
Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Familial AD and Genetics
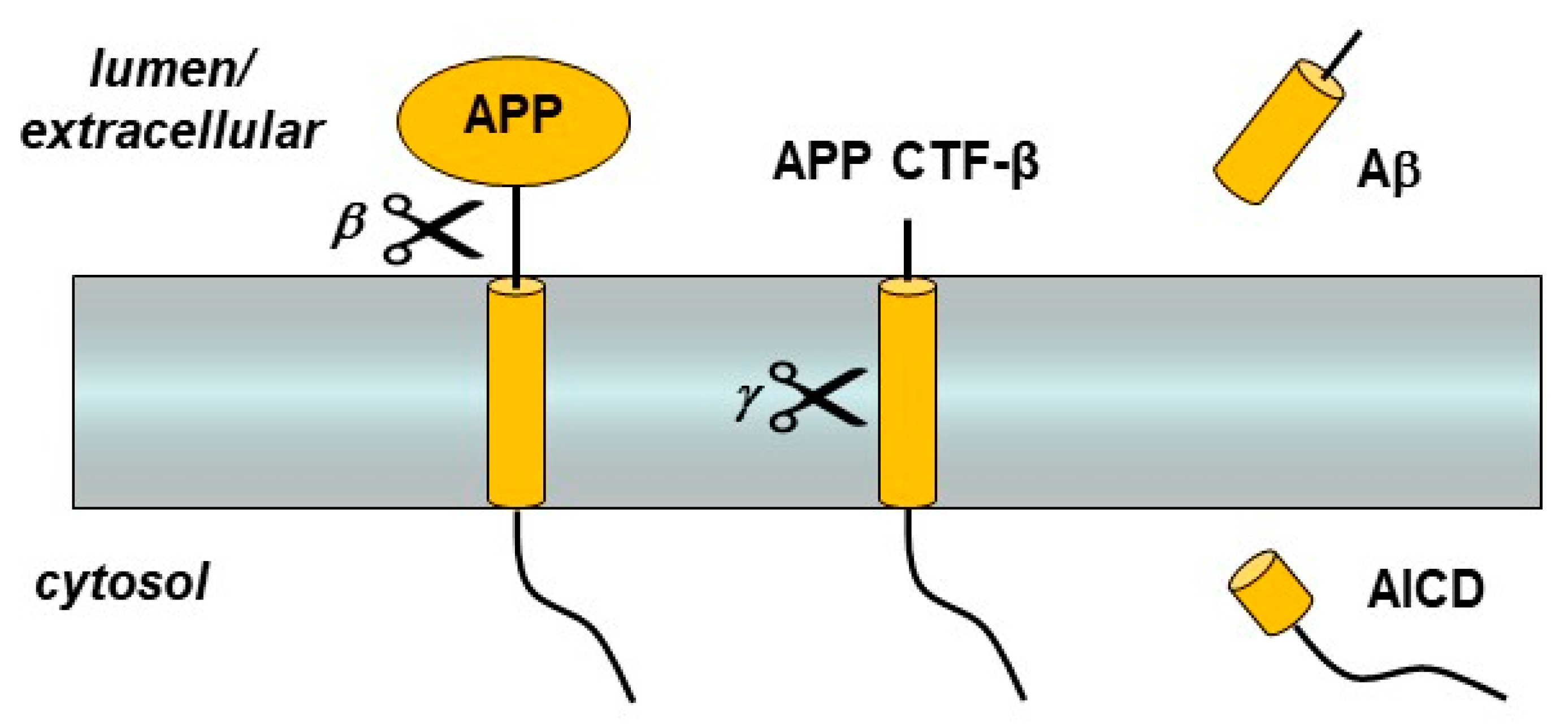
3. Presenilin and the γ-Secretase Complex
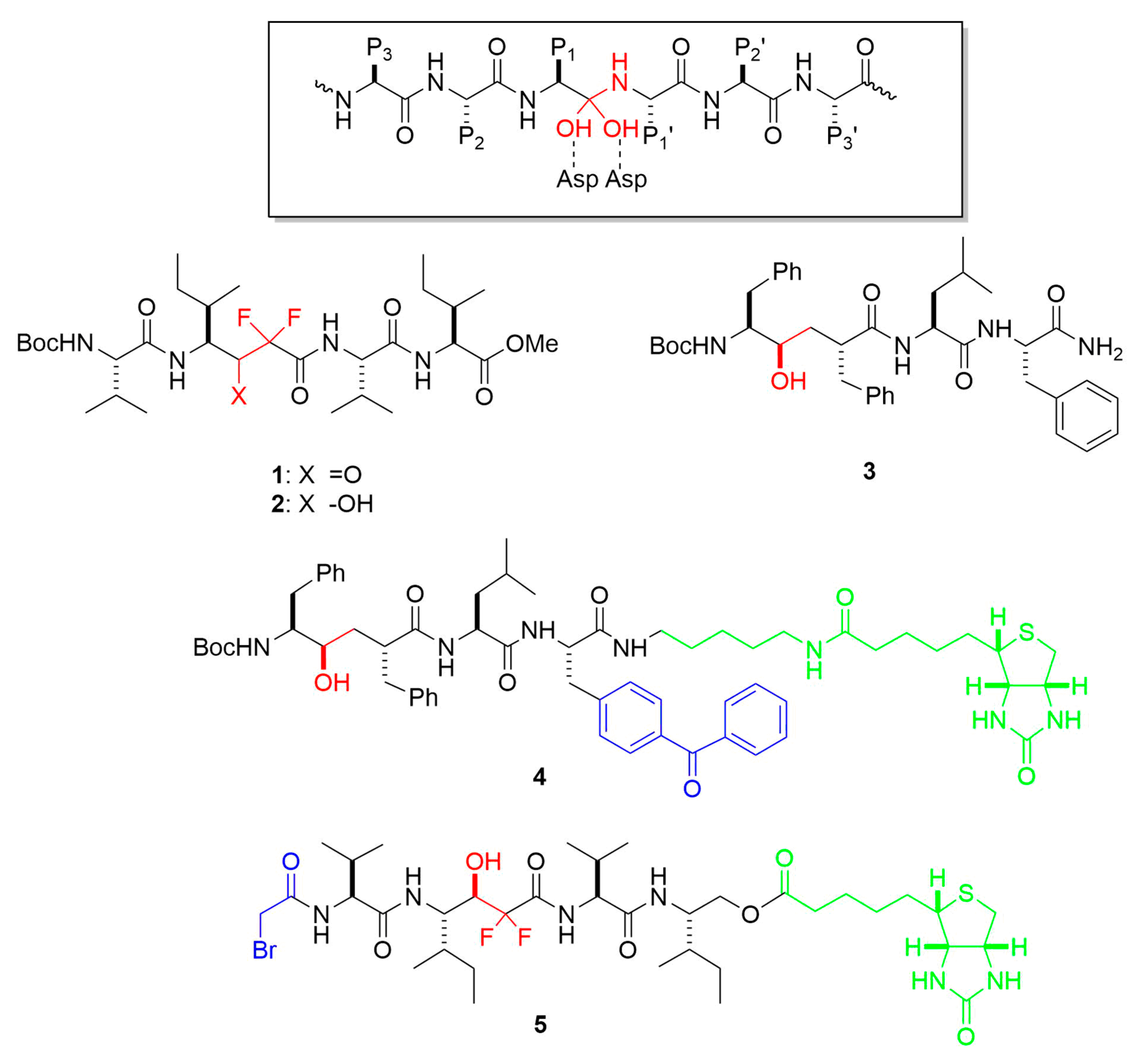
4. Chemical Probes
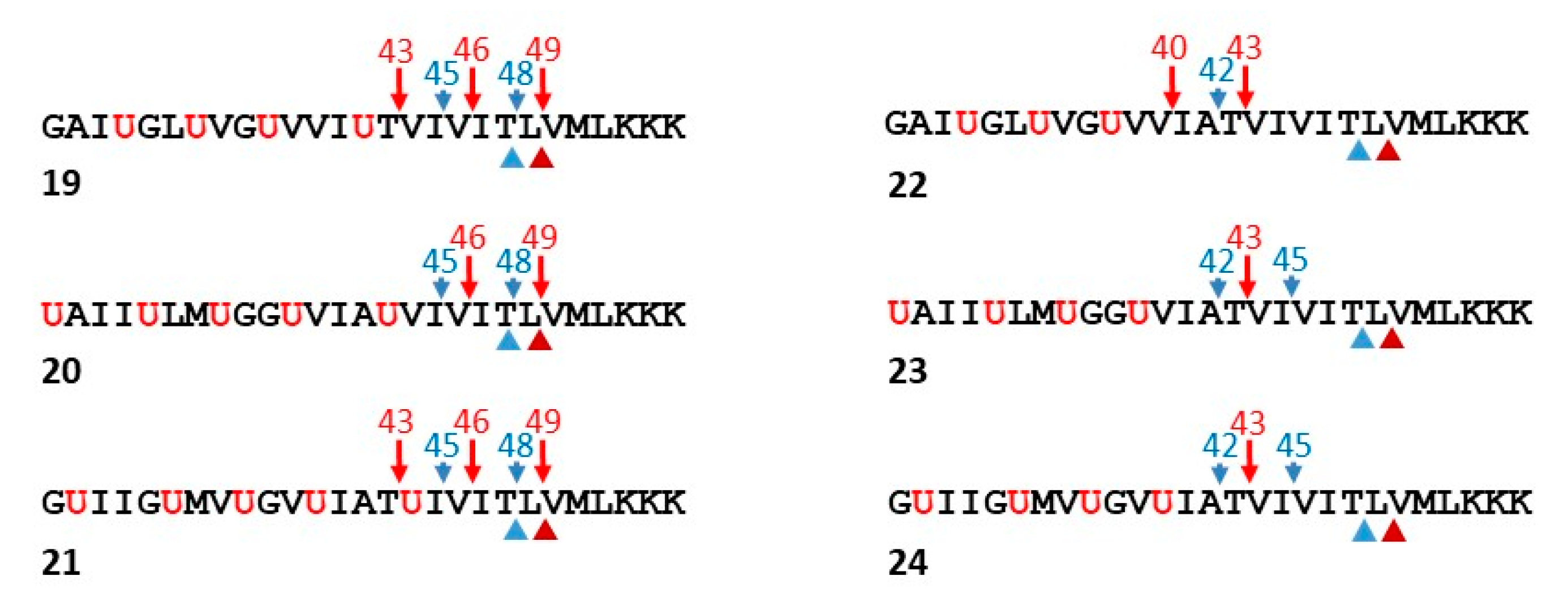
5. Structural Probes
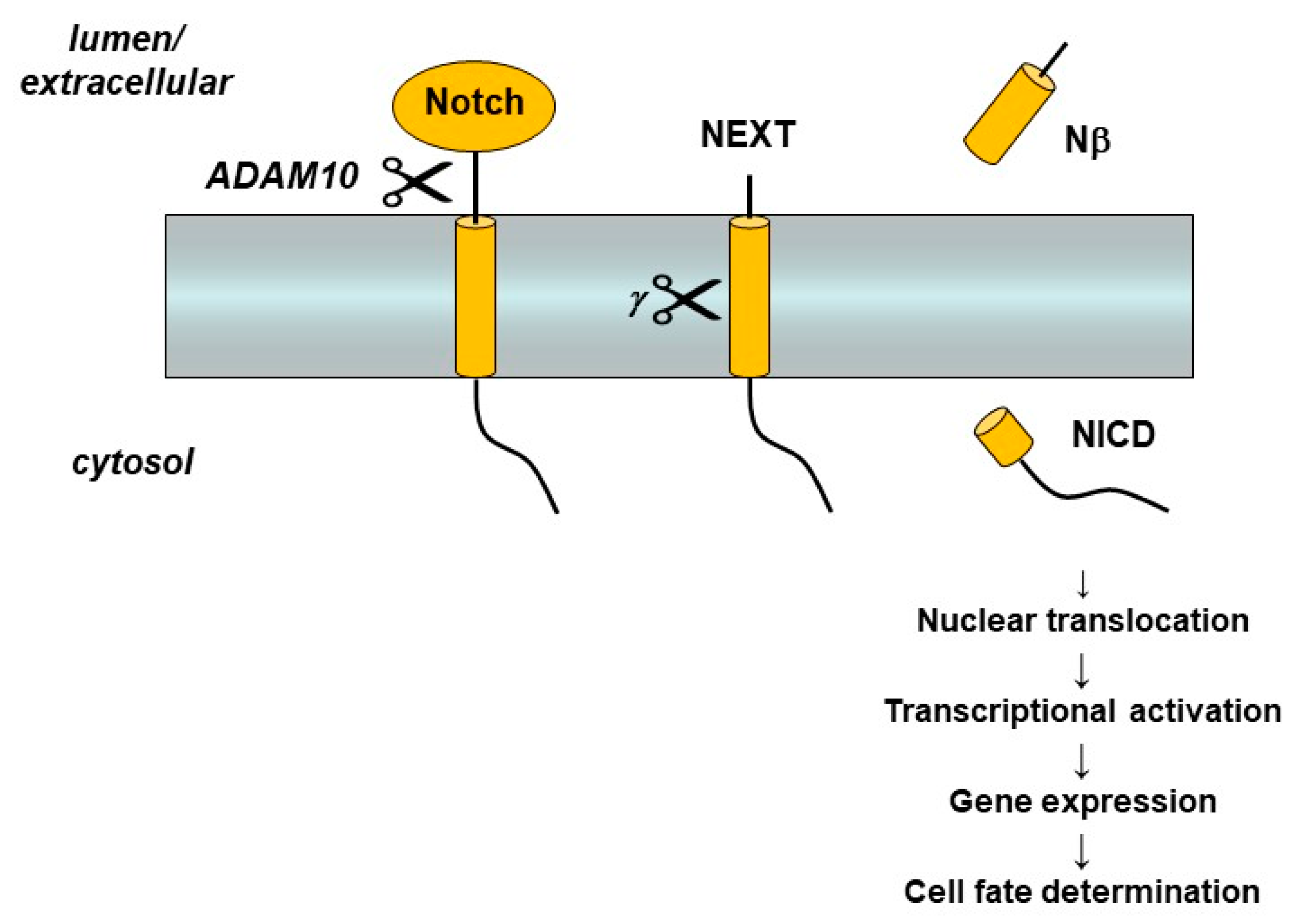
6. Functional Probes
7. γ-Secretase Inhibitors: Therapeutic Potential
8. γ-Secretase Modulators: Therapeutic Potential
9. Summary and Perspective
Funding
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Eng. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Ann. Rev. Pathol. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Marttinen, M.; Takalo, M.; Natunen, T.; Wittrahm, R.; Gabbouj, S.; Kemppainen, S.; Leinonen, V.; Tanila, H.; Haapasalo, A.; Hiltunen, M. Molecular Mechanisms of Synaptotoxicity and Neuroinflammation in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 963. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 1991, 353, 844–846. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The role of APP processing by BACE1, the β-secretase, in Alzheimer’s disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef]
- Wolfe, M.S. γ-Secretase in biology and medicine. Sem. Cell Dev. Biol. 2009, 20, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Available online: http://www.alzforum.org/mutations (accessed on 12 January 2021).
- Selkoe, D.J. Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu. Rev. Cell Biol. 1994, 10, 373–403. [Google Scholar] [CrossRef] [PubMed]
- Laudon, H.; Hansson, E.M.; Melen, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; von Heijne, G.; Naslund, J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef]
- Spasic, D.; Tolia, A.; Dillen, K.; Baert, V.; De Strooper, B.; Vrijens, S.; Annaert, W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 2006, 281, 26569–26577. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Herreman, A.; Serneels, L.; Annaert, W.; Collen, D.; Schoonjans, L.; De Strooper, B. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2000, 2, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nadeau, P.; Song, W.; Donoviel, D.; Yuan, M.; Bernstein, A.; Yankner, B.A. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat. Cell. Biol. 2000, 2, 463–465. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Citron, M.; Diehl, T.S.; Xia, W.; Donkor, I.O.; Selkoe, D.J. A substrate-based difluoro ketone selectively inhibits Alzheimer’s γ-secretase activity. J. Med. Chem. 1998, 41, 6–9. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Moore, C.L.; Leatherwood, D.D.; Ostaszewski, B.; Donkor, I.O.; Selkoe, D.J. Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 1999, 38, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Shearman, M.S.; Beher, D.; Clarke, E.E.; Lewis, H.D.; Harrison, T.; Hunt, P.; Nadin, A.; Smith, A.L.; Stevenson, G.; Castro, J.L. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 2000, 39, 8698–8704. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Esler, W.P.; Kimberly, W.T.; Ostaszewski, B.L.; Diehl, T.S.; Moore, C.L.; Tsai, J.-Y.; Rahmati, T.; Xia, W.; Selkoe, D.J.; Wolfe, M.S. Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2000, 2, 428–434. [Google Scholar] [CrossRef]
- Li, Y.M.; Xu, M.; Lai, M.T.; Huang, Q.; Castro, J.L.; DiMuzio-Mower, J.; Harrison, T.; Lellis, C.; Nadin, A.; Neduvelil, J.G.; et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405, 689–694. [Google Scholar] [CrossRef]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Thinakaran, G.; Harris, C.L.; Ratovitski, T.; Davenport, F.; Slunt, H.H.; Price, D.L.; Borchelt, D.R.; Sisodia, S.S. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem. 1997, 272, 28415–28422. [Google Scholar] [CrossRef]
- Capell, A.; Grunberg, J.; Pesold, B.; Diehlmann, A.; Citron, M.; Nixon, R.; Beyreuther, K.; Selkoe, D.J.; Haass, C. The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J. Biol. Chem. 1998, 273, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, F.; Levesque, G.; Nishimura, M.; Zhang, D.M.; Levesque, L.; Rogaeva, E.; Xu, D.; Liang, Y.; Duthie, M.; et al. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains β-catenin. J. Biol. Chem. 1998, 273, 16470–16475. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the γ-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of γ-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef]
- Gu, Y.; Misonou, H.; Sato, T.; Dohmae, N.; Takio, K.; Ihara, Y. Distinct intramembrane cleavage of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch. J. Biol. Chem. 2001, 276, 35235–35238. [Google Scholar] [CrossRef]
- Yu, C.; Kim, S.H.; Ikeuchi, T.; Xu, H.; Gasparini, L.; Wang, R.; Sisodia, S.S. Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in γ-secretase processing of the APP and Notch1 transmembrane domains. J. Biol. Chem. 2001, 276, 43756–43760. [Google Scholar] [CrossRef]
- Weidemann, A.; Eggert, S.; Reinhard, F.B.; Vogel, M.; Paliga, K.; Baier, G.; Masters, C.L.; Beyreuther, K.; Evin, G. A novel var epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 2002, 41, 2825–2835. [Google Scholar] [CrossRef]
- Sato, T.; Dohmae, N.; Qi, Y.; Kakuda, N.; Misonou, H.; Mitsumori, R.; Maruyama, H.; Koo, E.H.; Haass, C.; Takio, K.; et al. Potential link between amyloid β-protein 42 and C-terminal fragment gamma 49–99 of β-amyloid precursor protein. J. Biol. Chem. 2003, 278, 24294–24301. [Google Scholar] [CrossRef]
- Sastre, M.; Steiner, H.; Fuchs, K.; Capell, A.; Multhaup, G.; Condron, M.M.; Teplow, D.B.; Haass, C. Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001, 2, 835–841. [Google Scholar] [CrossRef]
- Qi-Takahara, Y.; Morishima-Kawashima, M.; Tanimura, Y.; Dolios, G.; Hirotani, N.; Horikoshi, Y.; Kametani, F.; Maeda, M.; Saido, T.C.; Wang, R.; et al. Longer forms of amyloid β protein: Implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 2005, 25, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Morishima-Kawashima, M.; Ishiura, S.; Ihara, Y. Aβ46 is processed to Aβ40 and Aβ43, but not to Aβ42, in the low density membrane domains. J. Biol. Chem. 2008, 283, 733–738. [Google Scholar] [CrossRef]
- Zhao, G.; Cui, M.Z.; Mao, G.; Dong, Y.; Tan, J.; Sun, L.; Xu, X. γ-Cleavage is dependent on ζ-cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J. Biol. Chem. 2005, 280, 37689–37697. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.A.; Klutkowski, J.A.; Freret, T.; Wolfe, M.S. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J. Biol. Chem. 2014, 289, 31043–31052. [Google Scholar] [CrossRef]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimer Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. γ-Secretase: Proteasome of the membrane? Nat. Rev. Cell Mol. Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef]
- Selkoe, D.; Kopan, R. Notch and Presenilin: Regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 2003, 26, 565–597. [Google Scholar] [CrossRef]
- Wong, P.C.; Zheng, H.; Chen, H.; Becher, M.W.; Sirinathsinghji, D.J.; Trumbauer, M.E.; Chen, H.Y.; Price, D.L.; Van der Ploeg, L.H.; Sisodia, S.S. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 1997, 387, 288–292. [Google Scholar] [CrossRef]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Moore, C.L.; Leatherwood, D.D.; Diehl, T.S.; Selkoe, D.J.; Wolfe, M.S. Difluoro Ketone Peptidomimetics Suggest a Large S1 Pocket for Alzheimer’s γ-Secretase: Implications for Inhibitor Design. J. Med. Chem. 2000, 43, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.; Wolfe, M.S. Stereochemical analysis of (hydroxyethyl)urea peptidomimetic inhibitors of γ-secretase. J. Med. Chem. 2004, 47, 6485–6489. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Das, C.; Wolfe, M.S. Probing pockets S2-S4′ of the gamma-secretase active site with (hydroxyethyl)urea peptidomimetics. Bioorg. Med. Chem. Lett. 2004, 14, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- Güner, G.; Lichtenthaler, S.F. The substrate repertoire of γ-secretase/presenilin. Sem. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-βforming tripeptide cleavage mechanism of γ-secretase. eLife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Esler, W.P.; Kimberly, W.T.; Ostaszewski, B.L.; Ye, W.; Diehl, T.S.; Selkoe, D.J.; Wolfe, M.S. Activity-dependent isolation of the presenilin/γ-secretase complex reveals nicastrin and a γ substrate. Proc. Natl. Acad. Sci. USA 2002, 99, 2720–2725. [Google Scholar] [CrossRef]
- Fraering, P.C.; Ye, W.; Strub, J.M.; Dolios, G.; LaVoie, M.J.; Ostaszewski, B.L.; Van Dorsselaer, A.; Wang, R.; Selkoe, D.J.; Wolfe, M.S. Purification and Characterization of the Human γ-Secretase Complex. Biochemistry 2004, 43, 9774–9789. [Google Scholar] [CrossRef]
- Das, C.; Berezovska, O.; Diehl, T.S.; Genet, C.; Buldyrev, I.; Tsai, J.Y.; Hyman, B.T.; Wolfe, M.S. Designed helical peptides inhibit an intramembrane protease. J. Am. Chem. Soc. 2003, 125, 11794–11795. [Google Scholar] [CrossRef]
- Bihel, F.; Das, C.; Bowman, M.J.; Wolfe, M.S. Discovery of a subnanomolar helical D-tridecapeptide inhibitor of γ-secretase. J. Med. Chem. 2004, 47, 3931–3933. [Google Scholar] [CrossRef]
- Kornilova, A.Y.; Bihel, F.; Das, C.; Wolfe, M.S. The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3230–3235. [Google Scholar] [CrossRef]
- Bhattarai, S.; Devkota, S.; Meneely, K.M.; Xing, M.; Douglas, J.T.; Wolfe, M.S. Design of Substrate Transmembrane Mimetics as Structural Probes for γ-Secretase. J. Am. Chem. Soc. 2020, 142, 3351–3355. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Philip, A.T.; Devkota, S.; Malvankar, S.; Bhattarai, S.; Meneely, K.M.; Williams, T.D.; Wolfe, M.S. Designed Helical Peptides as Functional Probes for γ-Secretase. Biochemistry 2019, 58, 4398–4407. [Google Scholar] [CrossRef]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; de Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 2001, 76, 173–181. [Google Scholar] [CrossRef]
- Barten, D.M.; Guss, V.L.; Corsa, J.A.; Loo, A.T.; Hansel, S.B.; Zheng, M.; Munoz, B.; Srinivasan, K.; Wang, B.; Robertson, B.J.; et al. Dynamics of β-Amyloid Reductions in Brain, Cerebrospinal Fluid and Plasma of β-Amyloid Precursor Protein Transgenic Mice Treated with a γ-Secretase Inhibitor. J. Pharmacol. Exp. Ther. 2004, 27, 27. [Google Scholar] [CrossRef]
- Searfoss, G.H.; Jordan, W.H.; Calligaro, D.O.; Galbreath, E.J.; Schirtzinger, L.M.; Berridge, B.R.; Gao, H.; Higgins, M.A.; May, P.C.; Ryan, T.P. Adipsin: A biomarker of gastrointestinal toxicity mediated by a functional γsecretase inhibitor. J. Biol. Chem. 2003, 278, 46107–46116. [Google Scholar] [CrossRef]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef]
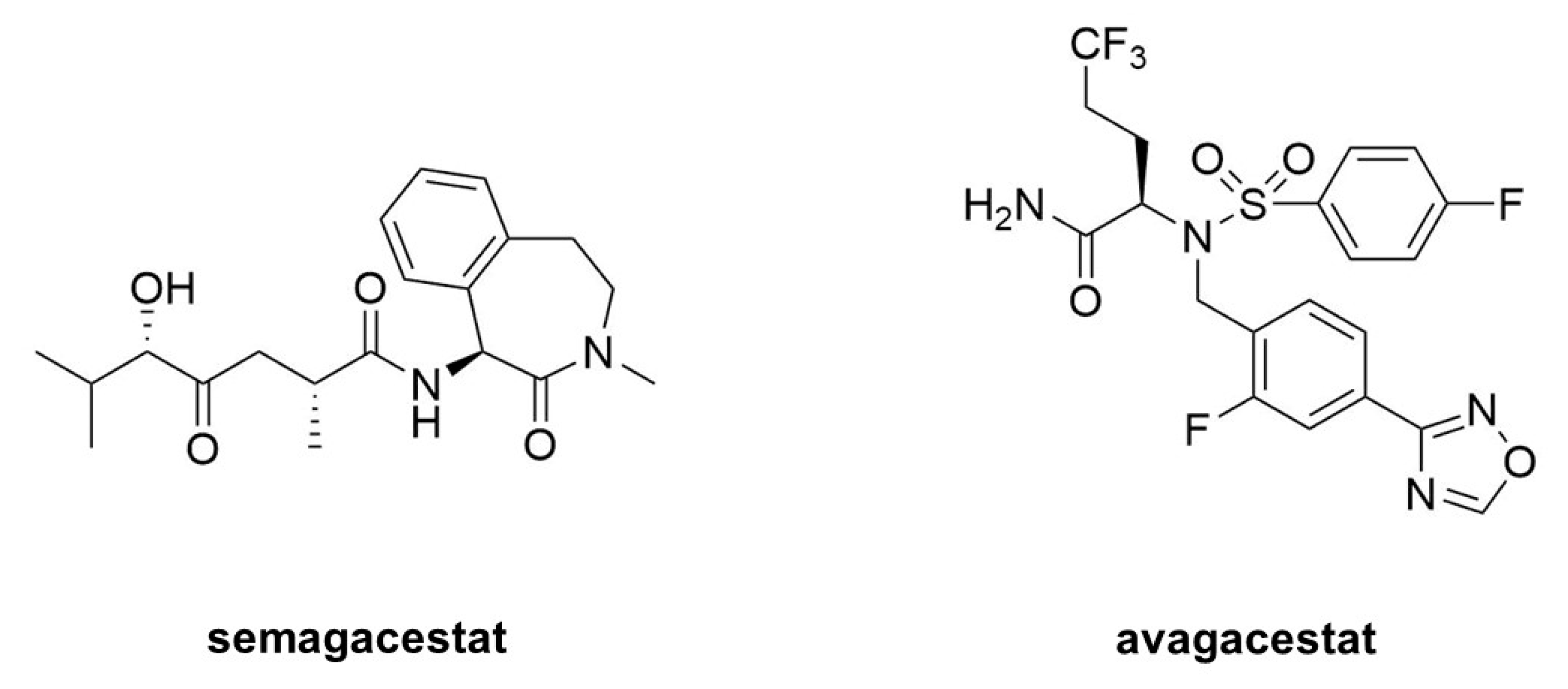
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Eng. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits β-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449. [Google Scholar] [CrossRef] [PubMed]
- Fraering, P.C.; Ye, W.; Lavoie, M.J.; Ostaszewski, B.L.; Selkoe, D.J.; Wolfe, M.S. γ-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide binding site. J. Biol. Chem. 2005, 280, 41987–41996. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.C.; Kreft, A.F.; Harrison, B.; Abou-Gharbia, M.; Antane, M.; Aschmies, S.; Atchison, K.; Chlenov, M.; Cole, D.C.; Comery, T.; et al. Discovery of begacestat, a Notch-1-sparing γ-secretase inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 2008, 51, 7348–7351. [Google Scholar] [CrossRef]
- Gillman, K.W.; Starrett, J.E.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable γ-Secretase Inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef]
- Crump, C.J.; Castro, S.V.; Wang, F.; Pozdnyakov, N.; Ballard, T.E.; Sisodia, S.S.; Bales, K.R.; Johnson, D.S.; Li, Y.M. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 2012, 51, 7209–7211. [Google Scholar] [CrossRef]
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential Effects between γ-Secretase Inhibitors and Modulators on Cognitive Function in Amyloid Precursor Protein-Transgenic and Nontransgenic Mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef]
- Lanz, T.A.; Karmilowicz, M.J.; Wood, K.M.; Pozdnyakov, N.; Du, P.; Piotrowski, M.A.; Brown, T.M.; Nolan, C.E.; Richter, K.E.; Finley, J.E.; et al. Concentration-dependent modulation of amyloid-β in vivo and in vitro using the γ-secretase inhibitor, LY-450139. J. Pharmacol. Exp. Ther. 2006, 319, 924–933. [Google Scholar] [CrossRef]
- Barnwell, E.; Padmaraju, V.; Baranello, R.; Pacheco-Quinto, J.; Crosson, C.; Ablonczy, Z.; Eckman, E.; Eckman, C.B.; Ramakrishnan, V.; Greig, N.H.; et al. Evidence of a Novel Mechanism for Partial γ-Secretase Inhibition Induced Paradoxical Increase in Secreted Amyloid βProtein. PLoS ONE 2014, 9, e91531. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef]
- Bursavich, M.G.; Harrison, B.A.; Blain, J.F. γ-Secretase Modulators: New Alzheimer’s Drugs on the Horizon? J. