
A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
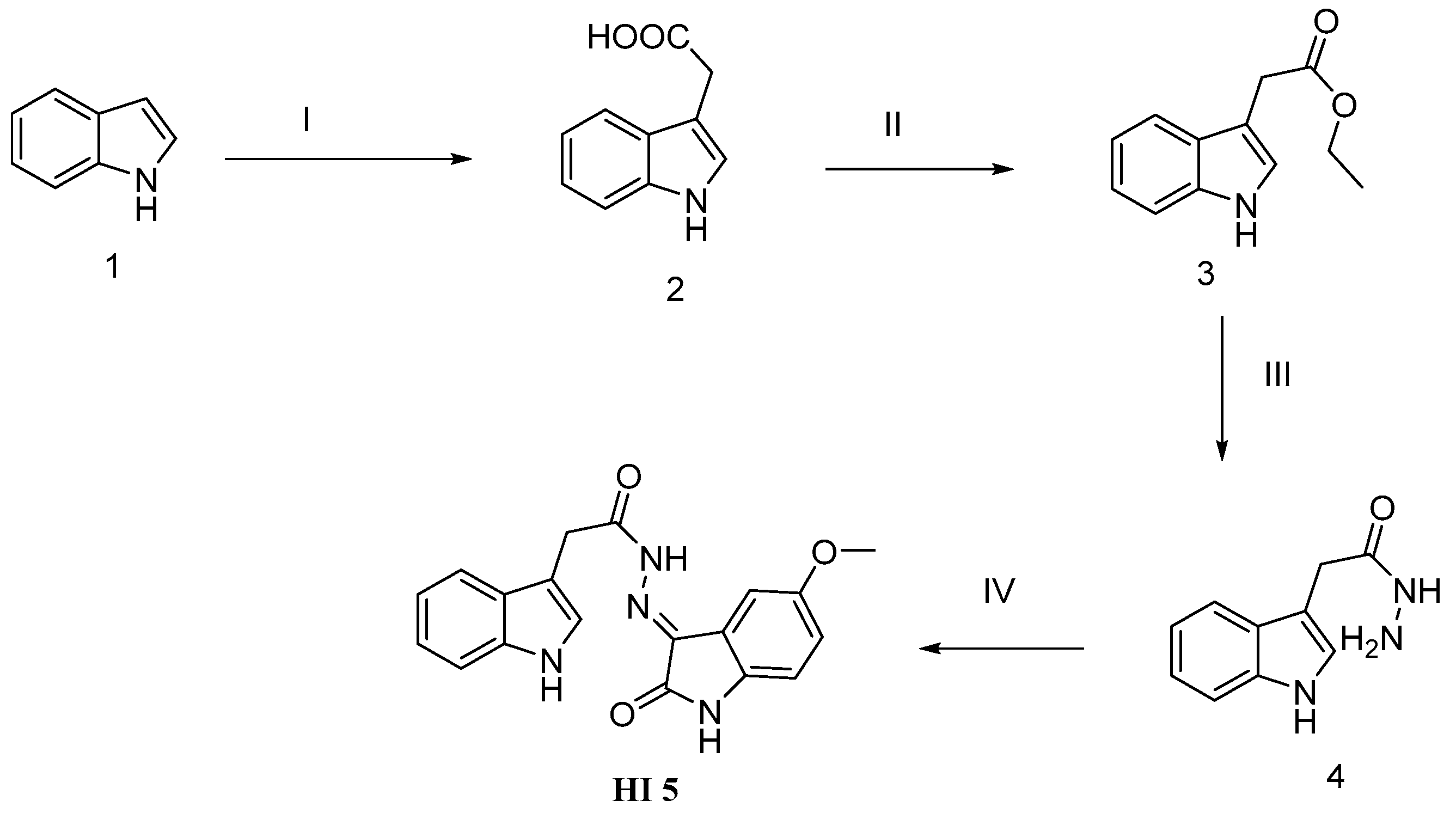
2.1. Chemistry
2.2. Biological Screening
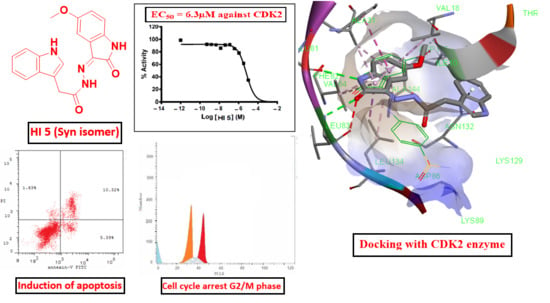
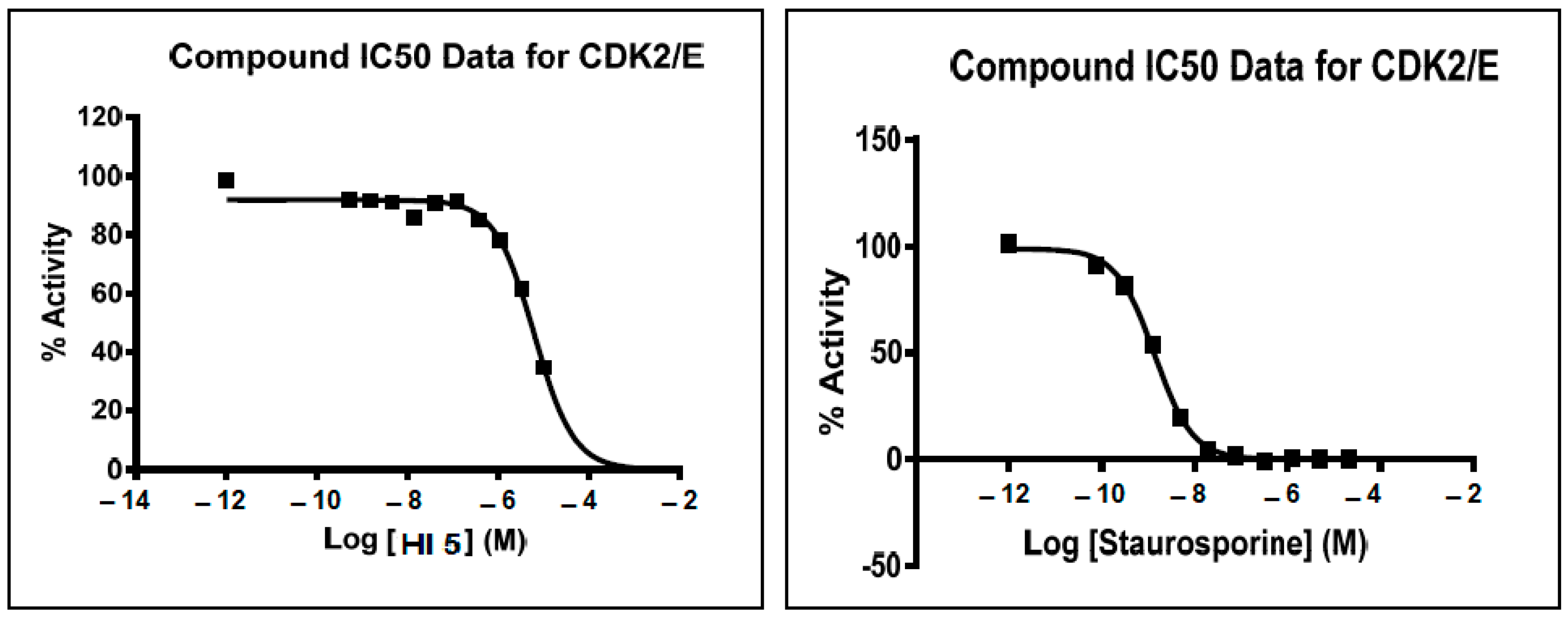
2.2.1. CDK2 Inhibitory Activity
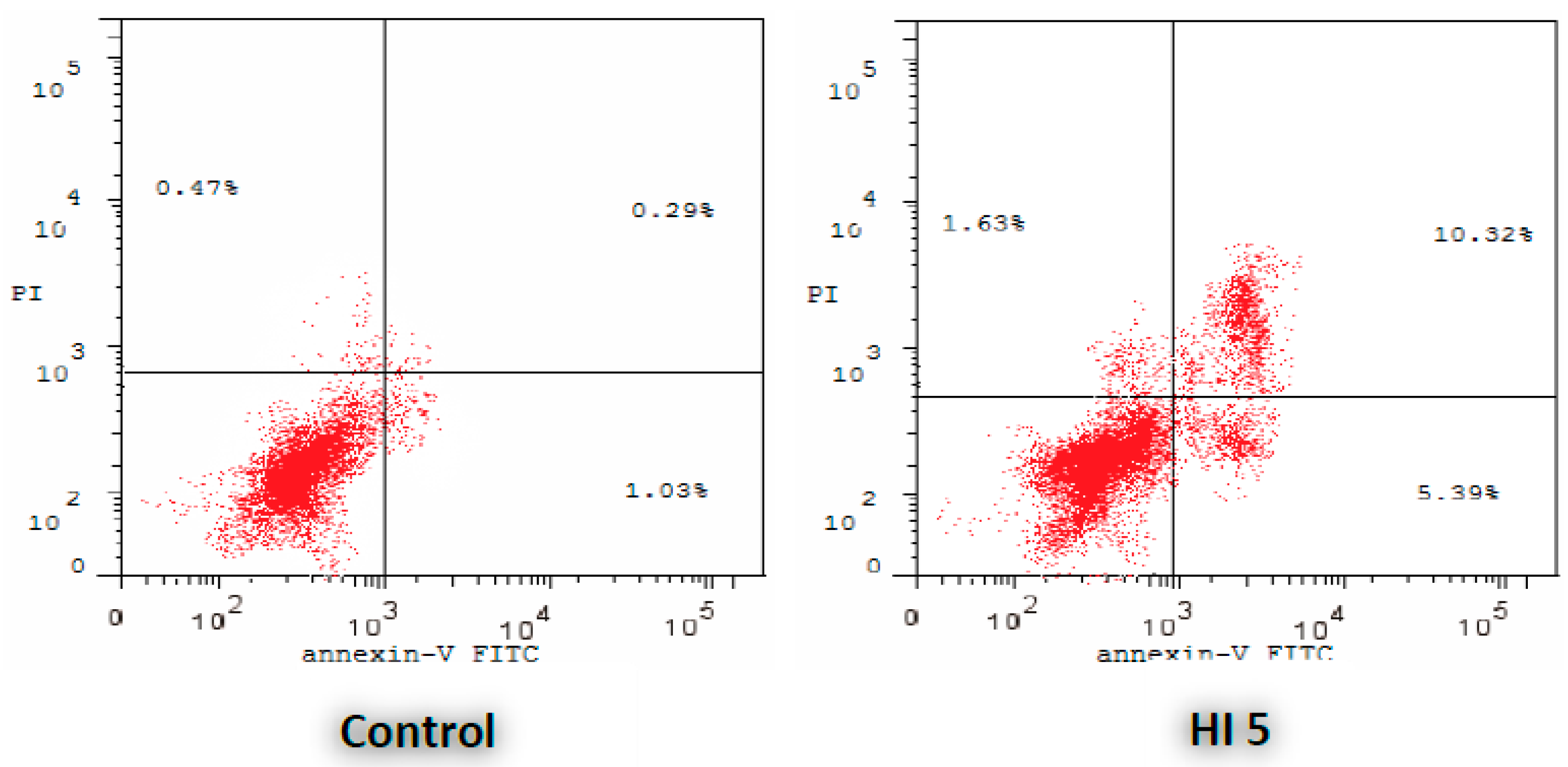
2.2.2. Annexin V-FITC Apoptosis Assay
2.2.3. Antiproliferative Activities toward Breast Cancer Cell Lines
2.2.4. The Effect on the Apoptotic and Anti-Apoptotic Marker Levels
2.2.5. Cell Cycle Assay
2.3. In Silico Studies
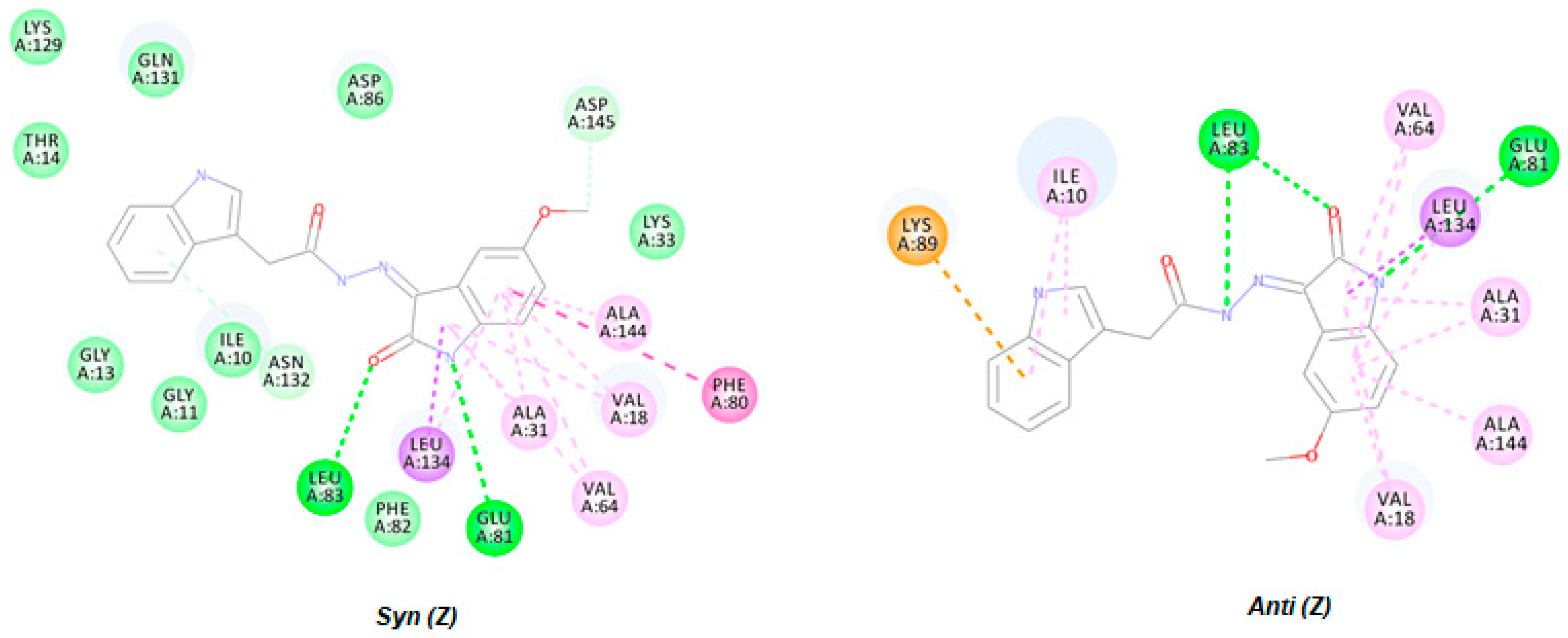
2.3.1. Virtual Docking of HI 5
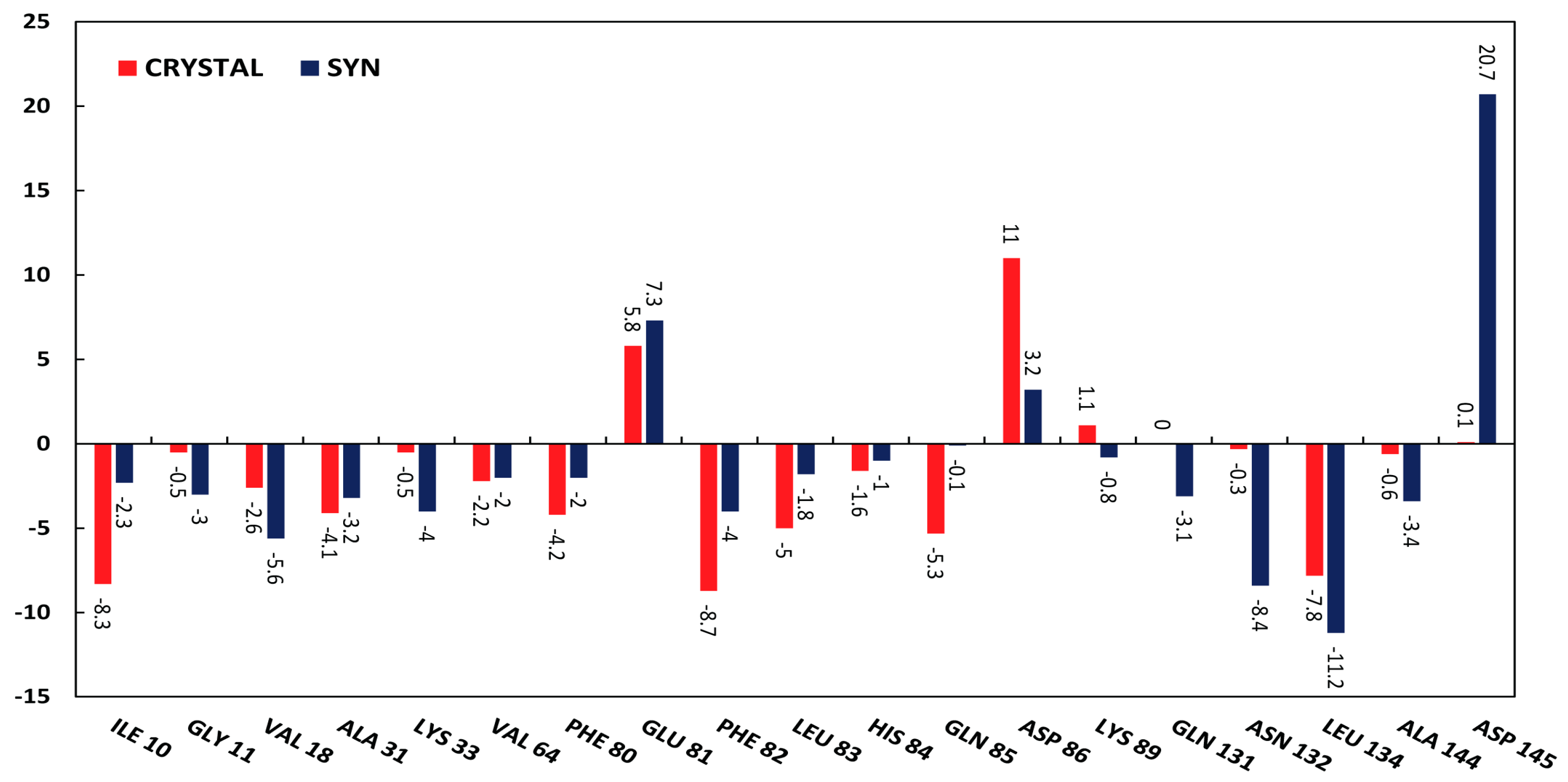
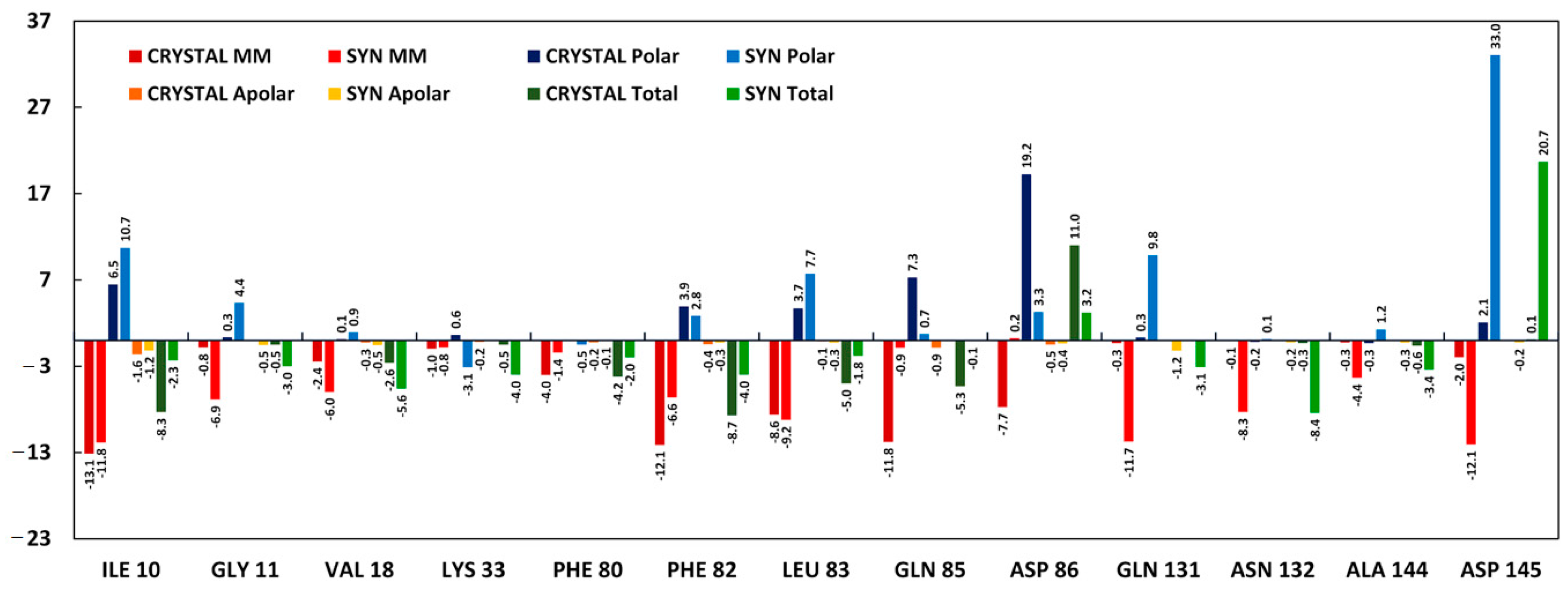
2.3.2. Molecular Dynamics Simulations
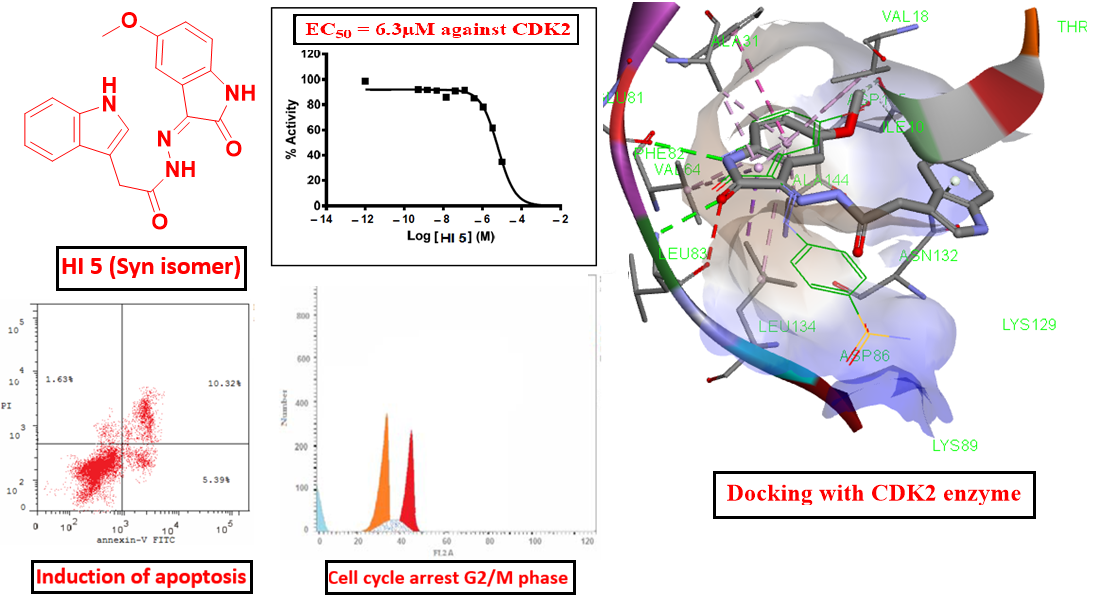
3. Discussion
3.1. Chemistry
3.2. Biological Screening
3.2.1. CDK2 Inhibitory Activity
3.2.2. Annexin V-FITC Apoptosis Assay
3.2.3. Antiproliferative Activities toward Breast Cancer Cell Lines
3.2.4. The Effect on the Apoptotic and Anti-Apoptotic Marker Levels
3.2.5. Cell Cycle Assay
3.3. In Silico Studies
3.3.1. Virtual Docking of HI 5
3.3.2. Molecular Dynamics Simulations
4. Materials and Methods
4.1. Chemistry
4.2. Antiproliferative Activities Toward Breast and Resistant Ovarian Cancer Cell Lines
4.3. Cytotoxicity Assay
4.4. Flow Cytometric Analysis of Apoptosis and Cell Cycle Distribution
4.5. ELISA Immunoassay
4.6. In Vitro CDK2/Cyclin A2 and c-Met Activity
4.7. In Silico Studies
4.7.1. Virtual Docking of HI 5
4.7.2. Molecular DYNAMICS simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Hilton, M.B.; Kaldis, P. p21 Inhibits Cdk1 in the Absence of Cdk2 to Maintain the G1/S Phase DNA Damage Checkpoint. Mol. Biol. Cell 2008, 19, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qian, H.; Pan, Y.; Chen, J.-Z. Cyclin-dependent kinase-2 as a target for cancer therapy: Progress in the development of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2015, 22, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Carroll, J.S.; Prall, O.W.; Musgrove, E.A.; Sutherland, R.L. A pure estrogen antagonist inhibits cyclin e-cdk2 activity in mcf-7 breast cancer cells and induces accumulation of p130-e2f4 complexes characteristic of quiescence. J. Biol. Chem. 2000, 275, 38221–38229. [Google Scholar] [CrossRef]
- Teixeira, C.; Pratt, M.A.C. Cdk2 is a target for retinoic acid-mediated growth inhibition in mcf-7 human breast cancer cells. Mol. Endocrinol. 1997, 11, 1191–1202. [Google Scholar] [CrossRef]
- Chohan, T.A.; Qayyum, A.; Rehman, K.; Tariq, M.; Akash, M.S.H. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed. Pharm. 2018, 107, 1326–1341. [Google Scholar] [CrossRef]
- García-Reyes, B.; Kretz, A.L.; Ruff, J.P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The emerging role of cyclin-dependent kinases (cdks) in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, R.; Zhang, J.; Zhang, J.; Lin, Z. Cyclin-dependent kinase 5 acts as a promising biomarker in clear cell renal cell carcinoma. BMC Cancer 2019, 19, 698. [Google Scholar] [CrossRef]
- Wiernik, P.H. Alvocidib (flavopiridol) for the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2016, 25, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Marwaha, R.K.; Marwaha, M.G.; Jyoti, J.; Nandal, R.; Sharma, A.K. Flavopiridol as cyclin dependent kinase (cdk) inhibitor: A review. New J. Chem. 2018, 42, 18500–18507. [Google Scholar] [CrossRef]
- Criscitiello, C.; Viale, G.; Esposito, A.; Curigliano, G. Dinaciclib for the treatment of breast cancer. Expert Opin. Investig. Drugs 2014, 23, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.L.; Yao, S.L.; Small, K.; et al. Randomized phase ii trial of the cyclin-dependent kinase inhibitor dinaciclib (mk-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Cuellar, R.A.; Berndt, N.; Lee, H.E.; Olesen, S.H.; Martin, M.P.; Jensen, J.T.; Georg, G.I.; Schonbrunn, E. Structural basis of wee kinases functionality and inactivation by diverse small molecule inhibitors. J. Med. Chem. 2017, 60, 7863–7875. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Shailubhai, K.; Bahleda, R.; Gazzah, A.; Varga, A.; Hollebecque, A.; Massard, C.; Spreafico, A.; Reni, M.; Soria, J.C. Phase i dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1257–1265. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Goodyear, S.; Sharma, M.C. Roscovitine regulates invasive breast cancer cell (mda-mb231) proliferation and survival through cell cycle regulatory protein cdk5. Exp. Mol. Pathol. 2007, 82, 25–32. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef]
- Moshinsky, D.J.; Bellamacina, C.R.; Boisvert, D.C.; Huang, P.; Hui, T.; Jancarik, J.; Kim, S.H.; Rice, A.G. Su9516: Biochemical analysis of cdk inhibition and crystal structure in complex with cdk2. Biochem. Biophys. Res. Commun. 2003, 310, 1026–1031. [Google Scholar] [CrossRef]
- Meschini, E. Purine-Based Dual Inhibitors of CDK2 and CDK7. Ph.D. Thesis, Newcastle University, Newcastle upon Tyne, UK, 2011. [Google Scholar]
- Anscombe, E.; Meschini, E.; Mora-Vidal, R.; Martin, M.P.; Staunton, D.; Geitmann, M.; Danielson, U.H.; Stanley, W.A.; Wang, L.Z.; Reuillon, T.; et al. Identification and characterization of an irreversible inhibitor of cdk2. Chem. Biol. 2015, 22, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Abdelazem, A.Z.; Park, B.S.; Yoo, K.H.; Sim, T.; Kwon, Y.J.; Lee, S.H. Ros1 kinase inhibitors for molecular-targeted therapies. Curr. Med. Chem. 2016, 23, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Bramson, H.N.; Corona, J.; Davis, S.T.; Dickerson, S.H.; Edelstein, M.; Frye, S.V.; Gampe, R.T., Jr.; Harris, P.A.; Hassell, A.; Holmes, W.D.; et al. Oxindole-based inhibitors of cyclin-dependent kinase 2 (cdk2): Design, synthesis, enzymatic activities, and x-ray crystallographic analysis. J. Med. Chem. 2001, 44, 4339–4358. [Google Scholar] [CrossRef] [PubMed]
- Garcia, H.H.; Brar, G.A.; Nguyen, D.H.; Bjeldanes, L.F.; Firestone, G.L. Indole-3-carbinol (i3c) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin e forms, and subcellular localization of the cdk2 protein complex. J. Biol. Chem. 2005, 280, 8756–8764. [Google Scholar] [CrossRef]
- Al-Sanea, M.M. Synthesis and biological evaluation of small molecule modulators of cdk8/cyclin c complex with phenylaminoquinoline scaffold. PeerJ 2020, 8, e8649. [Google Scholar] [CrossRef]
- Hsu, J.C.; Dev, A.; Wing, A.; Brew, C.T.; Bjeldanes, L.F.; Firestone, G.L. Indole-3-carbinol mediated cell cycle arrest of lncap human prostate cancer cells requires the induced production of activated p53 tumor suppressor protein. Biochem. Pharmacol. 2006, 72, 1714–1723. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Zakaria, A.; Park, B.S.; Kwon, Y.; Lee, S.H.; Lee, S.W.; Kim, I.T. Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules 2015, 20, 1031. [Google Scholar] [CrossRef]
- Alsayed, S.S.; Elshemy, H.A.; Abdelgawad, M.A.; Abdel-Latif, M.S.; Abdellatif, K.R. Design, synthesis and biological screening of some novel celecoxib and etoricoxib analogs with promising cox-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2017, 70, 173–183. [Google Scholar] [CrossRef]
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.S.; Abdel-Aziz, H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280. [Google Scholar] [CrossRef]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Ranson, M.; Skropeta, D. Cytotoxic and anticancer activities of isatin and its derivatives: A comprehensive review from 2000-2008. Anticancer Agents Med. Chem. 2009, 9, 397–414. [Google Scholar] [CrossRef]
- Narang, R.; Narasimhan, B.; Sharma, S. A review on biological activities and chemical synthesis of hydrazide derivatives. Curr. Med. Chem. 2012, 19, 569–612. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; El Kerdawy, A.M.; Aljaeed, N.; Ismael, O.E.; Ayyad, R.R.; Eldehna, W.M.; Abdel-Aziz, H.A.; Al-Ansary, G.H. Synthesis, biological evaluation and in silico studies of certain oxindole-indole conjugates as anticancer cdk inhibitors. Molecules 2020, 25, 2031. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Park, B.S.; Abdelazem, A.Z.; Selim, K.B.; Yoo, K.H.; Sim, T.; Tae, J.S.; Lee, S.H. Optimization of bipyridinyl pyrazole scaffolds via design, synthesis and screening of a new series of ros1 kinase-modulating compounds. Bull. Korean Chem. Soc. 2015, 36, 305–311. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Bua, S.; Lee, S.H.; Abdelgawad, M.A.; Roh, E.J.; Eldehna, W.M.; Supuran, C.T. Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino)benzenesulfonamides as carbonic anhydrase isoforms i and ii inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Abdelazem, A.Z.; Al-Sanea, M.M.; Park, H.M.; Lee, S.H. Synthesis of new diarylamides with pyrimidinyl pyridine scaffold and evaluation of their anti-proliferative effect on cancer cell lines. Bioorganic Med. Chem. Lett. 2016, 26, 1301–1304. [Google Scholar] [CrossRef]
- Ahmad, M.; Siddiqui, H.L.; Zia-ur-Rehman, M.; Parvez, M. Anti-oxidant and anti-bacterial activities of novel n’-arylmethylidene-2-(3, 4-dimethyl-5, 5-dioxidopyrazolo[4,3-c][1,2]benzothiazin-2(4h)-yl) acetohydrazides. Eur. J. Med. Chem. 2010, 45, 698–704. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; El-Gendy, A.O.; Kamel, G.M.; Azouz, A.A.; Bukhari, S.N.A. Discovery of a cox-2 selective inhibitor hit with anti-inflammatory activity and gastric ulcer protective effect. Future Med. Chem. 2017, 9, 1899–1912. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Belal, A.; Omar, H.A.; Hegazy, L.; Rateb, M.E. Synthesis, anti-breast cancer activity, and molecular modeling of some benzothiazole and benzoxazole derivatives. Arch. Pharm. 2013, 346, 534–541. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Labib, M.B.; Ali, W.A.M.; Kamel, G.; Azouz, A.A.; El-Nahass, E.S. Design, synthesis, analgesic, anti-inflammatory activity of novel pyrazolones possessing aminosulfonyl pharmacophore as inhibitors of cox-2/5-lox enzymes: Histopathological and docking studies. Bioorganic Chem. 2018, 78, 103–114. [Google Scholar] [CrossRef]
- Ali, G.M.E.; Ibrahim, D.A.; Elmetwali, A.M.; Ismail, N.S.M. Design, synthesis and biological evaluation of certain cdk2 inhibitors based on pyrazole and pyrazolo[1,5-a] pyrimidine scaffold with apoptotic activity. Bioorganic Chem. 2019, 86, 1–14. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Abdelgawad, M.A.; Elshemy, H.A.; Alsayed, S.S.; Kamel, G. Synthesis and anti-inflammatory evaluation of new 1,3,5-triaryl-4,5-dihydro-1h-pyrazole derivatives possessing an aminosulphonyl pharmacophore. Arch. Pharm. Res. 2015, 38, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine b (srb) assay in cell culture to investigate cell proliferation. Bio-Protocol 2016, 6, e1984. [Google Scholar] [CrossRef]
- Sabt, A.; Abdelhafez, O.M.; El-Haggar, R.S.; Madkour, H.M.F.; Eldehna, W.M.; El-Khrisy, E.E.A.M.; Abdel-Rahman, M.A.; Rashed, L.A. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and qsar studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 1095–1107. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K. Charmm-gui pdb manipulator for advanced modeling and simulations of proteins containing nonstandard residues. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 96, pp. 235–265. [Google Scholar]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa—A gromacs tool for high-throughput mm-pbsa calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Lundborg, M.; Lindahl, E. Automatic gromacs topology generation and comparisons of force fields for solvation free energy calculations. J. Phys. Chem. B 2015, 119, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. Acpype—Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the tip3p, spc, and spc/e water models at 298 k. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Enzyme Inhibitory Activity | |
| CDK2/A2 | c-Met | |
| HI 5 | 49 | 27 |
| Staurosporine | 100 | 100 |
| Comp. | Early Apoptosis (Lower Right (LR) %) | Late Apoptosis (Upper Right (UR) %) | Total (LR % + UR %) |
|---|---|---|---|
| HI 5 | 5.39 | 10.32 | 15.71 |
| Control | 1.03 | 0.29 | 1.32 |
| Comp. | IC50 (µM) a | ||
|---|---|---|---|
| MCF-7 | MDA-MB-231 | NCI-ADR | |
| HI 5 | 1.15 ± 0.04 | 10.54 ± 0.43 | 9.17 |
| Doxorubicin | 6.81 ± 0.22 | 10.29 ± 0.72 | ND b |
| Compound | Bax (pg/mg of Total Protein) | Bcl-2 (ng/mg of Total Protein) | Bax/Bcl-2 | Caspase-3 (pg/mg) | p53 (pg/mg) |
|---|---|---|---|---|---|
| HI 5 | 297.4 ± 11.1 | 2.56 ± 0.20 | 116.2 ± 0.8 | 387.5 ± 13.7 | 673.2 ± 41.0 |
| Control | 38.3 ± 2.2 | 4.65 ± 0.23 | 8.2 ± 0.7 | 35.92 ± 1.8 | 41.26 ± 2.7 |
| Comp. | %G0-G1 | %S | %G2-M | %Sub-G1 |
|---|---|---|---|---|
