
A New Class of Uracil–DNA Glycosylase Inhibitors Active against Human and Vaccinia Virus Enzyme

 ,
,  , ,
, ,
Abstract

1. Introduction
2. Results
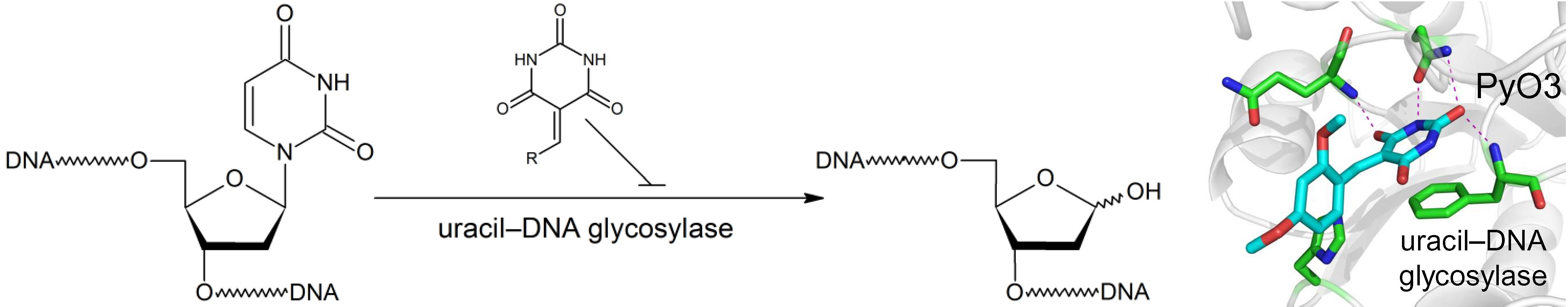
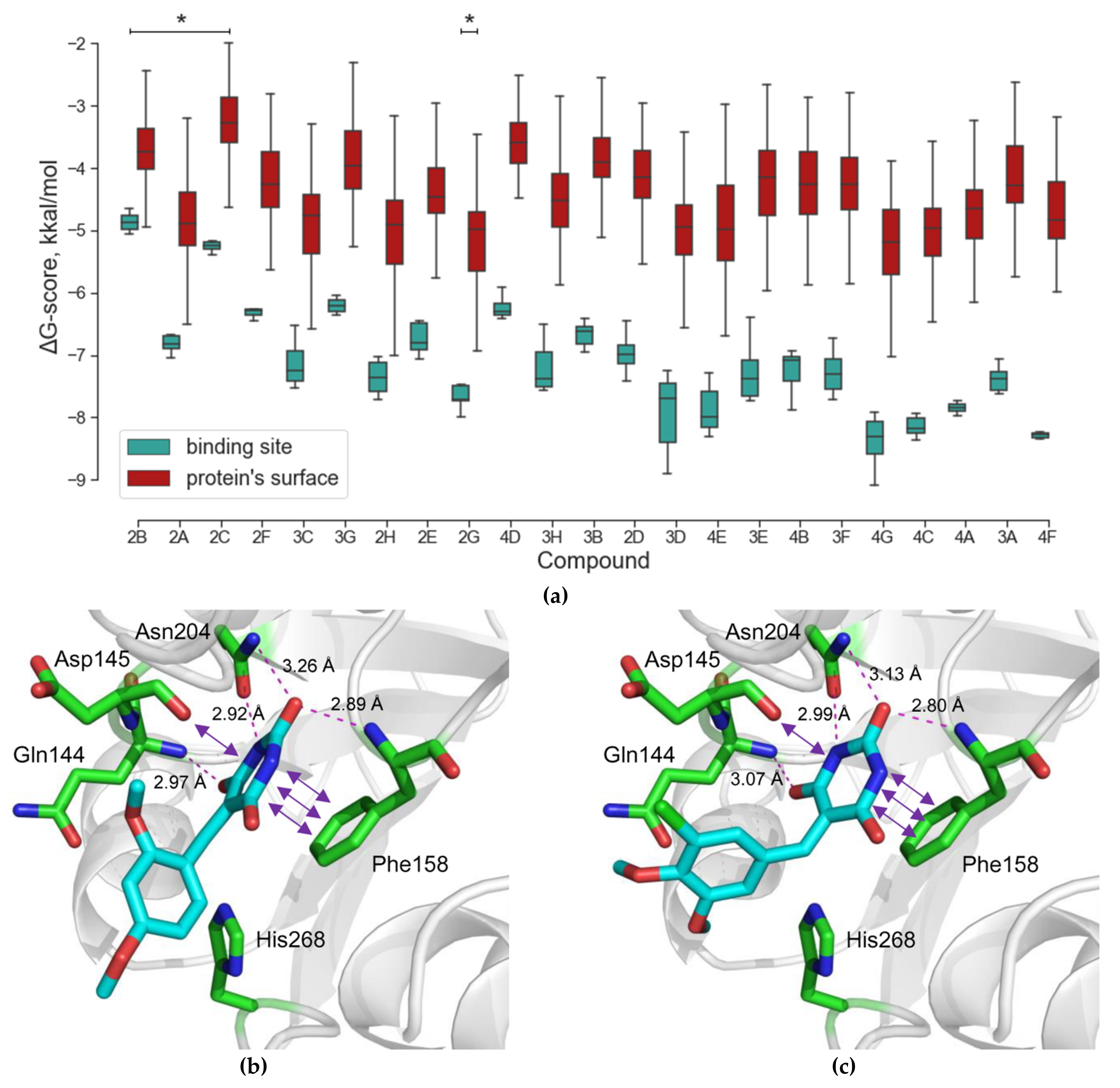
2.1. Virtual Screening
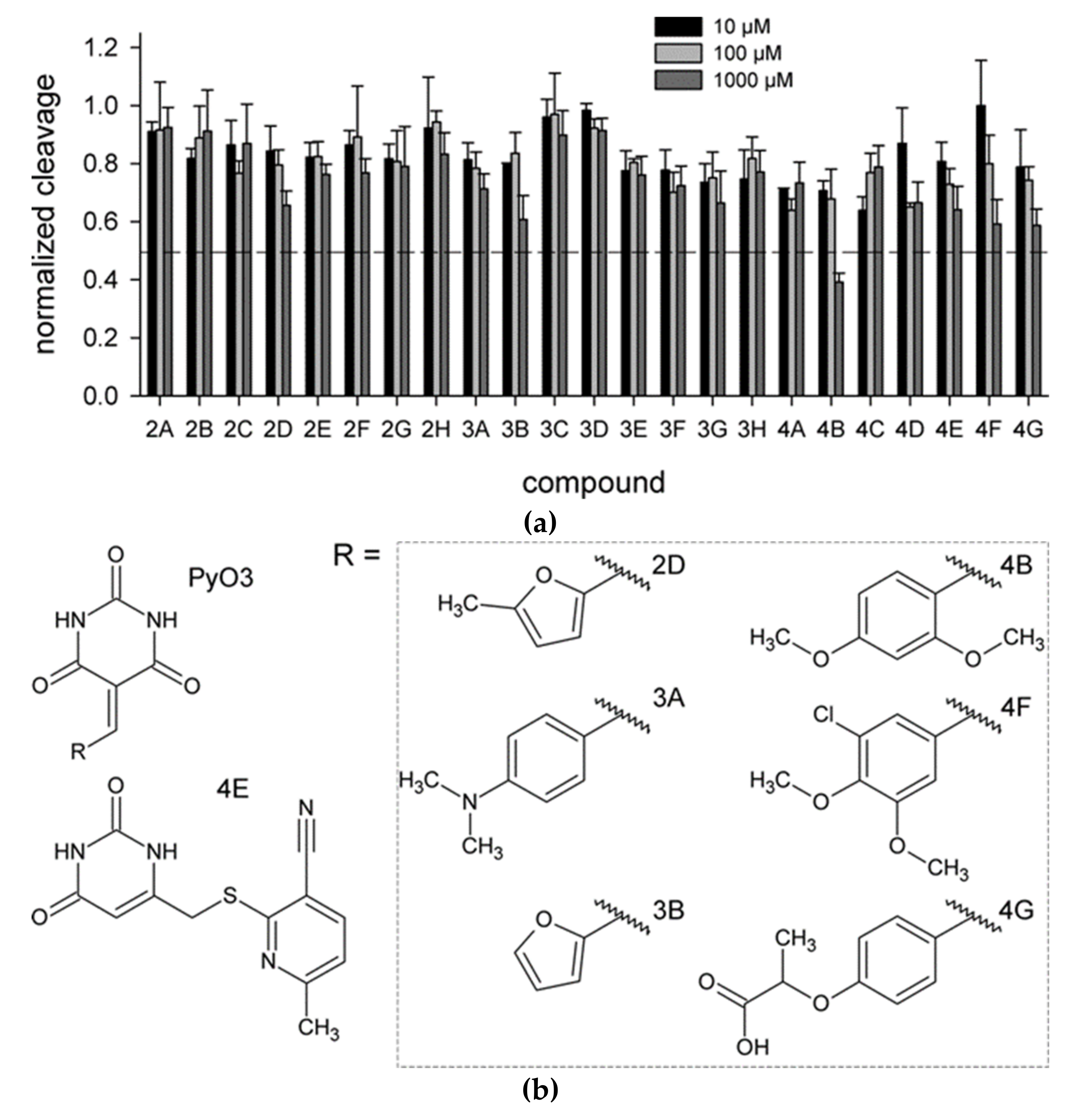
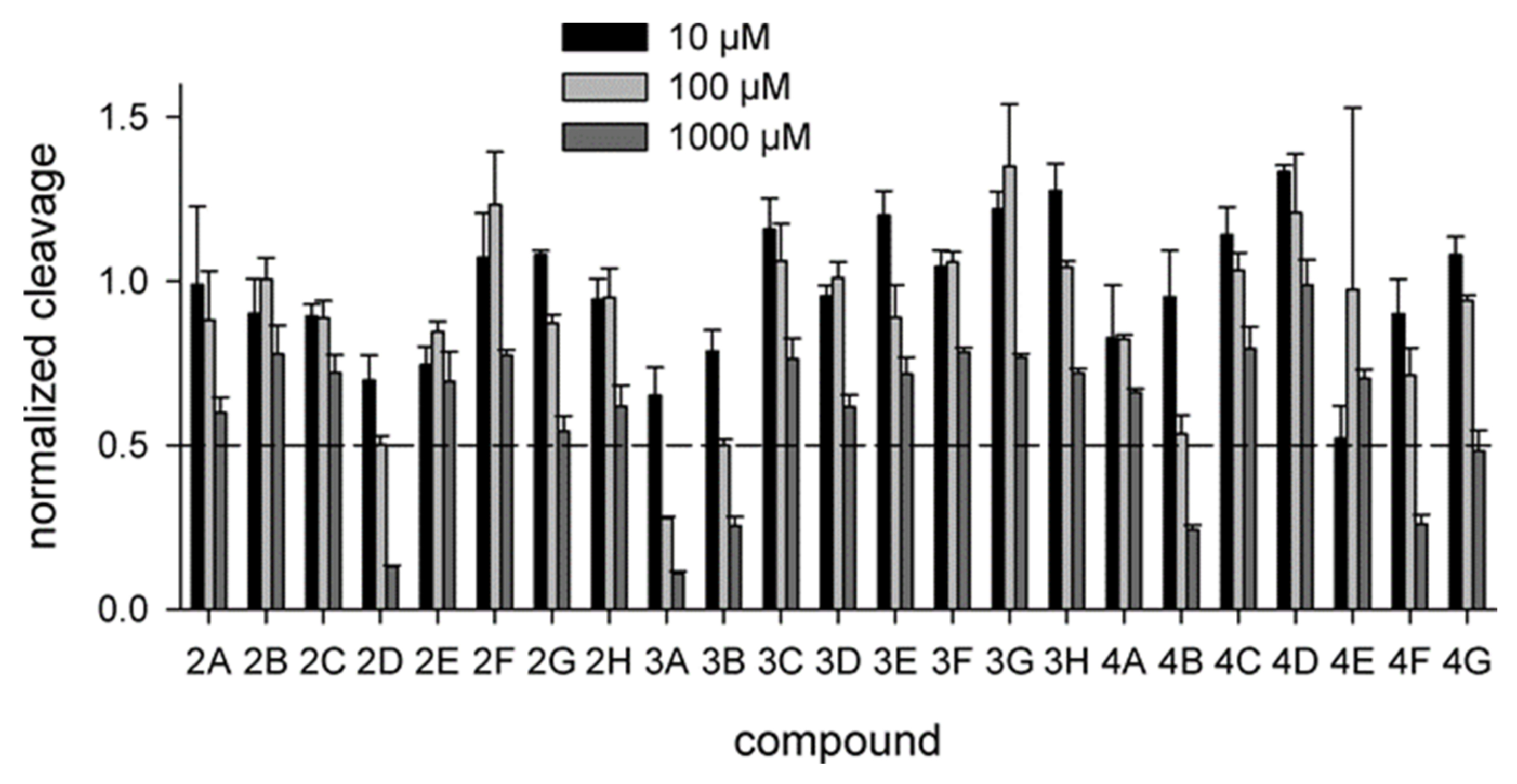
2.2. Biochemical Screening
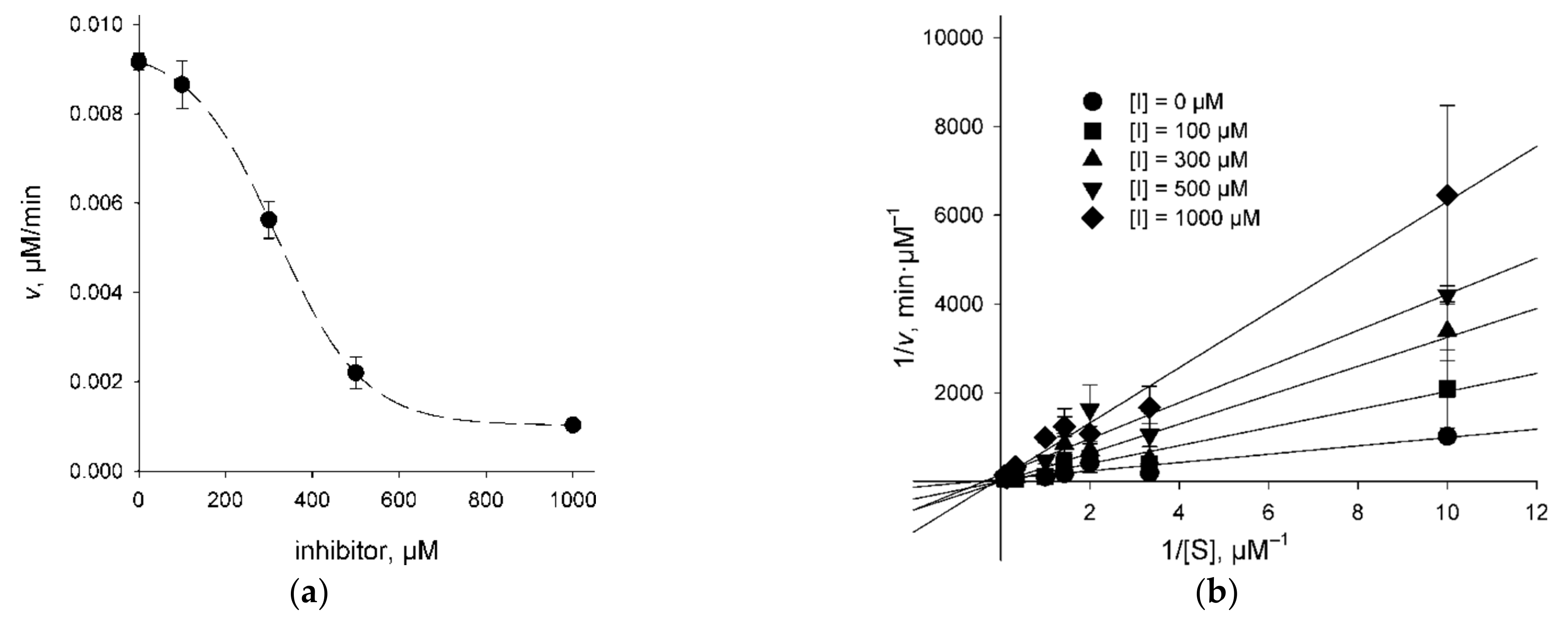
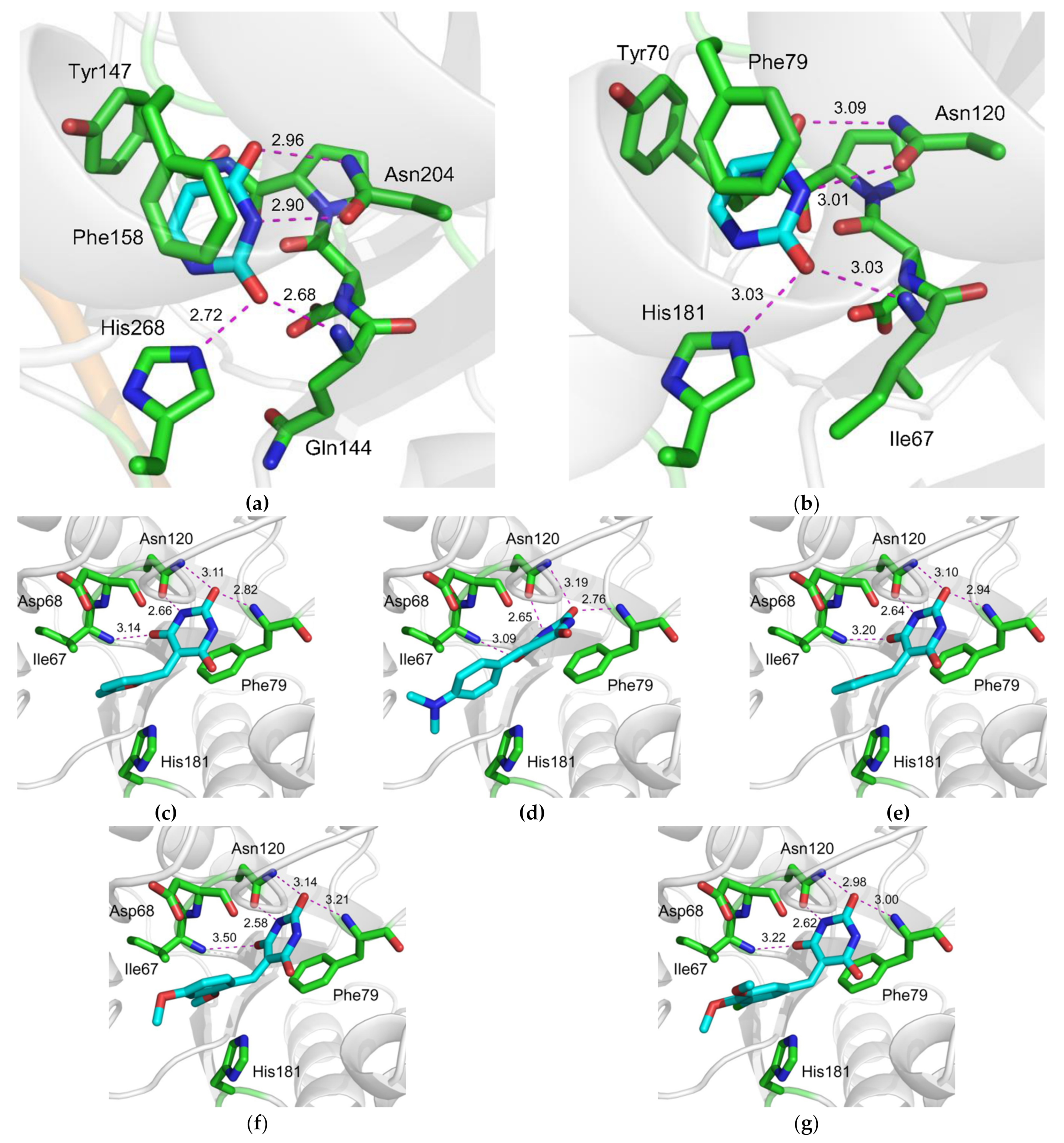
2.3. Character of UNG Inhibition
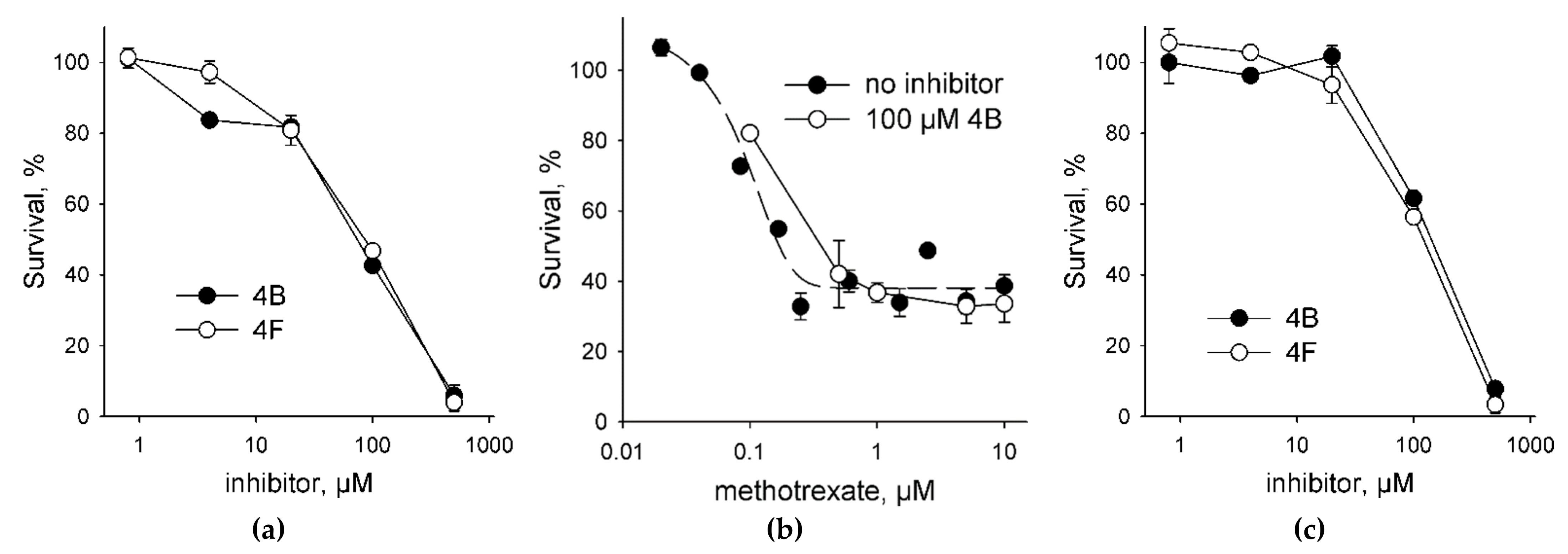
2.4. UNG Inhibitors Toxicity Is Independent of Methotrexate
2.5. Inhibition of Vaccinia Virus UNG
3. Discussion

4. Materials and Methods
4.1. Enzymes and Oligonucleotides
4.2. Virtual Screening
4.3. Uracil–DNA Glycosylase Assay
4.4. Inhibition Mechanism Determination
4.5. Cell Toxicity Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aravind, L.; Koonin, E.V. The α/β fold uracil DNA glycosylases: A common origin with diverse fates. Genome Biol. 2000, 1, research0007. [Google Scholar] [CrossRef] [PubMed]
- Visnes, T.; Doseth, B.; Pettersen, H.S.; Hagen, L.; Sousa, M.M.L.; Akbari, M.; Otterlei, M.; Kavli, B.; Slupphaug, G.; Krokan, H.E. Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Uracil in DNA—General mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006; p. 1118. [Google Scholar]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef]
- Bulgar, A.D.; Snell, M.; Donze, J.R.; Kirkland, E.B.; Li, L.; Yang, S.; Xu, Y.; Gerson, S.L.; Liu, L. Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia 2010, 24, 1795–1799. [Google Scholar] [CrossRef]
- Bulgar, A.D.; Weeks, L.D.; Miao, Y.; Yang, S.; Xu, Y.; Guo, C.; Markowitz, S.; Oleinick, N.; Gerson, S.L.; Liu, L. Removal of uracil by uracil DNA glycosylase limits pemetrexed cytotoxicity: Overriding the limit with methoxyamine to inhibit base excision repair. Cell Death Dis. 2012, 3, e252. [Google Scholar] [CrossRef]
- Yan, Y.; Qing, Y.; Pink, J.J.; Gerson, S.L. Loss of uracil DNA glycosylase selectively resensitizes p53-mutant and -deficient cells to 5-FdU. Mol. Cancer Res. 2018, 16, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Weeks, L.D.; Fu, P.; Gerson, S.L. Uracil-DNA glycosylase expression determines human lung cancer cell sensitivity to pemetrexed. Mol. Cancer Ther. 2013, 12, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Weeks, L.D.; Zentner, G.E.; Scacheri, P.C.; Gerson, S.L. Uracil DNA glycosylase (UNG) loss enhances DNA double strand break formation in human cancer cells exposed to pemetrexed. Cell Death Dis. 2014, 5, e1045. