
Dibenzofuran Derivatives Inspired from Cercosporamide as Dual Inhibitors of Pim and CLK1 Kinases

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
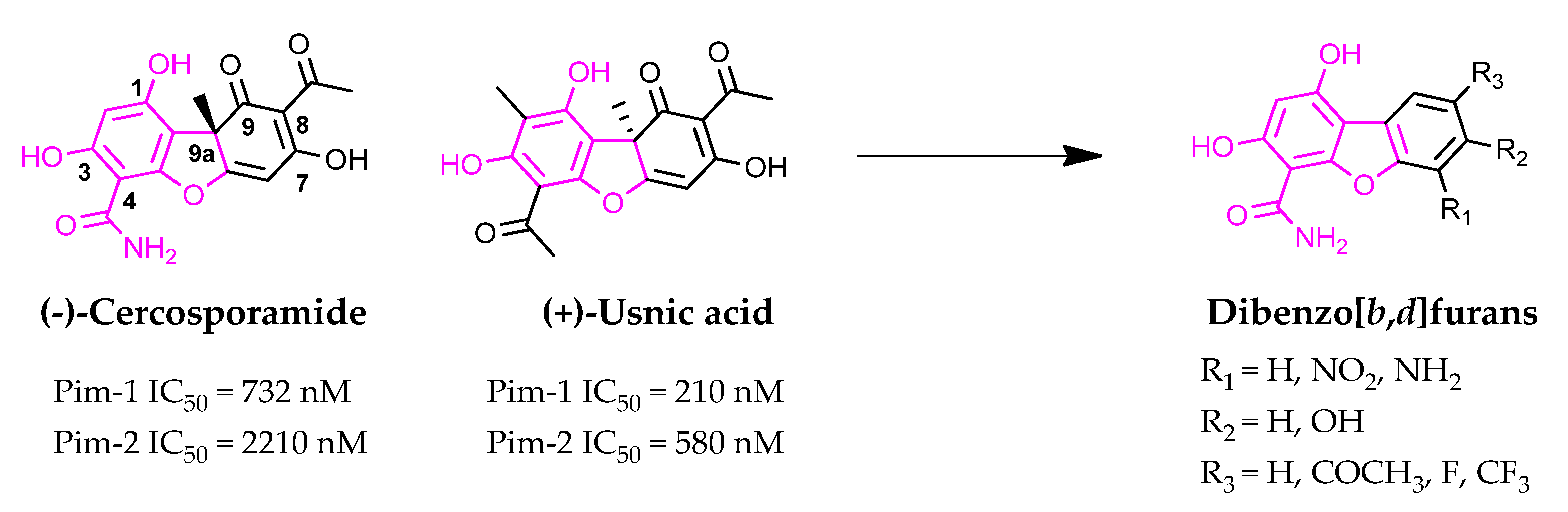
:1. Introduction
2. Results and Discussion
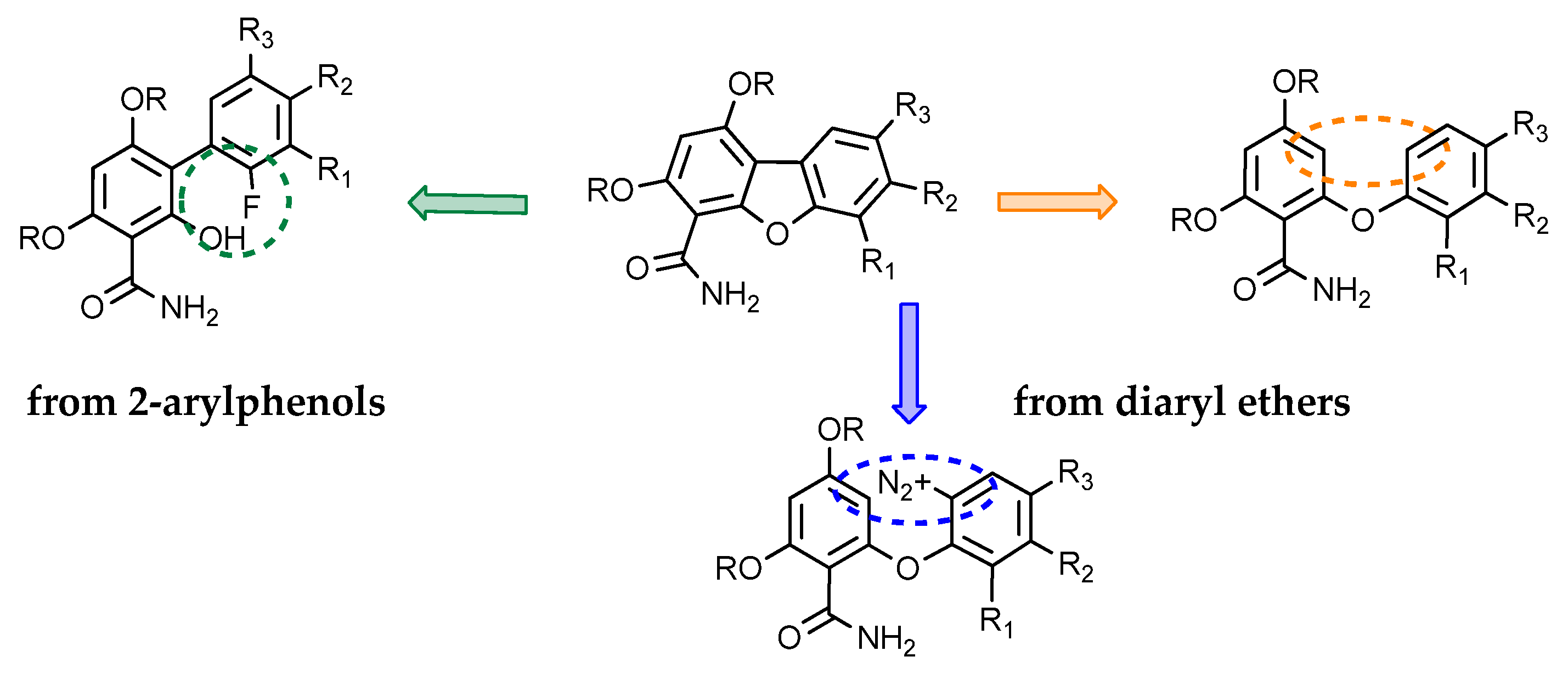
2.1. Synthesis
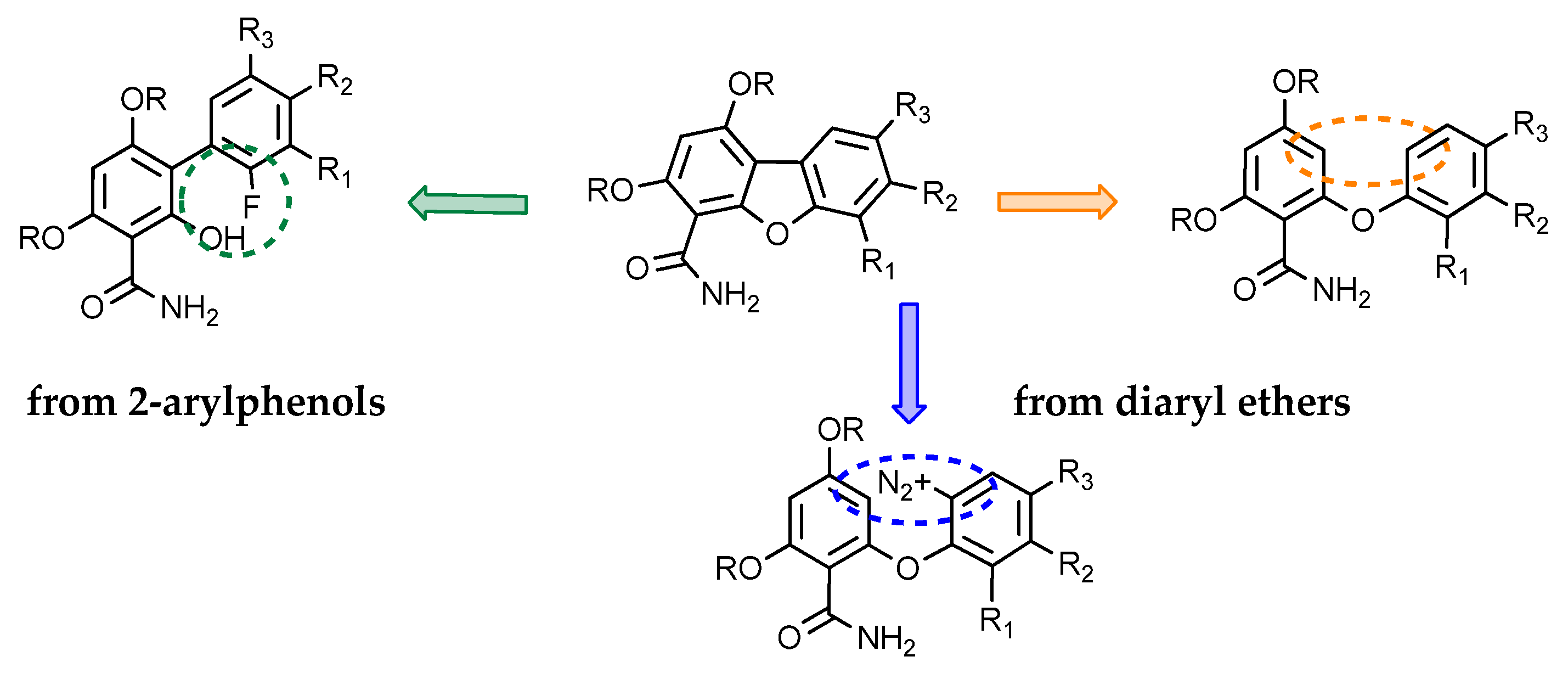
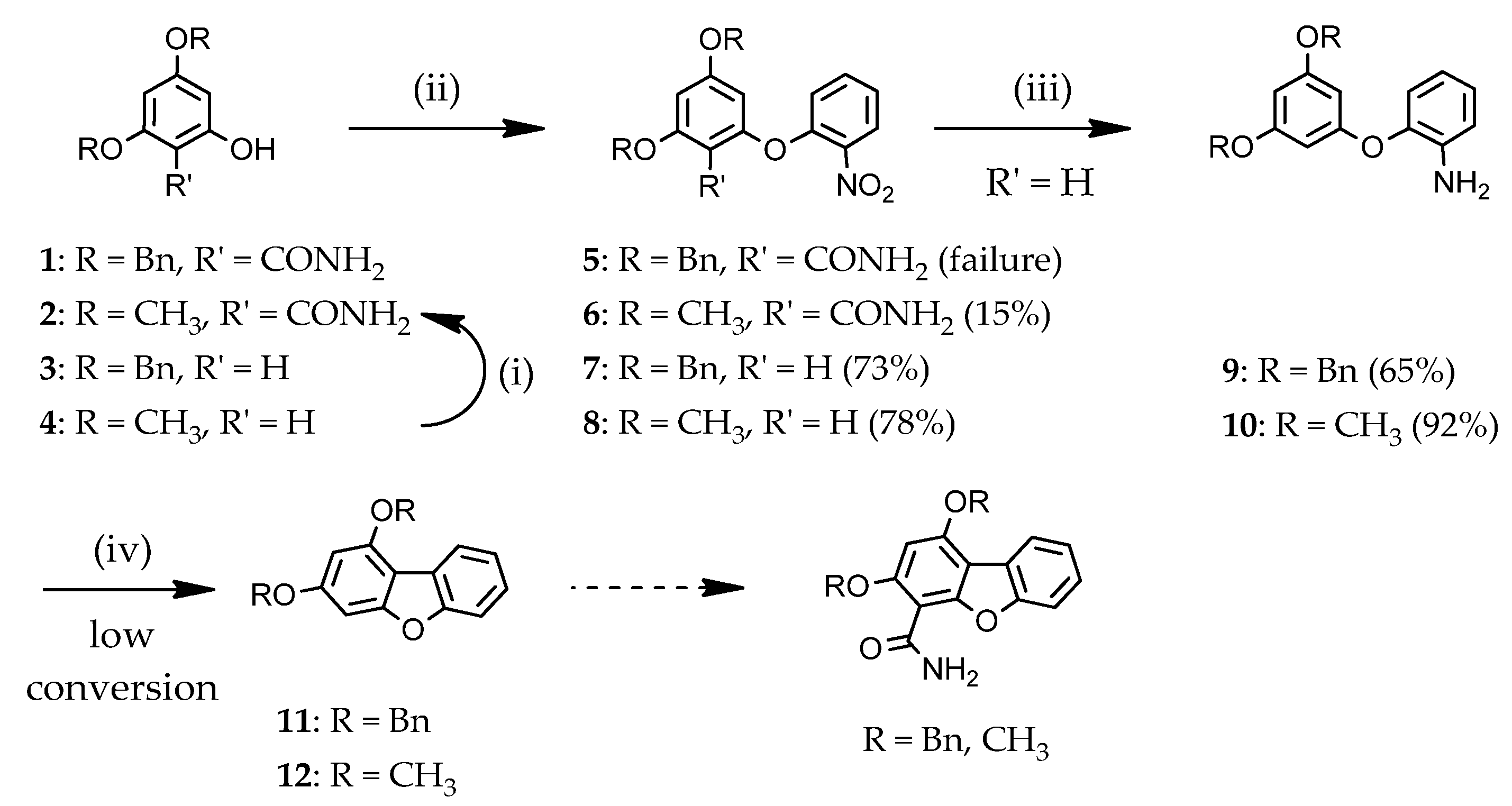
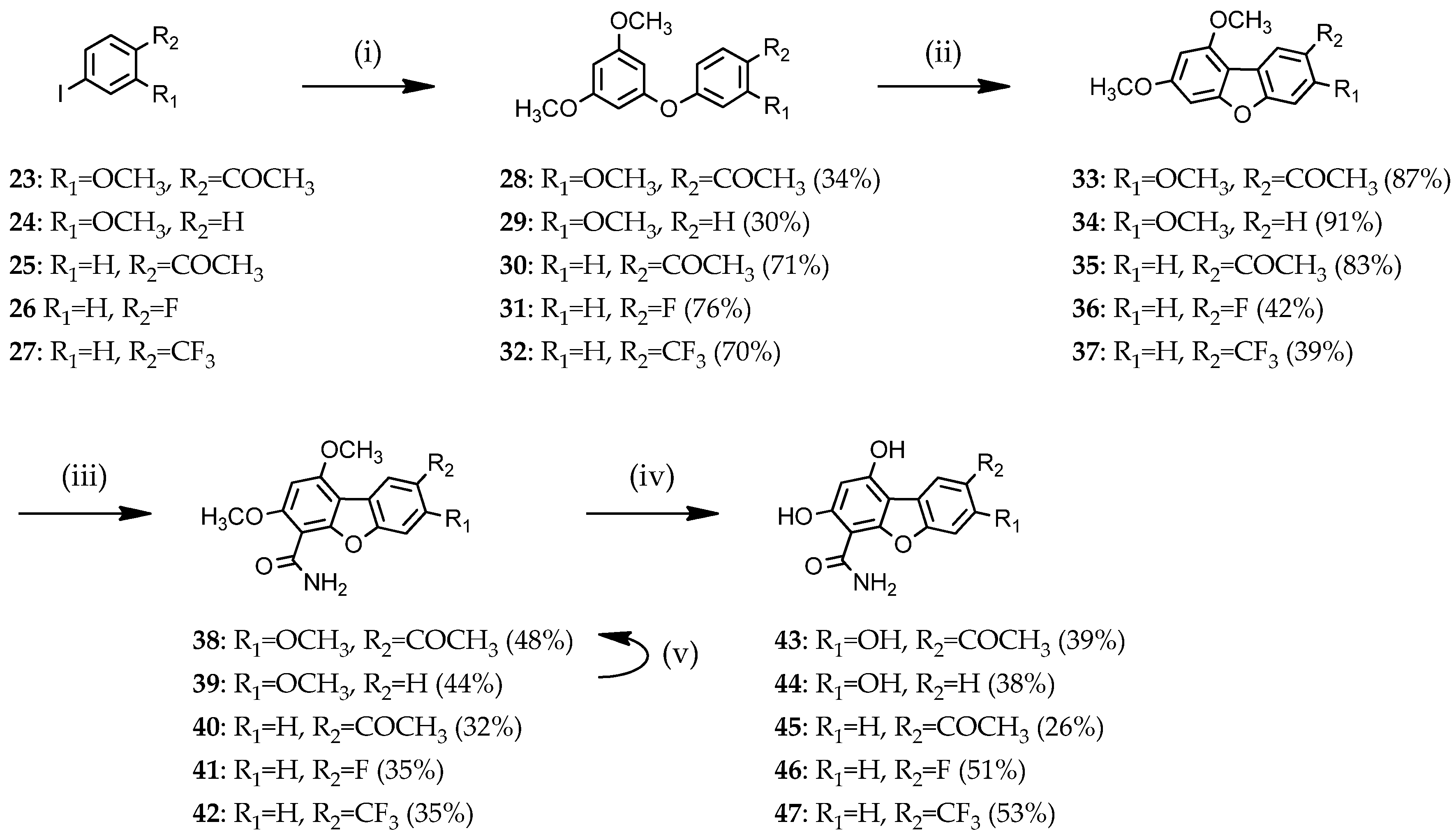
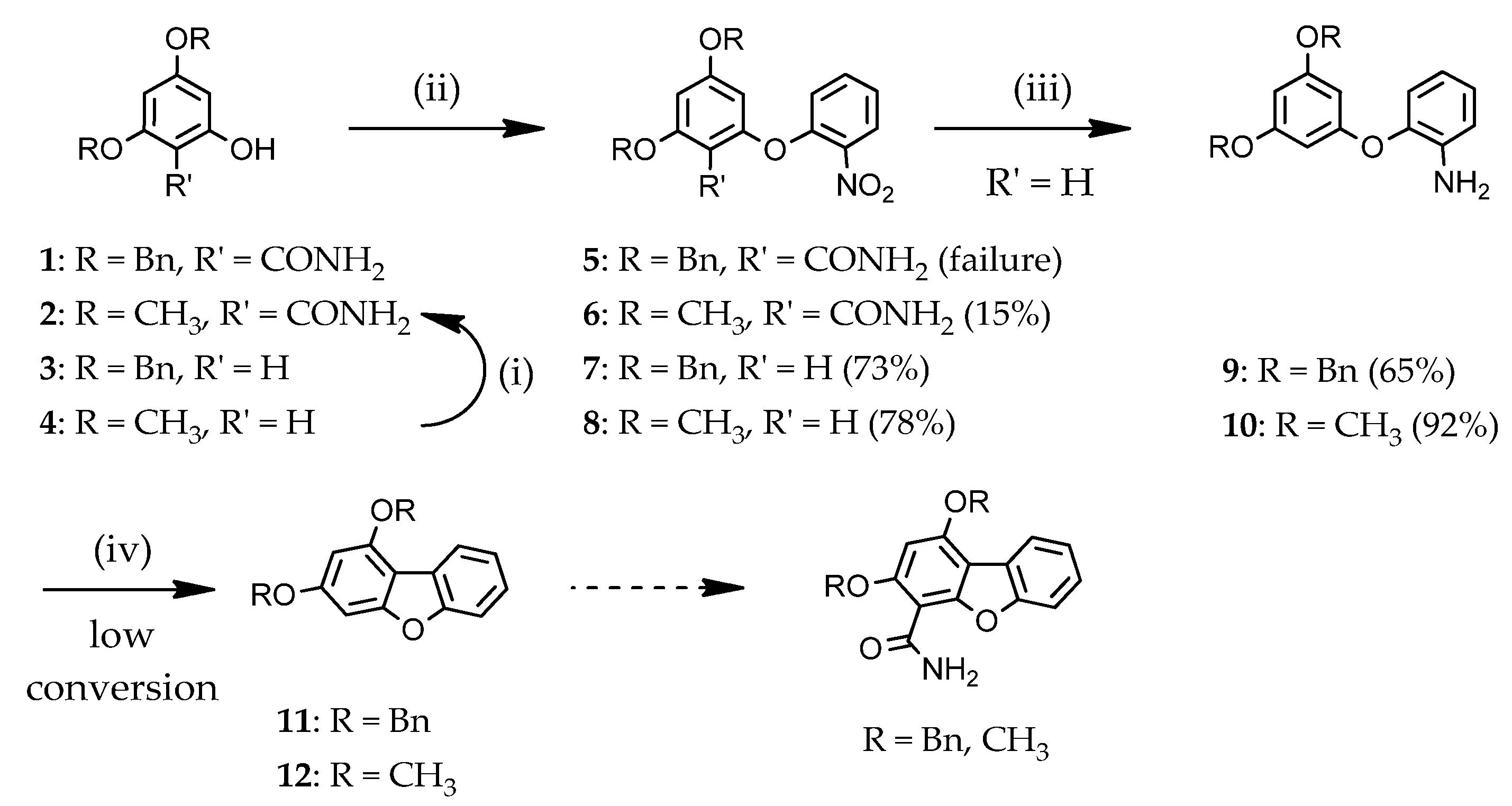
2.1.1. Access to Dibenzofuran Derivatives by Intramolecular C-C Bond Formation from Diaryl Ethers
2.1.2. Access to Dibenzofuran Derivatives by Intramolecular C-O Bond Formation from 2-Aryl Phenols
2.2. Kinase Assays
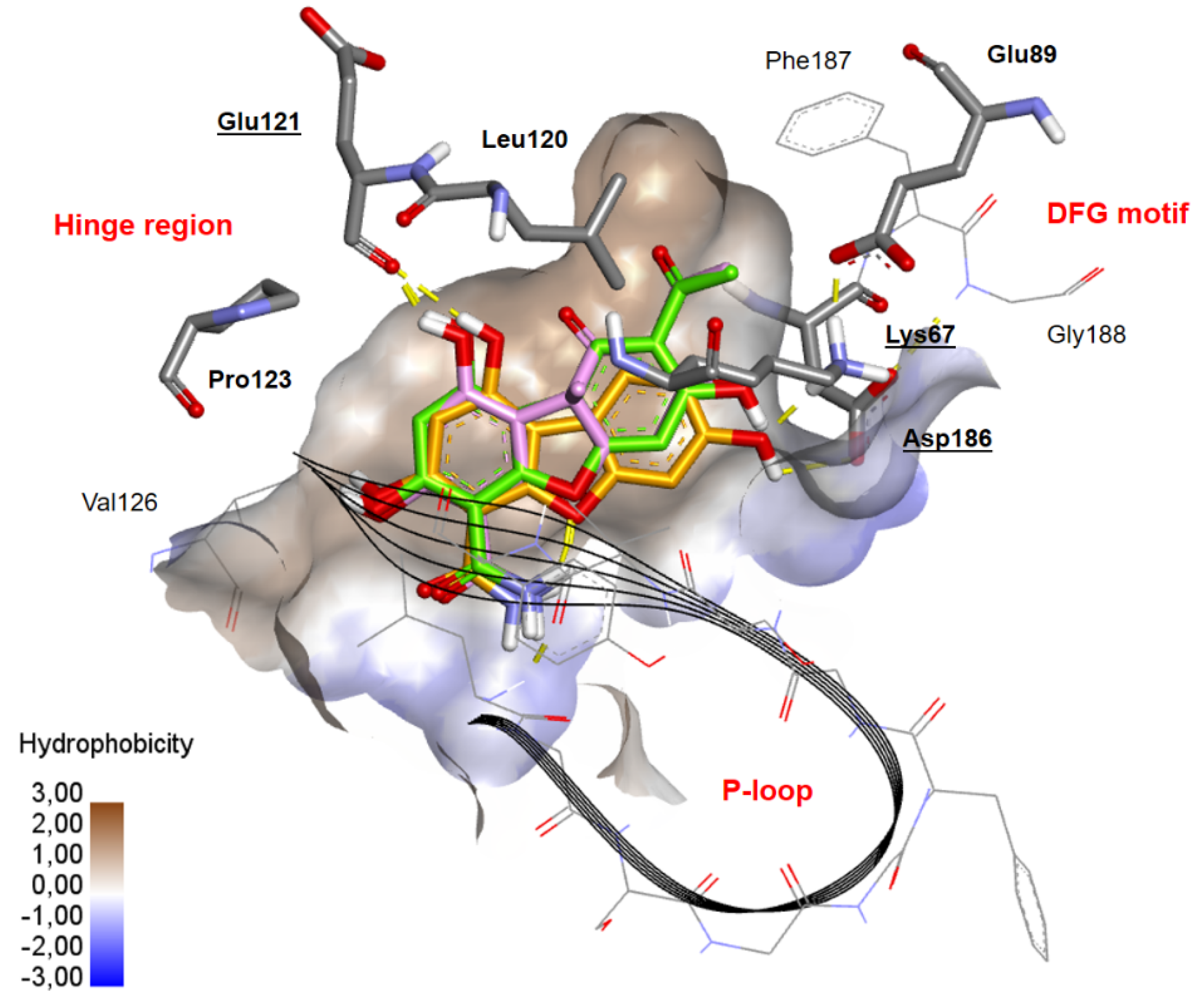
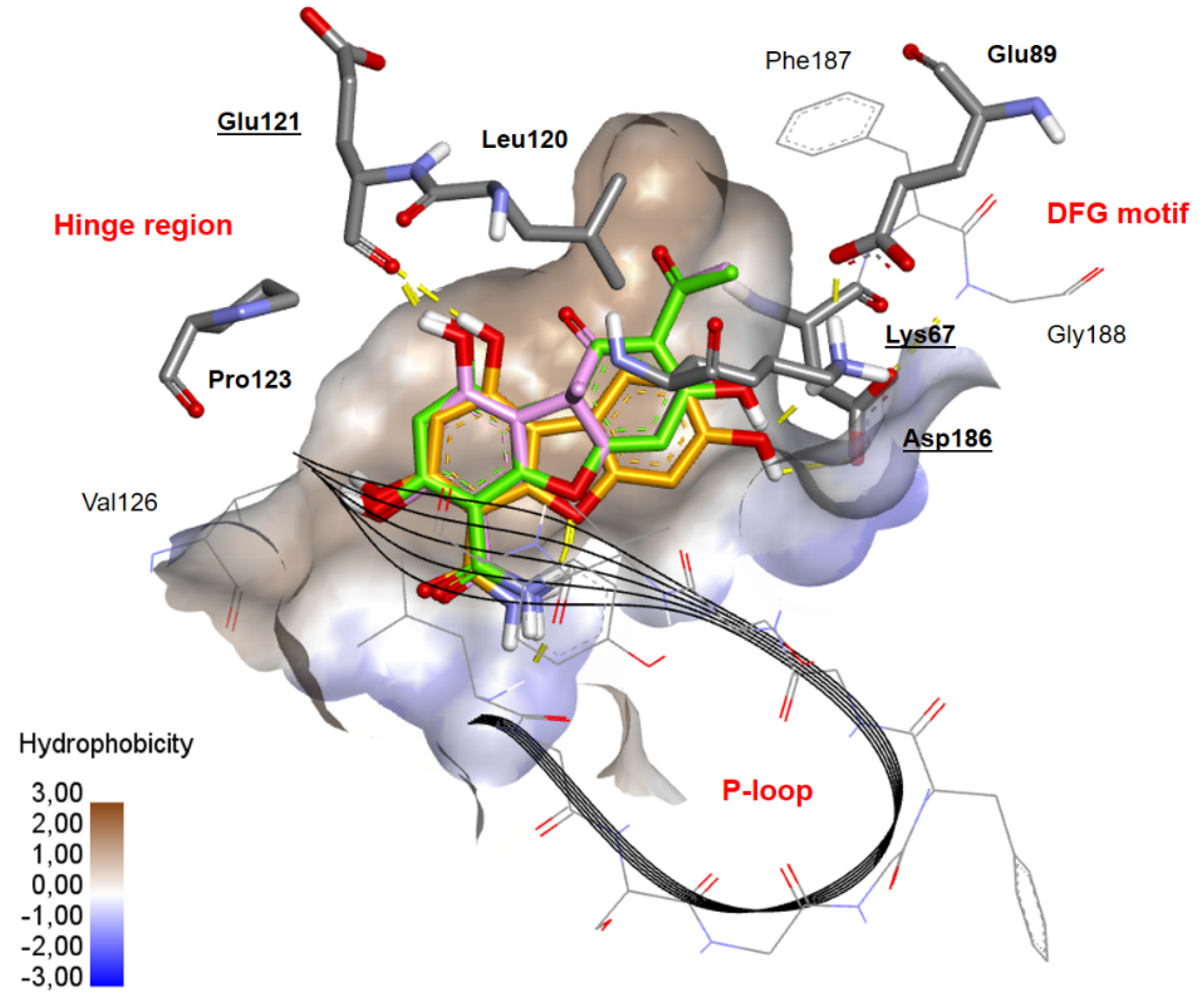
2.3. Docking Studies
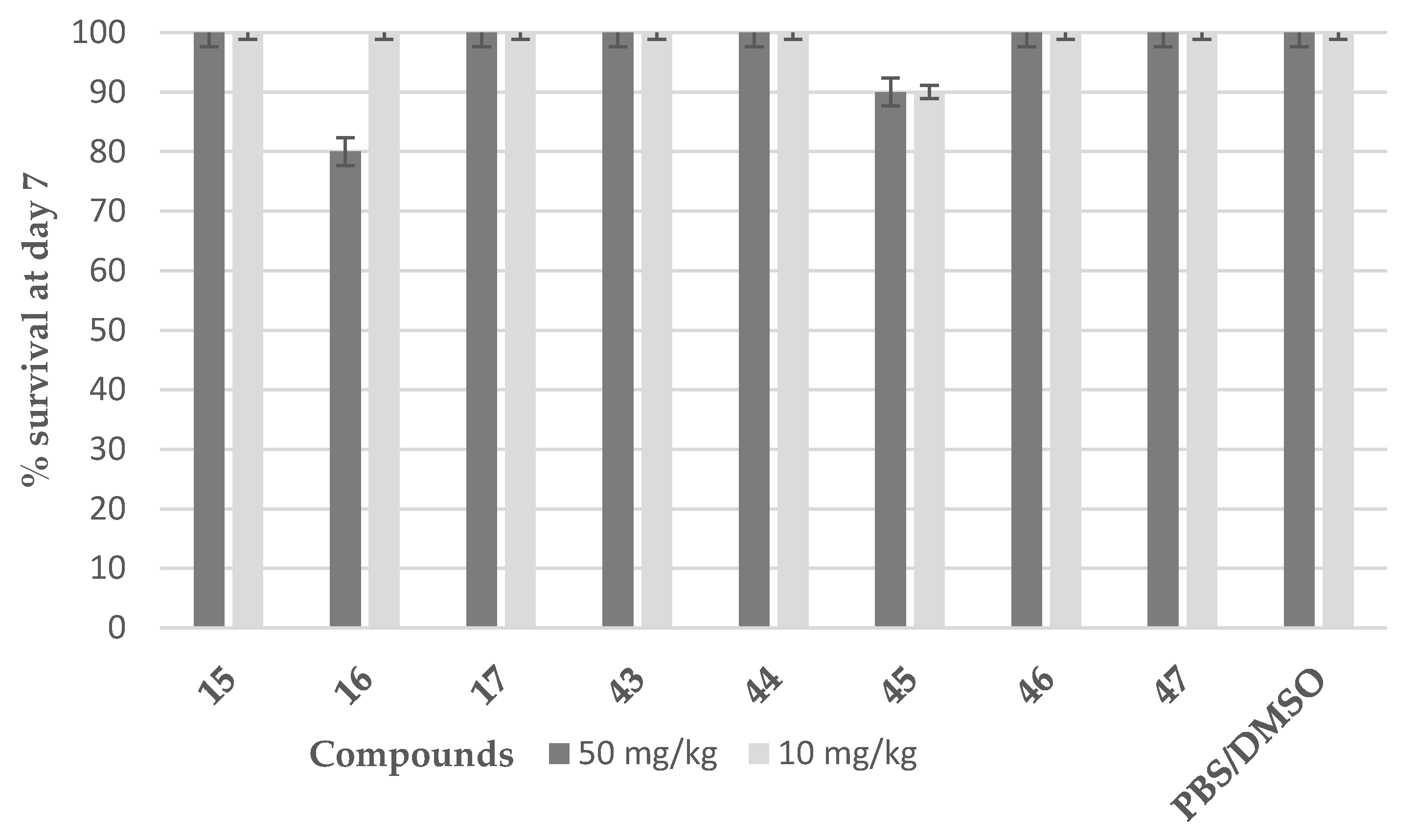
2.4. In Vitro Cell-Based Assays and Acute Toxicity Testing
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Procedures
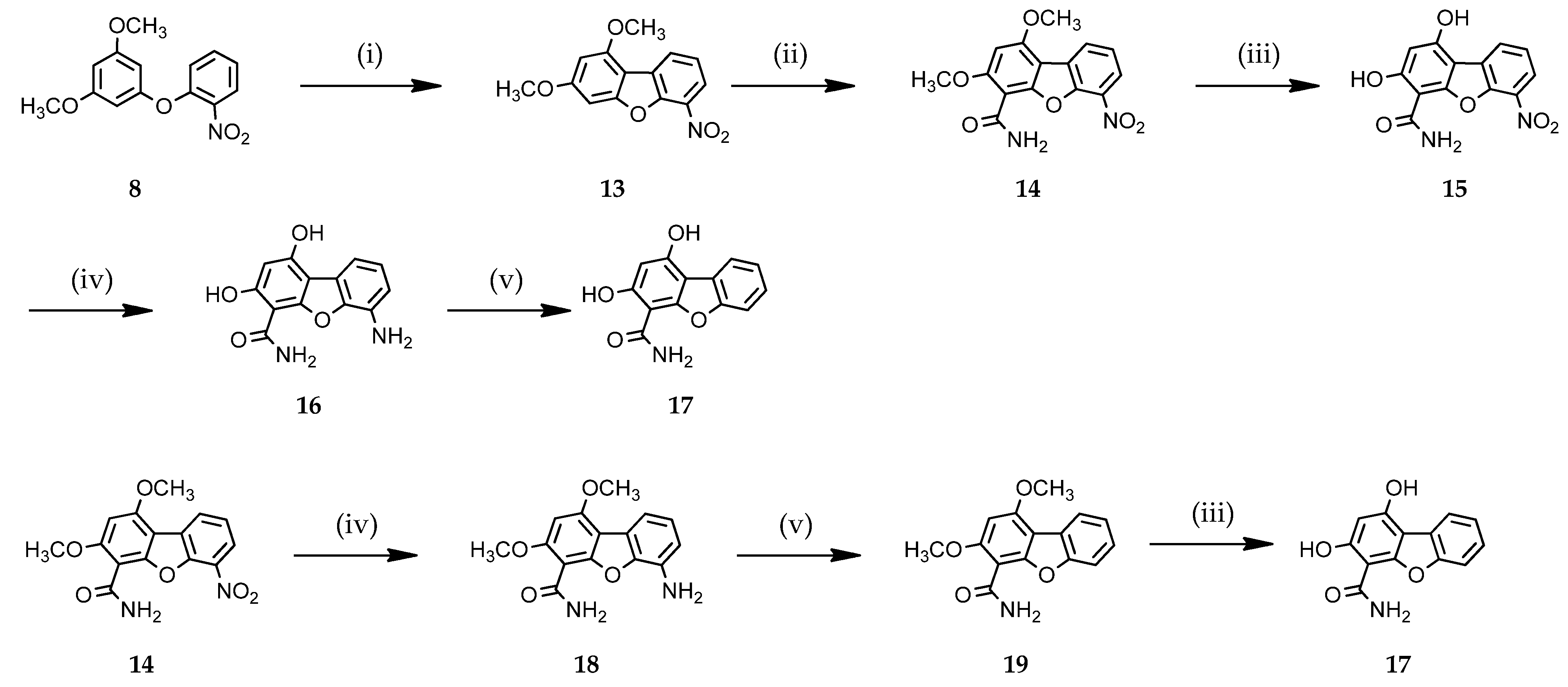
3.1.2. Access to Dibenzofuran Derivatives 14–19 by Intramolecular C-C Bond Formation from Diaryl Ethers
2-Hydroxy-4,6-dimethoxybenzamide (2)
2,4-Dimethoxy-6-(2′-nitrophenoxy)benzamide (6)
1,3-Dibenzyloxy-5-(2′-nitrophenoxy)benzene (7)
1,3-Dimethoxy-5-(2′-nitrophenoxy)benzene (8)
2-(3′,5′-Dibenzyloxyphenoxy)aniline (9)
2-(3′,5′-Dimethoxyphenoxy)aniline (10)
1,3-Dimethoxy-6-nitrodibenzo[b,d]furan (13)
1,3-Dimethoxy-6-nitrodibenzo[b,d]furan-4-carboxamide (14)
1,3-Dihydroxy-6-nitrodibenzo[b,d]furan-4-carboxamide (15)
6-Amino-1,3-dihydroxydibenzo[b,d]furan-4-carboxamide (16)
1,3-Dihydroxydibenzo[b,d]furan-4-carboxamide (17)
6-Amino-1,3-dimethoxydibenzo[b,d]furan-4-carboxamide (18)
1,3-Dimethoxydibenzo[b,d]furan-4-carboxamide (19)
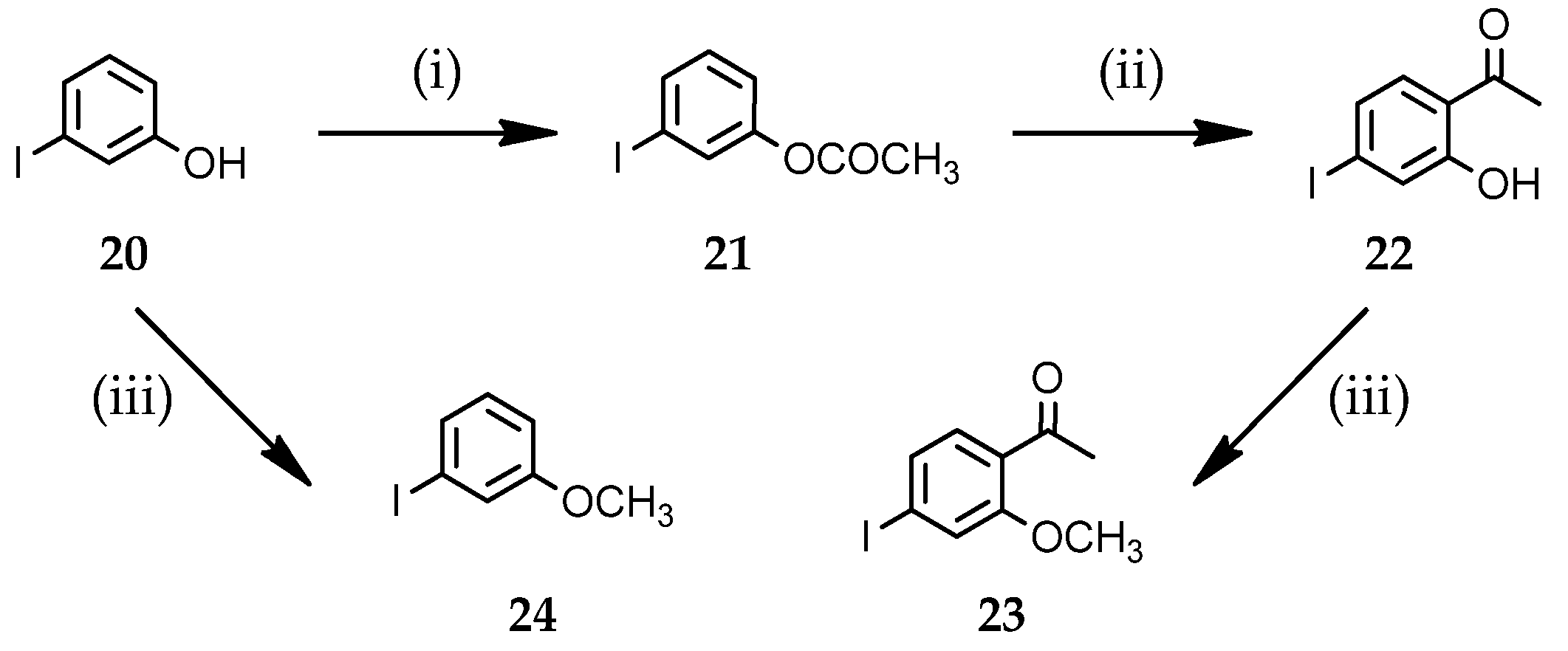
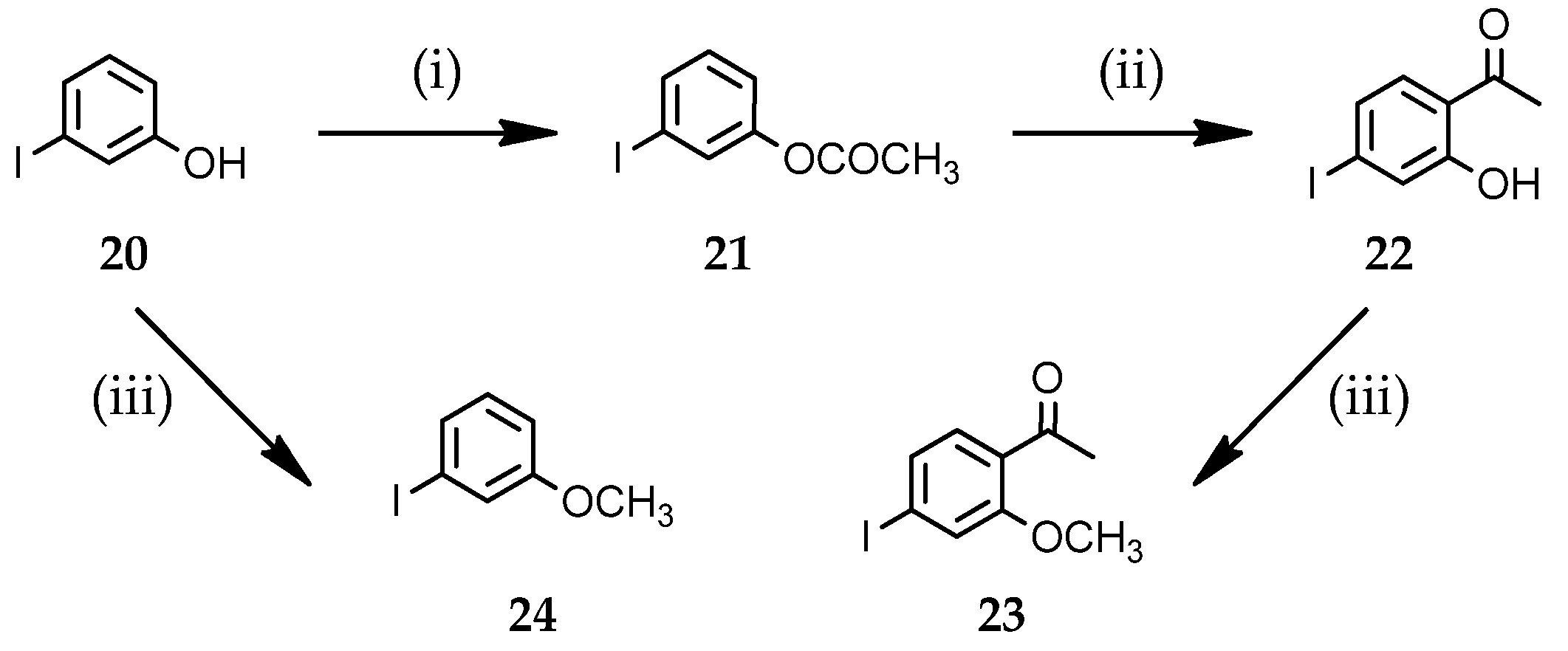
3.1.3. Preparation of Starting Iodo-Aryl Derivatives 21–24
3-Iodophenyl acetate (21)
1-(2-Hydroxy-4-iodophenyl)ethanone (22)
1-(4-Iodo-2-methoxyphenyl)ethanone (23)
1-Iodo-3-methoxybenzene (24)
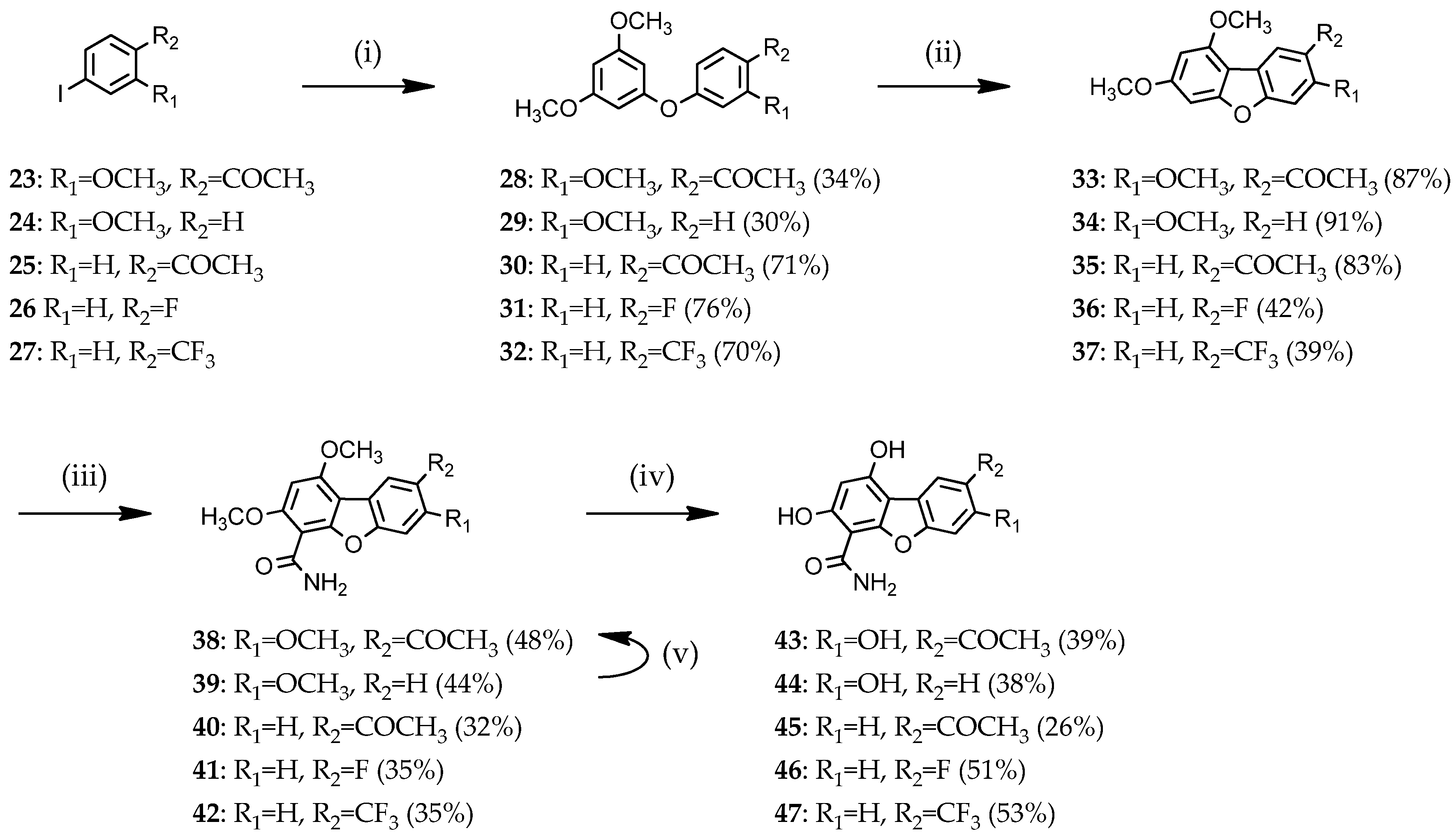
3.1.4. General Procedure for the Preparation of Diaryl Ethers 28–32
1-[4-(3′,5′-Dimethoxyphenoxy)-2-methoxyphenyl]ethanone (28)
1,3-Dimethoxy-5-(3′-methoxyphenoxy)benzene (29)
1-[4-(3′,5′-Dimethoxyphenoxy)phenyl]ethanone (30)
1-(4′-Fluorophenoxy)-3,5-dimethoxybenzene (31)
1,3-Dimethoxy-5-[4′-(trifluoromethyl)phenoxy]benzene (32)
3.1.5. General Procedure for the Preparation of 1,3-dimethoxydibenzofurans 33–37
1-(3,7,9-Trimethoxydibenzo[b,d]furan-2-yl)ethanone (33)
1,3,7-Trimethoxydibenzo[b,d]furan (34)
1-(7,9-Dimethoxydibenzo[b,d]furan-2-yl)ethanone (35)
8-Fluoro-1,3-dimethoxydibenzo[b,d]furan (36)
1,3-Dimethoxy-8-(trifluoromethyl)dibenzo[b,d]furan (37)
3.1.6. General Procedure for the Preparation of 1,3-dimethoxydibenzofuran-4-carboxamides 38–42
8-Acetyl-1,3,7-trimethoxydibenzo[b,d]furan-4-carboxamide (38)
1,3,7-Trimethoxydibenzo[b,d]furan-4-carboxamide (39)
8-Acetyl-1,3-dimethoxydibenzo[b,d]furan-4-carboxamide (40)
8-Fluoro-1,3-dimethoxydibenzo[b,d]furan-4-carboxamide (41)
1,3-Dimethoxy-8-(trifluoromethyl)dibenzo[b,d]furan-4-carboxamide (42)
3.1.7. General Procedure for the Preparation of 1,3-hydroxydibenzofuran-4-carboxamide 43–47
8-Acetyl-1,3,7-trihydroxydibenzo[b,d]furan-4-carboxamide (43)
1,3,7-Trihydroxydibenzo[b,d]furan-4-carboxamide (44)
8-Acetyl-1,3-dihydroxydibenzo[b,d]furan-4-carboxamide (45)
8-Fluoro-1,3-dihydroxydibenzo[b,d]furan-4-carboxamide (46)
1,3-Dihydroxy-8-(trifluoromethyl)dibenzo[b,d]furan-4-carboxamide (47)
3.1.8. Access to Dibenzofuran Derivative 19 by Intramolecular C-O Bond Formation from 2-Aryl Phenol 49
2-Hydroxy-3-iodo-4,6-dimethoxybenzamide (48)
2’-Fluoro-2-hydroxy-4,6-dimethoxybiphenyl-3-carboxamide (49)
1,3-Dimethoxydibenzo[b,d]furan-4-carboxamide (19)
3.2. Molecular Modeling
3.3. Biology
3.3.1. Mammalian Protein Kinase Assays
3.3.2. Cell Cultures and Reagents
3.3.3. In Vitro Cell-Based Assays
3.3.4. Evaluation of Compounds’ Cytotoxicity Using the In Vivo Galleria mellonella Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sugawara, F.; Strobel, S.; Strobel, G.; Larsen, R.D.; Berglund, D.L.; Gray, G.; Takahashi, N.; Coval, S.J.; Stout, T.J.; Clardy, J. The structure and biological activity of cercosporamide from Cercosporidium henningsii. J. Org. Chem. 1991, 56, 909–910. [Google Scholar] [CrossRef]
- Hoffman, A.M.; Mayer, S.G.; Strobel, G.A.; Hess, W.M.; Sovocool, G.W.; Grange, A.H.; Harper, J.K.; Arif, A.M.; Grant, D.M.; Kelley-Swift, E.G. Purification, identification and activity of phomodione, a furandione from an endophytic Phoma species. Phytochemistry 2008, 69, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-W.; Xu, B.-G.; Wang, J.-Y.; Su, Z.-Z.; Lin, F.-C.; Zhang, C.-L.; Kubicek, C.P. Bioactive metabolites from Phoma species, an endophytic fungus from the Chinese medicinal plant Arisaema Erubescens. Appl. Microbiol. Biotechnol. 2012, 93, 1231–1239. [Google Scholar] [CrossRef]
- Hosoya, T.; Ohsumi, J.; Hamano, K.; Ono, Y.; Miura, M. Method for Producing Cercosporamide. U.S. Patent 2010/0152467, 2010. [Google Scholar]
- Sussman, A.; Huss, K.; Chio, L.-C.; Heidler, S.; Shaw, M.; Ma, D.; Zhu, G.; Campbell, R.M.; Park, T.-S.; Kulanthaivel, P.; et al. Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-through put screening. Eukaryot. Cell 2004, 3, 932–943. [Google Scholar] [CrossRef] [Green Version]
- LaFayette, S.L.; Collins, C.; Zaas, A.K.; Schell, W.A.; Betancourt-Quiroz, M.; Gunatilaka, A.A.L.; Perfect, J.R.; Cowen, L.E. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010, 6, e1001069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konicek, B.W.; Stephens, J.R.; McNulty, A.M.; Robichaud, N.; Peery, R.B.; Dumstorf, C.A.; Dowless, M.S.; Iversen, P.W.; Parsons, S.; Ellis, K.E.; et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011, 71, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Lam, F.; Proud, C.; Wang, S. Targeting Mnks for cancer therapy. Oncotarget 2012, 3, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Altman, J.K.; Szilard, A.; Konicek, B.W.; Iversen, P.W.; Kroczynska, B.; Glaser, H.; Sassano, A.; Vakana, E.; Graff, J.R.; Platanias, L.C. Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood 2013, 121, 3675–3681. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sun, L.; Su, X.; Guo, S. Inhibition of eukaryotic initiation factor 4E phosphorylation by cercosporamide selectively suppresses angiogenesis, growth and survival of human hepatocellular carcinoma. Biomed. Pharmacother. 2016, 84, 237–243. [Google Scholar] [CrossRef]
- Grzmil, M.; Seebacher, J.; Hess, D.; Behe, M.; Schibli, R.; Moncayo, G.; Frank, S.; Hemmings, B.A. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell. Signal. 2016, 28, 1412–1421. [Google Scholar] [CrossRef]
- Hoeksma, J.; van der Zon, G.C.M.; ten Dijke, P.; den Hertog, J. Cercosporamide inhibits bone morphogenetic protein receptor type I kinase activity in zebrafish. Dis. Model. Mech. 2020, 13, dmm045971. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Arita, T.; Satoh, S.; Araki, K.; Kuroha, M.; Ohsumi, J. (−)-Cercosporamide derivatives as novel antihyperglycemic agents. Bioorg. Med. Chem. Lett. 2009, 19, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Arita, T.; Satoh, S.; Wakabayashi, K.; Hayashi, S.; Matsui, Y.; Araki, K.; Kuroha, M.; Ohsumi, J. Discovery of a novel selective PPARγ modulator from (−)-cercosporamide derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 2095–2098. [Google Scholar] [CrossRef]
- Bazin, M.-A.; Bodero, L.; Tomasoni, C.; Rousseau, B.; Roussakis, C.; Marchand, P. Synthesis and antiproliferative activity of benzofuran-based analogs of cercosporamide against non-small cell lung cancer cell lines. Eur. J. Med. Chem. 2013, 69, 823–832. [Google Scholar] [CrossRef]
- Dao, V.H.; Ourliac-Garnier, I.; Bazin, M.-A.; Jacquot, C.; Baratte, B.; Ruchaud, S.; Bach, S.; Grovel, O.; Le Pape, P.; Marchand, P. Benzofuro [3,2-d] pyrimidines inspired from cercosporamide CaPkc1 inhibitor: Synthesis and evaluation of fluconazole susceptibility restoration. Bioorg. Med. Chem. Lett. 2018, 28, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Antoine, M.; Schuster, T.; Seipelt, I.; Aicher, B.; Teifel, M.; Günther, E.; Gerlach, M.; Marchand, P. Efficient synthesis of novel disubstituted pyrido[3,4-b]pyrazines for the design of protein kinase inhibitors. Med. Chem. Commun. 2016, 7, 224–229. [Google Scholar] [CrossRef]
- Winfield, H.J.; Cahill, M.M.; O′Shea, K.D.; Pierce, L.T.; Robert, T.; Ruchaud, S.; Bach, S.; Marchand, P.; McCarthy, F.O. Synthesis and anticancer activity of novel bisindolylhydroxymaleimide derivatives with potent GSK-3 kinase inhibition. Bioorg. Med. Chem. 2018, 26, 4209–4224. [Google Scholar] [CrossRef]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Bonnet, P.; Souab, M.; Robert, T.; Ruchaud, S.; Bach, S.; Berthelot, P.; Gouilleux, F.; et al. Structure-based design of novel quinoxaline-2-carboxylic acids and analogues as Pim-1 inhibitors. Eur. J. Med. Chem. 2018, 154, 101–109. [Google Scholar] [CrossRef]
- Loidreau, Y.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Loaëc, N.; Meijer, L.; Besson, T.; Marchand, P. Exploring kinase inhibition properties of 9H-pyrimido[5,4-b]- and [4,5-b]indol-4-amine derivatives. Pharmaceuticals 2020, 13, 89. [Google Scholar] [CrossRef]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Robert, T.; Bach, S.; Ibrahim, S.; Raoul, W.; Croix, C.; Berthelot, P.; Guillon, J.; et al. New quinoxaline derivatives as dual Pim-1/2 kinase inhibitors: Design, synthesis and biological evaluation. Molecules 2021, 26, 867. [Google Scholar] [CrossRef]
- Bazin, M.-A.; Cojean, S.; Pagniez, F.; Bernadat, G.; Cavé, C.; Ourliac-Garnier, I.; Nourrisson, M.-R.; Morgado, C.; Picot, C.; Leclercq, O.; et al. In vitro identification of imidazo[1,2-a]pyrazine-based antileishmanial agents and evaluation of L. major casein kinase 1 inhibition. Eur. J. Med. Chem. 2021, 210, 112956. [Google Scholar] [CrossRef]
- Love, B.E. Isolation and synthesis of polyoxygenated dibenzofurans possessing biological activity. Eur. J. Med. Chem. 2015, 97, 377–387. [Google Scholar] [CrossRef]
- Oramas-Royo, S.; Pantoja, K.D.; Amesty, Á.; Romero, C.; Lorenzo-Castrillejo, I.; Machín, F.; Estévez-Braun, A. Synthesis and antibacterial activity of new symmetric polyoxygenated dibenzofurans. Eur. J. Med. Chem. 2017, 141, 178–187. [Google Scholar] [CrossRef]
- Heng, S.; Harris, K.M.; Kantrowitz, E.R. Designing inhibitors against fructose 1,6-bisphosphatase: Exploring natural products for novel inhibitor scaffolds. Eur. J. Med. Chem. 2010, 45, 1478–1484. [Google Scholar] [CrossRef] [Green Version]
- Millot, M.; Dieu, A.; Tomasi, S. Dibenzofurans and derivatives from lichens and ascomycetes. Nat. Prod. Rep. 2016, 33, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Huang, M.; Cao, Y.; Gong, P.; Liu, W.; Jin, S.; Wen, J.; Jing, Y.; Liu, D.; Zhao, L. Usnic acid is a novel Pim-1 inhibitor with the abilities of inhibiting growth and inducing apoptosis in human myeloid leukemia cells. RSC Adv. 2016, 6, 24091–24096. [Google Scholar] [CrossRef]
- Wang, S.; Zang, J.; Huang, M.; Guan, L.; Xing, K.; Zhang, J.; Liu, D.; Zhao, L. Discovery of novel (+)-usnic acid derivatives as potential anti-leukemia agents with pan-pim kinases inhibitory activity. Bioorg. Chem. 2019, 89, 102971. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Alnabulsi, S.; Al-Hurani, E.A. Pim kinase inhibitors in cancer: Medicinal chemistry insights into their activity and selectivity. Drug Discov. Today 2020, 25, 2062–2069. [Google Scholar] [CrossRef]
- Arrouchi, H.; Lakhlili, W.; Ibrahimi, A. A review on PIM kinases in tumors. Bioinformation 2019, 15, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 kinase as cancer drug target: An update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, N.K.; Sabina, E.P. A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy. Life Sci. 2020, 255, 117866. [Google Scholar] [CrossRef]
- Harshita, P.S.; Yasaswi, P.S.; Jyothi, V.; Jyostna, T.S. Pim-1 kinase: A novel target for cancer chemotherapy—A review. IJPSR 2020, 11, 2528–2538. [Google Scholar] [CrossRef]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef]
- Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel pan-pim kinase inhibitors with imidazopyridazine and thiazolidinedione structure exert potent antitumor activities. Front. Pharmacol. 2021, 12, 672536. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, J.; Si, C.; Ma, W. Synthesis of dibenzofurans by palladium-catalysed tandem denitrification/C-H activation. Synlett 2011, 2011, 3023–3025. [Google Scholar] [CrossRef]
- Panda, N.; Mattan, I.; Nayak, D.K. Synthesis of dibenzofurans via C–H activation of o -iodo diaryl ethers. J. Org. Chem. 2015, 80, 6590–6597. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, K.; Zheng, H.; Ding, R.; Yin, F.; Wang, N.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. A one-pot synthesis of dibenzofurans from 6-diazo-2-cyclohexenones. Org. Lett. 2015, 17, 5744–5747. [Google Scholar] [CrossRef]
- Cho, J.Y.; Roh, G.; Cho, E.J. Visible-light-promoted synthesis of dibenzofuran derivatives. J. Org. Chem. 2018, 83, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Camargo Solórzano, P.; Brigante, F.; Pierini, A.B.; Jimenez, L.B. Photoinduced synthesis of dibenzofurans: Intramolecular and intermolecular comparative methodologies. J. Org. Chem. 2018, 83, 7867–7877. [Google Scholar] [CrossRef]
- Wassmundt, F.W.; Pedemonte, R.P. An improved synthesis of dibenzofurans by a free-radical cyclization. J. Org. Chem. 1995, 60, 4991–4994. [Google Scholar] [CrossRef]
- Gajera, J.M.; Gopalan, B.; Yadav, P.S.; Patil, S.D.; Gharat, L.A. Synthesis of multi-substituted dibenzo[b,d]furan. J. Heterocycl. Chem. 2008, 45, 797–801. [Google Scholar] [CrossRef]
- Fang, L.; Fang, X.; Gou, S.; Lupp, A.; Lenhardt, I.; Sun, Y.; Huang, Z.; Chen, Y.; Zhang, Y.; Fleck, C. Design, synthesis and biological evaluation of d-ring opened galantamine analogs as multifunctional anti-Alzheimer agents. Eur. J. Med. Chem. 2014, 76, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalysed ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef]
- Brewster, R.Q.; Groening, T. p-nitrodiphenyl ether: Ether, p-nitrophenyl phenyl. In Organic Syntheses; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; Volume 14, p. 66. ISBN 978-0-471-26422-4. [Google Scholar]
- Kelly, S.M.; Lipshutz, B.H. Chemoselective reductions of nitroaromatics in water at room temperature. Org. Lett. 2014, 16, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Krug, M.; Wichapong, K.; Erlenkamp, G.; Sippl, W.; Schächtele, C.; Totzke, F.; Hilgeroth, A. Discovery of 4-benzylamino-substituted α-carbolines as a novel class of receptor tyrosine kinase inhibitors. ChemMedChem 2011, 6, 63–72. [Google Scholar] [CrossRef]
- Liégault, B.; Lee, D.; Huestis, M.P.; Stuart, D.R.; Fagnou, K. Intramolecular Pd(II)-catalyzed oxidative biaryl synthesis under air: Reaction development and scope. J. Org. Chem. 2008, 73, 5022–5028. [Google Scholar] [CrossRef] [PubMed]
- Bartholomäus, R.; Dommershausen, F.; Thiele, M.; Karanjule, N.S.; Harms, K.; Koert, U. Total synthesis of the postulated structure of fulicineroside. Chem. Eur. J. 2013, 19, 7423–7436. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, B.; Liu, B.; Huang, M.; Li, D.; Guan, L.; Zang, J.; Liu, D.; Zhao, L. Design and synthesis of novel 6-hydroxy-4-methoxy-3-methylbenzofuran-7-carboxamide derivatives as potent mnks inhibitors by fragment-based drug design. Bioorg. Med. Chem. 2018, 26, 4602–4614. [Google Scholar] [CrossRef]
- Jepsen, T.H.; Donor, D.-F. Synthesis of dibenzothiophene, dibenzofuran and carbazole donor–acceptor chromophores. Synthesis 2013, 45, 1115–1120. [Google Scholar] [CrossRef]
- Boyer, J.L.; Krum, J.E.; Myers, M.C.; Fazal, A.N.; Wigal, C.T. Synthetic utility and mechanistic implications of the fries rearrangement of hydroquinone diesters in boron trifluoride complexes. J. Org. Chem. 2000, 65, 4712–4714. [Google Scholar] [CrossRef]
- Liu, L.-X.; Wang, X.-Q.; Yan, J.-M.; Li, Y.; Sun, C.-J.; Chen, W.; Zhou, B.; Zhang, H.-B.; Yang, X.-D. Synthesis and antitumor activities of novel dibenzo[b,d]furan–imidazole hybrid compounds. Eur. J. Med. Chem. 2013, 66, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fitzgerald, A.E.; Mani, N.S. Facile assembly of fused benzo[4,5]furo heterocycles. J. Org. Chem. 2008, 73, 2951–2954. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A bioluminescent and homogeneous ADP monitoring assay for kinases. ASSAY Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.F.; Meijer, L. Dual-specificity, tyrosine phosphorylation-regulated kinases (DYRKs) and Cdc2-like kinases (CLKs) in human disease, an overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Dinh, H.; Semenec, L.; Kumar, S.S.; Short, F.L.; Cain, A.K. Microbiology’s next top model: Galleria in the molecular age. Pathog. Dis. 2021, 79, ftab006. [Google Scholar] [CrossRef]
- Bennett, C.J.; Caldwell, S.T.; McPhail, D.B.; Morrice, P.C.; Duthie, G.G.; Hartley, R.C. Potential therapeutic antioxidants that combine the radical scavenging ability of myricetin and the lipophilic chain of vitamin E to effectively inhibit microsomal lipid peroxidation. Bioorg. Med. Chem. 2004, 12, 2079–2098. [Google Scholar] [CrossRef]
- Chen, F.C.; Chang, T. Synthesis of 7-halogenoflavone and related compounds. J. Chem. Soc. 1958, 146–150. [Google Scholar] [CrossRef]
- Jin, X.; Davies, R.P. Copper-catalysed aromatic-finkelstein reactions with amine-based ligand systems. Catal. Sci. Technol. 2017, 7, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- Tripos Associates, Inc. SYBYL-X 1.3; Tripos Associates, Inc.: St. Louis, MO, USA, 2013. [Google Scholar]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Morishita, D.; Takami, M.; Yoshikawa, S.; Katayama, R.; Sato, S.; Kukimoto-Niino, M.; Umehara, T.; Shirouzu, M.; Sekimizu, K.; Yokoyama, S.; et al. Cell-permeable carboxyl-terminal P27Kip1 peptide exhibits anti-tumor activity by inhibiting Pim-1 kinase. J. Biol. Chem. 2011, 286, 2681–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes. Dassault Systèmes BIOVIA, Discovery Studio Visualizer, v19.1.0.18287; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]

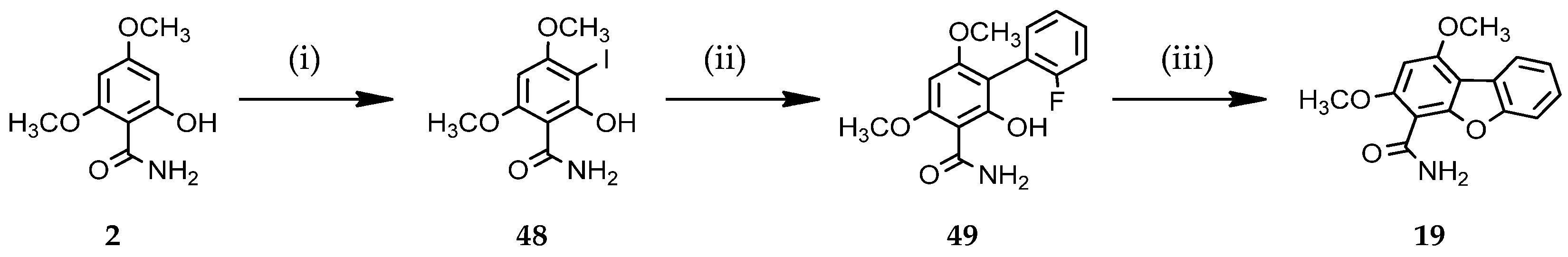


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
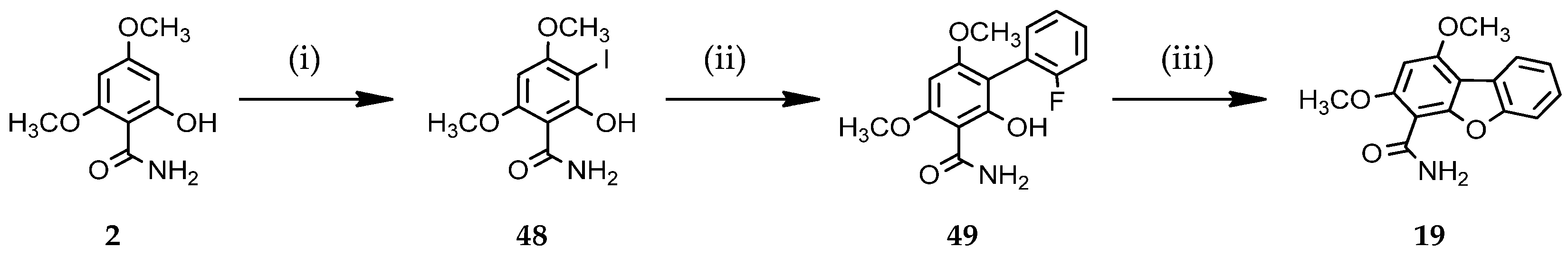{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Kinase Enzymatic IC50 (µM) 1,2 | |||||||||
| Entry | Compound | Series | R1 | R2 | R3 | CDK5/ p25 | CDK9/ CyclinT | Pim-1 | CLK1 |
| 1 | Cerco 3 | 5.60 | 0.22 | 0.73 | >10 | ||||
| 2 | 14 | A | NO2 | H | H | >10 | >10 | 3.21 | >10 |
| 3 | 15 | B | NO2 | H | H | >10 | >10 | 0.18 | 1.22 |
| 4 | 18 | A | NH2 | H | H | >10 | >10 | >10 | >10 |
| 5 | 16 | B | NH2 | H | H | >10 | >10 | 0.13 | 0.45 |
| 6 | 19 | A | H | H | H | >10 | >10 | >10 | 0.75 |
| 7 | 17 | B | H | H | H | >10 | >10 | 0.23 | 0.26 |
| 8 | 38 | A | H | OCH3 | COCH3 | >10 | >10 | >10 | >10 |
| 9 | 43 | B | H | OH | COCH3 | >10 | >10 | 0.82 | 0.29 |
| 10 | 39 | A | H | OCH3 | H | >10 | >10 | >10 | >10 |
| 11 | 44 | B | H | OH | H | >10 | >10 | 0.06 | 0.026 |
| 12 | 40 | A | H | H | COCH3 | >10 | >10 | >10 | >10 |
| 13 | 45 | B | H | H | COCH3 | >10 | >10 | 0.28 | 0.14 |
| 14 | 41 | A | H | H | F | >10 | >10 | >10 | 0.85 |
| 15 | 46 | B | H | H | F | >10 | 0.92 | 1.20 | 0.62 |
| 16 | 42 | A | H | H | CF3 | >10 | >10 | >10 | >10 |
| 17 | 47 | B | H | H | CF3 | >10 | >10 | 2.35 | >10 |
 | ||||||
|---|---|---|---|---|---|---|
| IC50 (µM) 1 | ||||||
| Entry | Compound | R1 | R2 | R3 | Pim-1 | Pim-2 |
| 1 | Cerco 2 | 0.73 | 1.43 | |||
| 2 | 15 | NO2 | H | H | 0.18 | 0.33 |
| 3 | 16 | NH2 | H | H | 0.13 | 0.085 |
| 4 | 17 | H | H | H | 0.23 | 0.20 |
| 5 | 43 | H | OH | COCH3 | 0.82 | 0.14 |
| 6 | 44 | H | OH | H | 0.06 | 0.035 |
| 7 | 45 | H | H | COCH3 | 0.28 | 0.12 |
| 8 | 46 | H | H | F | 1.2 | 0.25 |
| 9 | 47 | H | H | CF3 | 2.35 | 0.37 |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) 1,2 | ||||||||||
| Entry | Compound | R1 | R2 | R3 | MV4-11 | KU812 | K562 | MCF-7 | HeLa | L929 |
| 1 | 15 | NO2 | H | H | 30.7 ± 1.1 | 78.4 ± 2.1 | >100 | >100 | 24.7 ± 6.7 | 49.2 ± 1.0 |
| 2 | 16 | NH2 | H | H | 10.1 ± 0.8 | >100 | >100 | >100 | 14.6 ± 7.2 | >100 |
| 3 | 17 | H | H | H | 14.3 ± 1.1 | >100 | >100 | >100 | 9.5 ± 4.1 | >100 |
| 4 | 43 | H | OH | COCH3 | 52.2 ± 1.7 | >100 | >100 | >100 | 57.1 ± 7.6 | >100 |
| 5 | 44 | H | OH | H | 2.6 ± 0.4 | 42.1 ± 1.3 | 75.7 ± 11.8 | 52.5 ± 1.2 | 10.2 ± 1.2 | 28.4 ± 1.1 |
| 6 | 45 | H | H | COCH3 | 8.8 ± 0.9 | 69.4 ± 12.9 | >100 | >100 | 59.6 ± 7.6 | >100 |
| 7 | 46 | H | H | F | 16.1 ± 1.6 | >100 | >100 | - | - | - |
| 8 | 47 | H | H | CF3 | >100 | >100 | >100 | - | - | - |
| 9 | Cerco 3 | 31.5 ± 4.7 | >100 | >100 | 44.3 ± 2.1 | 7.5 ± 3.1 | - | |||
| 10 | Doxo 3 | - | - | - | 0.50 ± 0.02 | 2.7 ± 0.2 | 2.4 ± 0.4 | |||
| 11 | SGI-1776 3 | 0.030 ± 0.003 | 3.5 ± 0.6 | 3.7 ± 1.5 | - | - | - | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dao, V.H.; Ourliac-Garnier, I.; Logé, C.; McCarthy, F.O.; Bach, S.; da Silva, T.G.; Denevault-Sabourin, C.; Thiéfaine, J.; Baratte, B.; Robert, T.; et al. Dibenzofuran Derivatives Inspired from Cercosporamide as Dual Inhibitors of Pim and CLK1 Kinases. Molecules 2021, 26, 6572. https://doi.org/10.3390/molecules26216572
Dao VH, Ourliac-Garnier I, Logé C, McCarthy FO, Bach S, da Silva TG, Denevault-Sabourin C, Thiéfaine J, Baratte B, Robert T, et al. Dibenzofuran Derivatives Inspired from Cercosporamide as Dual Inhibitors of Pim and CLK1 Kinases. Molecules. 2021; 26(21):6572. https://doi.org/10.3390/molecules26216572
Chicago/Turabian StyleDao, Viet Hung, Isabelle Ourliac-Garnier, Cédric Logé, Florence O. McCarthy, Stéphane Bach, Teresinha Gonçalves da Silva, Caroline Denevault-Sabourin, Jérôme Thiéfaine, Blandine Baratte, Thomas Robert, and et al. 2021. "Dibenzofuran Derivatives Inspired from Cercosporamide as Dual Inhibitors of Pim and CLK1 Kinases" Molecules 26, no. 21: 6572. https://doi.org/10.3390/molecules26216572
APA StyleDao, V. H., Ourliac-Garnier, I., Logé, C., McCarthy, F. O., Bach, S., da Silva, T. G., Denevault-Sabourin, C., Thiéfaine, J., Baratte, B., Robert, T., Gouilleux, F., Brachet-Botineau, M., Bazin, M.-A., & Marchand, P. (2021). Dibenzofuran Derivatives Inspired from Cercosporamide as Dual Inhibitors of Pim and CLK1 Kinases. Molecules, 26(21), 6572. https://doi.org/10.3390/molecules26216572