and their application as enantioselective phase-transfer catalysts. Res. Chem. Intermed. 2017, 44, 1627–1645. [Google Scholar] [CrossRef]
- Rapi, Z.; Nemcsok, T.; Bagi, P.; Keglevich, G.; Bakó, P. Synthesis of chiral crown ethers derived from d-galactose and their application in enantioselective reactions. Tetrahedron 2019, 75, 3993–4004. [Google Scholar] [CrossRef]
- Nemcsok, T.; Rapi, Z.; Bagi, P.; Hou, G.Y.; Orbán, I.; Keglevich, G.; Bakó, P. Enantioselective cyclopropanation of conjugated cyanosulfones using carbohydrate-based crown ether catalysts. Tetrahedron 2020, 76, 130965. [Google Scholar] [CrossRef]
- Bakó, P.; Kiss, T.; Tőke, L. Chiral azacrown ethers derived from D-glucose as catalysts for enantioselective Michael addition. Tetrahedron Lett. 1997, 38, 7259–7262. [Google Scholar] [CrossRef]
- Makó, A.; Szöllősy, Á.; Keglevich, G.; Menyhárd, D.K.; Bakó, P.; Tőke, L. Synthesis of methyl-α-d-glucopyranoside-based azacrown ethers and their application in enantioselective reactions. Monats. Chem. 2008, 139, 525–535. [Google Scholar] [CrossRef]
- Bakó, P.; Bajor, Z.; Tőke, L.J. Synthesis of novel chiral crown ethers derived from d-glucose and their application to an enantioselective Michael reaction. Chem. Soc. Perkin Trans. I 1999, 24, 3651–3655. [Google Scholar] [CrossRef]
- Bakó, P.; Czinege, E.; Bakó, T.; Czugler, M.; Tőke, L. Asymmetric C–C bond forming reactions with chiral crown catalysts derived from d-glucose and d-galactose. Tetrahedron Asymmetry 1999, 10, 4539–4551. [Google Scholar] [CrossRef]
- Bakó, P.; Bakó, T.; Mészáros, A.; Keglevich, G.; Szöllősy, Á.; Bodor, S.; Makó, A.; Tőke, L. Phase Transfer Catalysed Asym-metric Epoxidation of Chalcones Using Chiral Crown Ethers Derived from D-Glucose and D-Mannose. Synlett 2004, 2004, 643–646. [Google Scholar] [CrossRef]
- Rapi, Z.; Démuth, B.; Keglevich, G.; Grün, A.; Drahos, L.; Sóti, P.L.; Bakó, P. Enantioselective Michael addition of malonates to aromatic nitroalkenes catalyzed by monosaccharide-based chiral crown ethers. Tetrahedron Asymmetry 2014, 25, 141–147. [Google Scholar] [CrossRef]
- Bakó, P.; Rapi, Z.; Grün, A.; Nemcsok, T.; Hegedűs, L.; Keglevich, G. Asymmetric Michael addition of malonates to enones catalyzed by an α-D-glucopyranoside-based brown ether. Synlett 2015, 26, 1847–1851. [Google Scholar] [CrossRef] [Green Version]
- Whistler, R.L.; Wolform, M.L. Methods in Carbohydrate Chemistry; Academic Press Inc.: New York, NY, USA, 1963; Volume 2, pp. 307–308. [Google Scholar]
- Lonnecker, A.T.; Lim, Y.H.; Felder, S.E.; Besset, C.J.; Wooley, K.L. Four Different Regioisomeric Polycarbonates Derived from One Natural Product, d-Glucose. Macromolecules 2016, 49, 7857–7867. [Google Scholar] [CrossRef]
- Deng, S.; Gangadharmath, U.; Chang, C.T. Sonochemistry: A Powerful Way of Enhancing the Efficiency of Carbohydrate Synthesis. J. Org. Chem. 2006, 71, 5179–5185. [Google Scholar] [CrossRef] [PubMed]
- Bakó, P.; Szöllősy, Á.; Bombicz, P.; Tőke, L. Asymmetric C-C Bond Forming Reactions by Chiral Crown Catalysts; Darzens Condensation and Nitroalkane Addition to the Double Bond. Synlett 1997, 291–292. [Google Scholar] [CrossRef]
- Bakó, P.; Rapi, Z.; Keglevich, G.; Szabó, T.; Soti, P.L.; Vigh, T.; Grun, A.; Holczbauer, T. Asymmetric C–C bond formation via Darzens condensation and Michael addition using monosaccharide-based chiral crown ethers. Tetrahedron Lett. 2011, 52, 1473–1476. [Google Scholar] [CrossRef]
- Rapi, Z.; Nemcsok, T.; Grün, A.; Pálvölgyi, Á.; Samu, G.; Hessz, D.; Kubinyi, M.; Kállay, M.; Keglevich, G.; Bakó, P. Asymmetric cyclopropanation reactions catalyzed by carbohydrate-based crown ethers. Tetrahedron 2018, 74, 3512–3526. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Time, h | Yield, % a | ee, % b |
|---|---|---|---|---|
| 1 | 1a | 1 | 69 | 52 |
| 2 | 1b | 1 | 68 | 29 |
| 3 | 1c | 1 | 66 | 19 |
| 4 | 2a | 1 | 74 | 62 c |
| 5 | 2b | 1 | 74 | 21 c |
| 6 | 2c | 1 | 38 | 29 |
| Entry | Catalyst | Time, h | Yield, % a | ee, % b |
|---|---|---|---|---|
| 1 | 1a | 4 | 79 | 75 |
| 2 | 1b | 4 | 87 | 24 |
| 3 | 1c | 24 | 70 | 21 |
| 4 | 2a | 1 | 82 | 92 c |
| 5 | 2b | 2 | 61 | 23 c |
| 6 | 2c | 72 | 73 | 3 |
| Entry | Catalyst | Time, h | Yield, % a | ee, % b |
|---|---|---|---|---|
| 1 | 1a | 72 | 49 | 42 |
| 2 | 1b | 48 | 42 | 28 |
| 3 | 1c | 120 | 33 | 21 |
| 4 | 2a | 3 | 70 | 99 c |
| 5 | 2b | 22 | 58 | 38 |
| 6 | 2c | 120 | 36 | 15 |
| Entry | Catalyst | Time, h | Yield, % a | ee, % b |
|---|---|---|---|---|
| 1 | 1a | 48 | 38 | 22 |
| 2 | 1b | 24 | 40 | 99 |
| 3 | 1c | 24 | 92 | 15 |
| 4 | 2a | 20 | 82 | 32 c |
| 5 | 2b | 24 | 97 | 70 |
| 6 | 2c | 24 | 81 | 58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orbán, I.; Varga, B.; Bagi, P.; Hegedűs, L.; Bakó, P.; Rapi, Z. Synthesis of Methyl 4,6-Di-O-ethyl-α-d-glucopyranoside-Based Azacrown Ethers and Their Effects in Asymmetric Reactions. Molecules 2021, 26, 4668. https://doi.org/10.3390/molecules26154668
Orbán I, Varga B, Bagi P, Hegedűs L, Bakó P, Rapi Z. Synthesis of Methyl 4,6-Di-O-ethyl-α-d-glucopyranoside-Based Azacrown Ethers and Their Effects in Asymmetric Reactions. Molecules. 2021; 26(15):4668. https://doi.org/10.3390/molecules26154668
Chicago/Turabian StyleOrbán, István, Bertalan Varga, Péter Bagi, László Hegedűs, Péter Bakó, and Zsolt Rapi. 2021. "Synthesis of Methyl 4,6-Di-O-ethyl-α-d-glucopyranoside-Based Azacrown Ethers and Their Effects in Asymmetric Reactions" Molecules 26, no. 15: 4668. https://doi.org/10.3390/molecules26154668
APA StyleOrbán, I., Varga, B., Bagi, P., Hegedűs, L., Bakó, P., & Rapi, Z. (2021). Synthesis of Methyl 4,6-Di-O-ethyl-α-d-glucopyranoside-Based Azacrown Ethers and Their Effects in Asymmetric Reactions. Molecules, 26(15), 4668. https://doi.org/10.3390/molecules26154668