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, J.; Kumar, P.; Krishna, P.S.M.; Manjunath, R.; Varshney, U. Importance of uracil DNA glycosylase in Pseudomonas aeruginosa and Mycobacterium smegmatis, G+C-rich bacteria, in mutation prevention, tolerance to acidified nitrite, and endurance in mouse macrophages. J. Biol. Chem. 2003, 278, 24350–24358. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Acosta, V.M.; Aguilar-Pereyra, F.; Vidal, A.E.; Navarro, M.; Ruiz-Pérez, L.M.; González-Pacanowska, D. Trypanosomes lacking uracil-DNA glycosylase are hypersensitive to antifolates and present a mutator phenotype. Int. J. Biochem. Cell Biol. 2012, 44, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.T.; Upton, C.; Higman, M.A.; Niles, E.G.; McFadden, G. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J. Virol. 1993, 67, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Ellison, K.S.; Peng, W.; McFadden, G. Mutations in active-site residues of the uracil-DNA glycosylase encoded by vaccinia virus are incompatible with virus viability. J. Virol. 1996, 70, 7965–7973. [Google Scholar] [CrossRef]
- Prichard, M.N.; Duke, G.M.; Mocarski, E.S. Human cytomegalovirus uracil DNA glycosylase is required for the normal temporal regulation of both DNA synthesis and viral replication. J. Virol. 1996, 70, 3018–3025. [Google Scholar] [CrossRef] [PubMed]
- De Silva, F.S.; Moss, B. Effects of vaccinia virus uracil DNA glycosylase catalytic site and deoxyuridine triphosphatase deletion mutations individually and together on replication in active and quiescent cells and pathogenesis in mice. Virol. J. 2008, 5, 145. [Google Scholar] [CrossRef]
- Priet, S.; Gros, N.; Navarro, J.-M.; Boretto, J.; Canard, B.; Quérat, G.; Sire, J. HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol. Cell 2005, 17, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Roche, M.; Gantier, M.P.; Begum, N.A.; Honjo, T.; Caradonna, S.; Williams, B.R.G.; Mak, J. X4 and R5 HIV-1 have distinct post-entry requirements for uracil DNA glycosylase during infection of primary cells. J. Biol. Chem. 2010, 285, 18603–18614. [Google Scholar] [CrossRef] [PubMed]
- Guenzel, C.A.; Hérate, C.; Le Rouzic, E.; Maidou-Peindara, P.; Sadler, H.A.; Rouyez, M.-C.; Mansky, L.M.; Benichou, S. Recruitment of the nuclear form of uracil DNA glycosylase into virus particles participates in the full infectivity of HIV-1. J. Virol. 2012, 86, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Focher, F.; Verri, A.; Spadari, S.; Manservigi, R.; Gambino, J.; Wright, G.E. Herpes simplex virus type 1 uracil-DNA glycosylase: Isolation and selective inhibition by novel uracil derivatives. Biochem. J. 1993, 292, 883–889. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pregnolato, M.; Ubiali, D.; Verri, A.; Focher, F.; Spadari, S.; Sun, H.; Zhi, C.; Wright, G.E. Synthesis and molecular modeling of novel HSV1 uracil-DNA glycosylase inhibitors. Nucleosides Nucleotides 1999, 18, 709–711. [Google Scholar] [CrossRef]
- Sun, H.; Zhi, C.; Wright, G.E.; Ubiali, D.; Pregnolato, M.; Verri, A.; Focher, F.; Spadari, S. Molecular modeling and synthesis of inhibitors of herpes simplex virus type 1 uracil-DNA glycosylase. J. Med. Chem. 1999, 42, 2344–2350. [Google Scholar] [CrossRef]
- Hendricks, U.; Crous, W.; Naidoo, K.J. Computational rationale for the selective inhibition of the herpes simplex virus type 1 uracil-DNA glycosylase enzyme. J. Chem. Inf. Model. 2014, 54, 3362–3372. [Google Scholar] [CrossRef] [PubMed]
- Suksangpleng, T.; Leartsakulpanich, U.; Moonsom, S.; Siribal, S.; Boonyuen, U.; Wright, G.E.; Chavalitshewinkoon-Petmitr, P. Molecular characterization of Plasmodium falciparum uracil-DNA glycosylase and its potential as a new anti-malarial drug target. Malar. J. 2014, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Cao, C.; Stivers, J.T.; Song, F.; Ichikawa, Y. The merits of bipartite transition-state mimics for inhibition of uracil DNA glycosylase. Bioorg. Chem. 2004, 32, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Krosky, D.J.; Seiple, L.; Stivers, J.T. Uracil-directed ligand tethering: An efficient strategy for uracil DNA glycosylase (UNG) inhibitor development. J. Am. Chem. Soc. 2005, 127, 17412–17420. [Google Scholar] [CrossRef] [PubMed]
- Krosky, D.J.; Bianchet, M.A.; Seiple, L.; Chung, S.; Amzel, L.M.; Stivers, J.T. Mimicking damaged DNA with a small molecule inhibitor of human UNG2. Nucleic Acids Res. 2006, 34, 5872–5879. [Google Scholar] [CrossRef]
- Chung, S.; Parker, J.B.; Bianchet, M.; Amzel, L.M.; Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat. Chem. Biol. 2009, 5, 407–413. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Chung, S.; Krosky, D.J.; Stivers, J.T. Synthesis and high-throughput evaluation of triskelion uracil libraries for inhibition of human dUTPase and UNG2. Bioorg. Med. Chem. 2006, 14, 5666–5672. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, X.; Wang, K.; Tan, W.; Li, H.; Tang, H. Real-time monitoring of uracil removal by uracil-DNA glycosylase using fluorescent resonance energy transfer probes. Anal. Biochem. 2007, 366, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Huang, Z.; Pu, F.; Ren, J.; Qu, X. A label-free, quadruplex-based functional molecular beacon (LFG4-MB) for fluorescence turn-on detection of DNA and nuclease. Chemistry 2011, 17, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Jiang, J.; Yu, R. A highly sensitive electrochemical platform for the assay of uracil-DNA glycosylase activity combined with enzymatic amplification. Anal. Sci. 2013, 29, 193–198. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Moiani, D.; Ahmed, Z.; Arvai, A.S.; Namjoshi, S.; Shin, D.S.; Fedorov, Y.; Selvik, E.J.; Jones, D.E.; Pink, J.; et al. An effective human uracil-DNA glycosylase inhibitor targets the open pre-catalytic active site conformation. Prog. Biophys. Mol. Biol. 2021, 163, 143–159. [Google Scholar] [CrossRef]
- Slupphaug, G.; Eftedal, I.; Kavli, B.; Bharati, S.; Helle, N.M.; Haug, T.; Levine, D.W.; Krokan, H.E. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 1995, 34, 128–138. [Google Scholar] [CrossRef]
- Myung, J.I.; Tang, Y.; Pitt, M.A. Evaluation and comparison of computational models. Methods Enzymol. 2009, 454, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Moe, E.; Leiros, I.; Riise, E.K.; Olufsen, M.; Lanes, O.; Smalås, A.O.; Willassen, N.P. Optimisation of the surface electrostatics as a strategy for cold adaptation of uracil-DNA N-glycosylase (UNG) from atlantic cod (Gadus morhua). J. Mol. Biol. 2004, 343, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Krusong, K.; Carpenter, E.P.; Bellamy, S.R.W.; Savva, R.; Baldwin, G.S. A comparative study of uracil-DNA glycosylases from human and herpes simplex virus type 1. J. Biol. Chem. 2006, 281, 4983–4992. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Drabløs, F.; Slupphaug, G. Uracil in DNA—Occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [PubMed]
- Stanitsa, E.S.; Arps, L.; Traktman, P. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J. Biol. Chem. 2006, 281, 3439–3451. [Google Scholar] [CrossRef]
- Czarnecki, M.W.; Traktman, P. The vaccinia virus DNA polymerase and its processivity factor. Virus Res. 2017, 234, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Slupphaug, G.; Mol, C.D.; Kavli, B.; Arvai, A.S.; Krokan, H.E.; Tainer, J.A. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature 1996, 384, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Mol, C.D.; Slupphaug, G.; Bharati, S.; Krokan, H.E.; Tainer, J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Walcher, G.; Jones, G.D.; Slupphaug, G.; Krokan, H.E.; Blackburn, G.M.; Tainer, J.A. Uracil-DNA glycosylase–DNA substrate and product structures: Conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. USA 2000, 97, 5083–5088. [Google Scholar] [CrossRef] [PubMed]
- Mechetin, G.V.; Endutkin, A.V.; Diatlova, E.A.; Zharkov, D.O. Inhibitors of DNA glycosylases as prospective drugs. Int. J. Mol. Sci. 2020, 21, 3118. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Tarbouriech, N.; Fender, P.; Contesto-Richefeu, C.; Peyrefitte, C.N.; Iseni, F. Crystal structure of the vaccinia virus uracil-DNA glycosylase in complex with DNA. J. Biol. Chem. 2015, 290, 17923–17934. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.B.; Bianchet, M.A.; Krosky, D.J.; Friedman, J.I.; Amzel, L.M.; Stivers, J.T. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 2007, 449, 433–437. [Google Scholar] [CrossRef]
- Goulian, M.; Bleile, B.; Tseng, B.Y. The effect of methotrexate on levels of dUTP in animal cells. J. Biol. Chem. 1980, 255, 10630–10637. [Google Scholar] [CrossRef]
- Goulian, M.; Bleile, B.; Tseng, B.Y. Methotrexate-induced misincorporation of uracil into DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 1956–1960. [Google Scholar] [CrossRef]
- Pettersen, H.S.; Visnes, T.; Vågbø, C.B.; Svaasand, E.K.; Doseth, B.; Slupphaug, G.; Kavli, B.; Krokan, H.E. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011, 39, 8430–8444. [Google Scholar] [CrossRef]
- Christenson, E.S.; Gizzi, A.; Cui, J.; Egleston, M.; Seamon, K.J.; DePasquale, M.; Orris, B.; Park, B.H.; Stivers, J.T. Inhibition of human uracil DNA glycosylase sensitizes a large fraction of colorectal cancer cells to 5-fluorodeoxyuridine and raltitrexed but not fluorouracil. Mol. Pharmacol. 2021, 99, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Kouzminova, E.A.; Kuzminov, A. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol. Microbiol. 2004, 51, 1279–1295. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.; Kouzminova, E.A.; Kuzminov, A. Synthetic lethality with the dut defect in Escherichia coli reveals layers of DNA damage of increasing complexity due to uracil incorporation. J. Bacteriol. 2008, 190, 5841–5854. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, H.; Rosewell, I.; Robins, P.; Skjelbred, C.F.; Andersen, S.; Slupphaug, G.; Daly, G.; Krokan, H.E.; Lindahl, T.; Barnes, D.E. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell 2000, 5, 1059–1065. [Google Scholar] [CrossRef]
- Kronenberg, G.; Harms, C.; Sobol, R.W.; Cardozo-Pelaez, F.; Linhart, H.; Winter, B.; Balkaya, M.; Gertz, K.; Gay, S.B.; Cox, D.; et al. Folate deficiency induces neurodegeneration and brain dysfunction in mice lacking uracil DNA glycosylase. J. Neurosci. 2008, 28, 7219–7230. [Google Scholar] [CrossRef]
- Grogan, B.C.; Parker, J.B.; Guminski, A.F.; Stivers, J.T. Effect of the thymidylate synthase inhibitors on dUTP and TTP pool levels and the activities of DNA repair glycosylases on uracil and 5-fluorouracil in DNA. Biochemistry 2011, 50, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Walla, M.; Wyatt, M.D. Uracil incorporation into genomic DNA does not predict toxicity caused by chemotherapeutic inhibition of thymidylate synthase. DNA Repair 2008, 7, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Sèle, C.; Gabel, F.; Gutsche, I.; Ivanov, I.; Burmeister, W.P.; Iseni, F.; Tarbouriech, N. Low-resolution structure of vaccinia virus DNA replication machinery. J. Virol. 2013, 87, 1679–1689. [Google Scholar] [CrossRef]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J. 1999, 18, 3834–3844. [Google Scholar] [CrossRef]
- Hagen, L.; Kavli, B.; Sousa, M.M.L.; Torseth, K.; Liabakk, N.B.; Sundheim, O.; Pena-Diaz, J.; Otterlei, M.; Hørning, O.; Jensen, O.N.; et al. Cell cycle-specific UNG2 phosphorylations regulate protein turnover, activity and association with RPA. EMBO J. 2008, 27, 51–61. [Google Scholar] [CrossRef]
- Nuth, M.; Huang, L.; Saw, Y.L.; Schormann, N.; Chattopadhyay, D.; Ricciardi, R.P. Identification of inhibitors that block vaccinia virus infection by targeting the DNA synthesis processivity factor D4. J. Med. Chem. 2011, 54, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Sommers, C.I.; Prichard, M.N.; Keith, K.A.; Noah, J.W.; Nuth, M.; Ricciardi, R.P.; Chattopadhyay, D. Identification of protein-protein interaction inhibitors targeting vaccinia virus processivity factor for development of antiviral agents. Antimicrob. Agents Chemother. 2011, 55, 5054–5062. [Google Scholar] [CrossRef]
- De Silva, F.S.; Moss, B. Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: Catalytic site mutations reduce virulence but not virus replication in cultured cells. J. Virol. 2003, 77, 159–166. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scaramozzino, N.; Sanz, G.; Crance, J.M.; Saparbaev, M.; Drillien, R.; Laval, J.; Kavli, B.; Garin, D. Characterisation of the substrate specificity of homogeneous vaccinia virus uracil-DNA glycosylase. Nucleic Acids Res. 2003, 31, 4950–4957. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, O.V.; Novikov, F.N.; Zeifman, A.A.; Stroylov, V.S.; Chilov, G.G. TSAR, a new graph–theoretical approach to computational modeling of protein side-chain flexibility: Modeling of ionization properties of proteins. Proteins 2011, 79, 2693–2710. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Medvedev, M.G.; Dmitrienko, A.O.; Gerasimov, I.; Svitanko, I.V.; Chilov, G.G. The role of human in the loop: Lessons from D3R challenge 4. J. Comput. Aided Mol. Des. 2020, 34, 121–130. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead Finder: An approach to improve accuracy of protein–ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Sagnou, M.; Novikov, F.N.; Ivanova, E.S.; Alexiou, P.; Stroylov, V.S.; Titov, I.Y.; Tatarskiy, V.V.; Vagida, M.S.; Pelecanou, M.; Shtil, A.A.; et al. Novel curcumin derivatives as P-glycoprotein inhibitors: Molecular modeling, synthesis and sensitization of multidrug resistant cells to doxorubicin. Eur. J. Med. Chem. 2020, 198, 112331. [Google Scholar] [CrossRef]
- Huitema, C.; Horsman, G. Analyzing enzyme kinetic data using the powerful statistical capabilities of R. biorXiv 2018, 316588. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Model | θrela | Vmax, μM/min | KM, μM | Ki, μM |
|---|---|---|---|---|---|
| 3A | competitive | 0.655 | 0.029 ± 0.003 | 1.6 ± 0.5 | 530 ± 270 |
| uncompetitive | best model | 0.034 ± 0.004 | 2.6 ± 0.7 | 1300 ± 500 | |
| 4B | competitive | best model | 0.019 ± 0.001 | 1.1 ± 0.3 | 140 ± 40 |
| uncompetitive | 2.53 × 10−5 | 0.024 ± 0.003 | 2.9 ± 0.7 | 550 ± 140 | |
| 4F | competitive | 0.8 | 0.053 ± 0.014 | 3.1 ± 2.2 | 200 ± 130 |
| uncompetitive | best model | 0.075 ± 0.028 | 6.4 ± 4.5 | 250 ± 140 | |
| 4G | competitive | best model | 0.042 ± 0.002 | 3.7 ± 0.6 | 3300 ± 2300 |
| uncompetitive | 0.312 | 0.043 ± 0.003 | 4.2 ± 0.7 | >10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grin, I.R.; Mechetin, G.V.; Kasymov, R.D.; Diatlova, E.A.; Yudkina, A.V.; Shchelkunov, S.N.; Gileva, I.P.; Denisova, A.A.; Stepanov, G.A.; Chilov, G.G.; et al. A New Class of Uracil–DNA Glycosylase Inhibitors Active against Human and Vaccinia Virus Enzyme. Molecules 2021, 26, 6668. https://doi.org/10.3390/molecules26216668
Grin IR, Mechetin GV, Kasymov RD, Diatlova EA, Yudkina AV, Shchelkunov SN, Gileva IP, Denisova AA, Stepanov GA, Chilov GG, et al. A New Class of Uracil–DNA Glycosylase Inhibitors Active against Human and Vaccinia Virus Enzyme. Molecules. 2021; 26(21):6668. https://doi.org/10.3390/molecules26216668
Chicago/Turabian StyleGrin, Inga R., Grigory V. Mechetin, Rustem D. Kasymov, Evgeniia A. Diatlova, Anna V. Yudkina, Sergei N. Shchelkunov, Irina P. Gileva, Alexandra A. Denisova, Grigoriy A. Stepanov, Ghermes G. Chilov, and et al. 2021. "A New Class of Uracil–DNA Glycosylase Inhibitors Active against Human and Vaccinia Virus Enzyme" Molecules 26, no. 21: 6668. https://doi.org/10.3390/molecules26216668
APA StyleGrin, I. R., Mechetin, G. V., Kasymov, R. D., Diatlova, E. A., Yudkina, A. V., Shchelkunov, S. N., Gileva, I. P., Denisova, A. A., Stepanov, G. A., Chilov, G. G., & Zharkov, D. O. (2021). A New Class of Uracil–DNA Glycosylase Inhibitors Active against Human and Vaccinia Virus Enzyme. Molecules, 26(21), 6668. https://doi.org/10.3390/molecules26216668