| HI 5 | 41.54 ± 0.8 | 22.17 ± 1.0 | 36.29 ± 1.2 | 17.34 ± 0.2 |
| Control | 57.26 ± 0.2 | 28.59 ± 0.5 | 14.15 ± 1.0 | 1.79 ± 0.8 |
| Compound | Van der Waals | Electrostatic | Polar Solvation | SASA | Binding Energy |
|---|---|---|---|---|---|
| Co-crystallized ligand 1 | −178.546 ± 10.295 | −20.344 ± 7.826 | 107.937 ± 11.584 | −15.750 ± 0.720 | −106.703 ± 3.411 |
| HI 5 (syn-isomer) | −186.753 ± 9.303 | −53.024 ± 6.399 | 165.947 ± 8.579 | −17.805 ± 0.788 | −91.636 ± 12.345 |
| HI 5 (anti-isomer) | −102.060 ± 25.028 | −28.865 ± 8.671 | 82.992 ± 56.836 | −10.440 ± 2.166 | −58.374 ± 47.381 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Sanea, M.M.; Obaidullah, A.J.; Shaker, M.E.; Chilingaryan, G.; Alanazi, M.M.; Alsaif, N.A.; Alkahtani, H.M.; Alsubaie, S.A.; Abdelgawad, M.A. A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights. Molecules 2021, 26, 412. https://doi.org/10.3390/molecules26020412
Al-Sanea MM, Obaidullah AJ, Shaker ME, Chilingaryan G, Alanazi MM, Alsaif NA, Alkahtani HM, Alsubaie SA, Abdelgawad MA. A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights. Molecules. 2021; 26(2):412. https://doi.org/10.3390/molecules26020412
Chicago/Turabian StyleAl-Sanea, Mohammad M., Ahmad J. Obaidullah, Mohamed E. Shaker, Garri Chilingaryan, Mohammed M. Alanazi, Nawaf A. Alsaif, Hamad M. Alkahtani, Sultan A. Alsubaie, and Mohamed A. Abdelgawad. 2021. "A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights" Molecules 26, no. 2: 412. https://doi.org/10.3390/molecules26020412
APA StyleAl-Sanea, M. M., Obaidullah, A. J., Shaker, M. E., Chilingaryan, G., Alanazi, M. M., Alsaif, N. A., Alkahtani, H. M., Alsubaie, S. A., & Abdelgawad, M. A. (2021). A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights. Molecules, 26(2), 412. https://doi.org/10.3390/molecules26020412