
Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors


Abstract
:1. Introduction
2. Results and Discussion
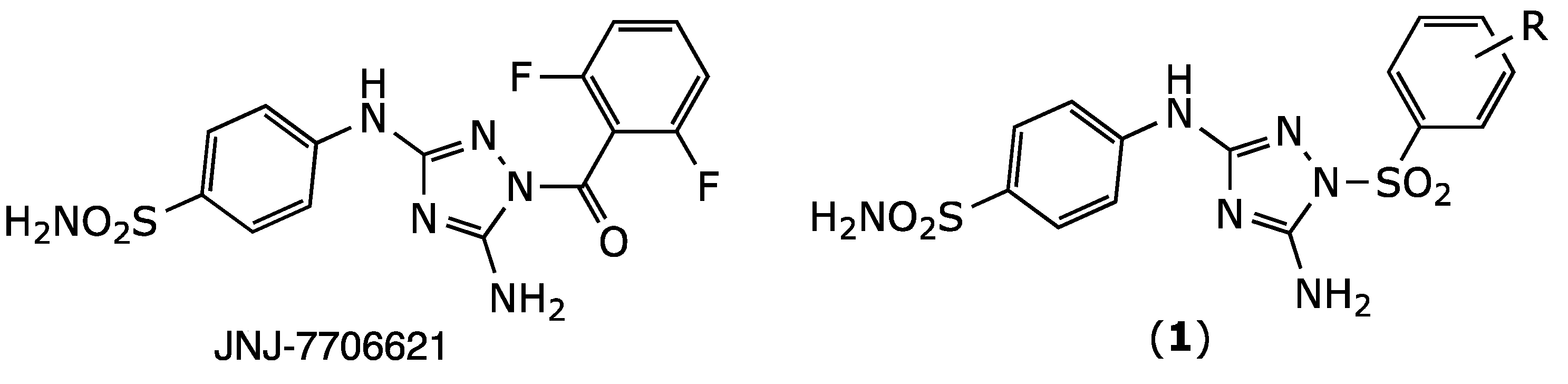
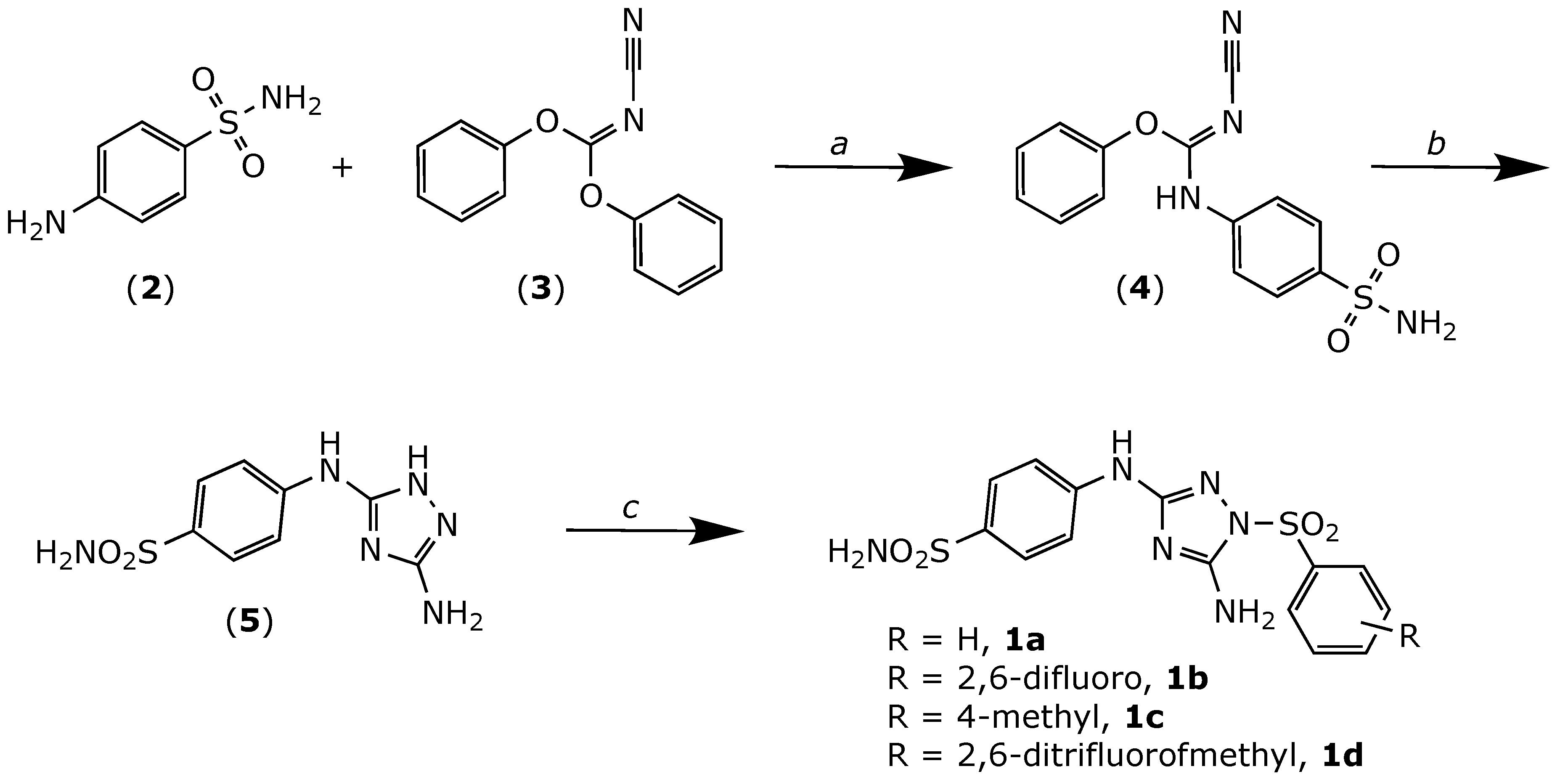
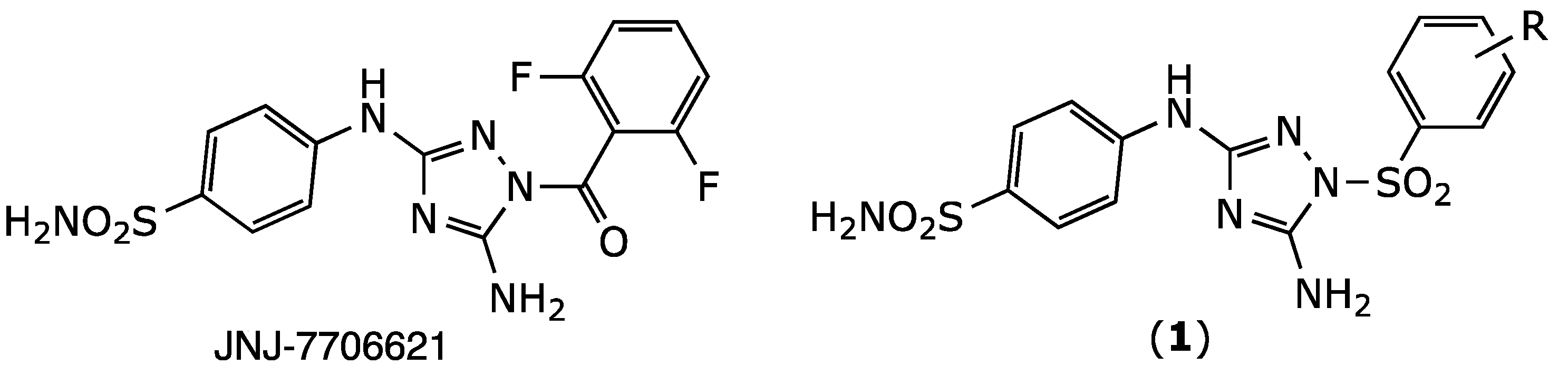
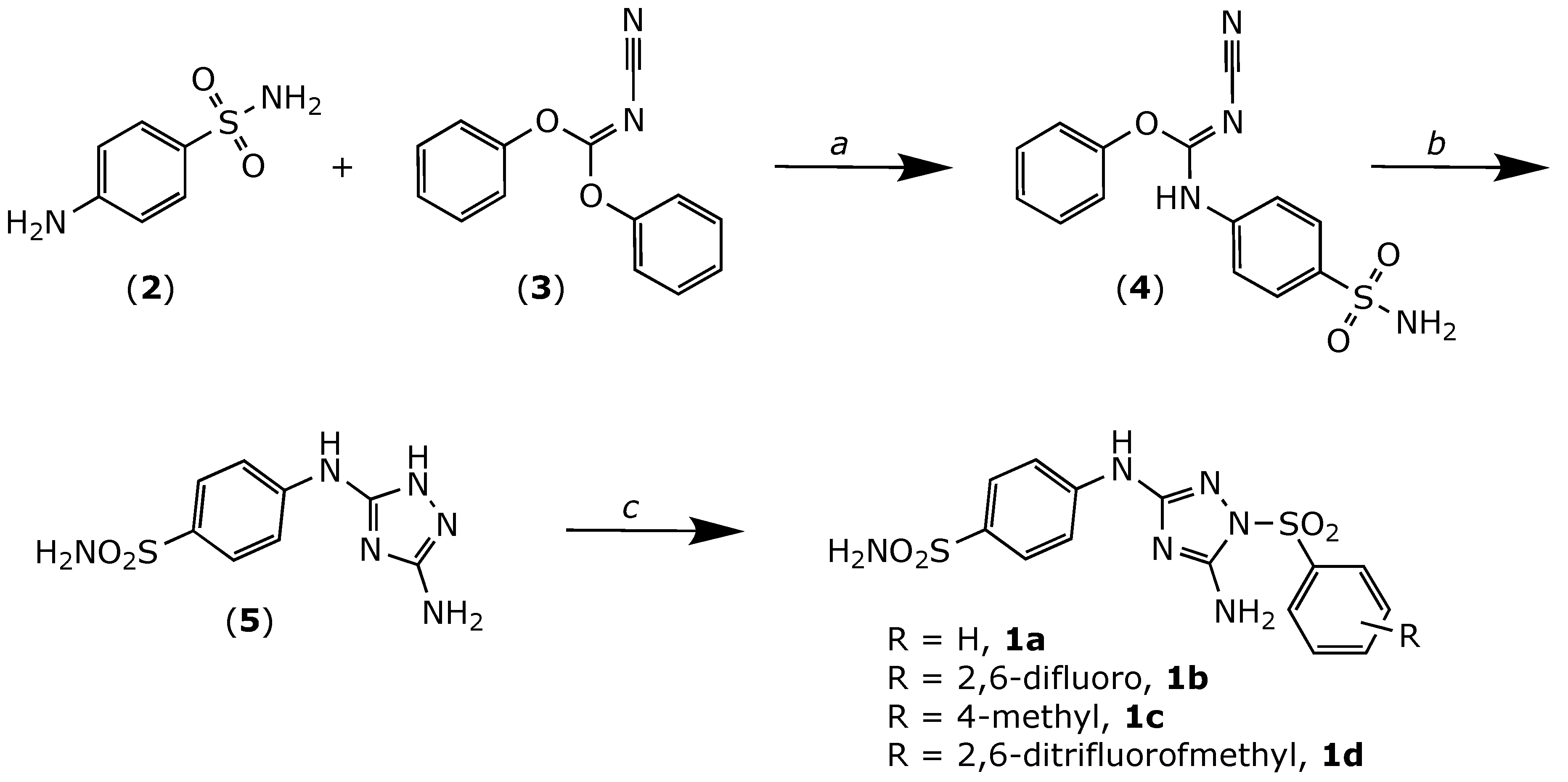
2.1. Chemistry
2.2. Biological Evaluation
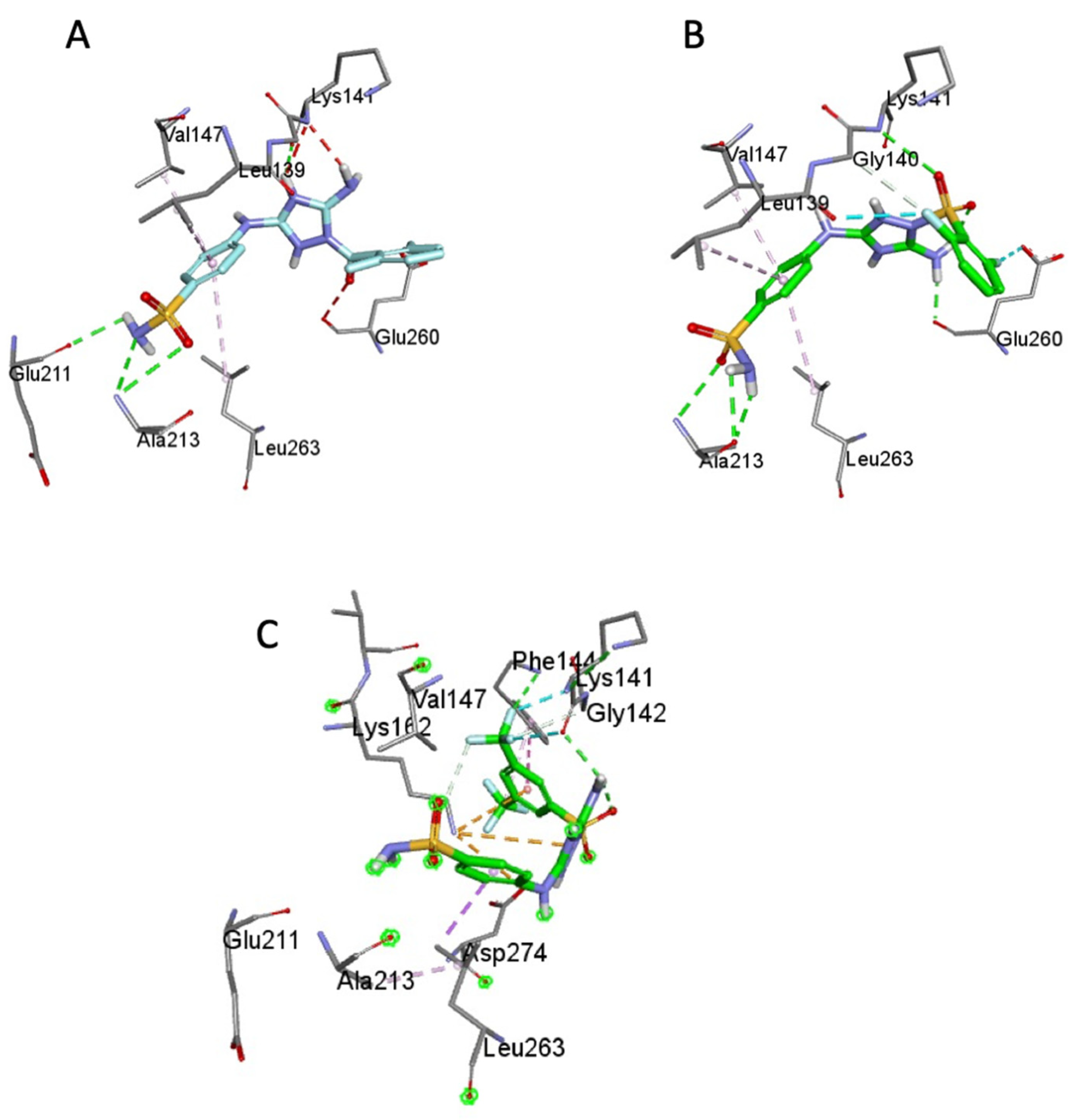
2.3. Molecular Modeling
3. Materials and Methods
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Neuse, C.J.; Lomas, O.C.; Schliemann, C.; Shen, Y.J.; Manier, S.; Bustoros, M.; Ghobrial, I.M. Genome instability in multiple myeloma. Leukemia 2020, 34, 2887–2897. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Assoro, A.B.; Haddad, T.; Galanis, E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 2015, 5, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitzen, J.J.; de Jonge, M.J.; Verweij, J. Aurora kinase inhibitors. Crit. Rev. Oncol. Hematol. 2010, 73, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, S.; Rugg, C.A.; Gruninger, R.H.; Lin, R.; Fuentes-Pesquera, A.; Connolly, P.J.; Wetter, S.K.; Hollister, B.; Kruger, W.W.; Napier, C.; et al. The in vitro and in vivo effects of JNJ-7706621: A dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005, 65, 9038–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodland, G.E.; Melhus, K.; Generalov, R.; Gilani, S.; Bertoni, F.; Dahle, J.; Syljuasen, R.G.; Patzke, S. The Dual Cell Cycle Kinase Inhibitor JNJ-7706621 Reverses Resistance to CD37-Targeted Radioimmunotherapy in Activated B Cell Like Diffuse Large B Cell Lymphoma Cell Lines. Front. Oncol. 2019, 9, 1301. [Google Scholar] [CrossRef] [PubMed]
- Vijayadas, K.N.; Davis, H.C.; Kotmale, A.S.; Gawade, R.L.; Puranik, V.G.; Rajamohanan, P.R.; Sanjayan, G.J. An unusual conformational similarity of two peptide folds featuring sulfonamide and carboxamide on the backbone. Chem. Commun. 2012, 48, 9747–9749. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Connolly, P.J.; Huang, S.; Wetter, S.K.; Lu, Y.; Murray, W.V.; Emanuel, S.L.; Gruninger, R.H.; Fuentes-Pesquera, A.R.; Rugg, C.A.; et al. 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: Synthesis and evaluation of biological activities. J. Med. Chem. 2005, 48, 4208–4211. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.R.; Peterson, L.B.; Imperiali, B.; Stains, C.I. Quantification of protein kinase enzymatic activity in unfractionated cell lysates using CSox-based sensors. Curr. Protoc. Chem. Biol. 2014, 6, 135–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.; Wang, L.; Xu, S.; Xu, J. Aurora-A kinase inhibitor scaffolds and binding modes. Drug Discov. Today 2011, 16, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
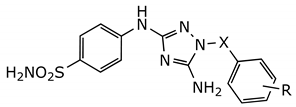 | |||
| Compound | X | R | IC50 (µM) 1 |
| JNJ-7706621 | CO | 2,6-difluoro | 0.016 ± 0.00 |
| 1a | SO2 | H | 0.13 ± 0.06 |
| 1b | SO2 | 2,6-difluoro | 0.21 ± 0.02 |
| 1c | SO2 | 4-Me | 0.23 ± 0.02 |
| 1d | SO2 | 2,6-ditrifluoromethyl | 1.78 ± 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah, O. Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors. Molecules 2021, 26, 5678. https://doi.org/10.3390/molecules26185678
Abdullah O. Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors. Molecules. 2021; 26(18):5678. https://doi.org/10.3390/molecules26185678
Chicago/Turabian StyleAbdullah, Omeima. 2021. "Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors" Molecules 26, no. 18: 5678. https://doi.org/10.3390/molecules26185678
APA StyleAbdullah, O. (2021). Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors. Molecules, 26(18), 5678. https://doi.org/10.3390/molecules26185678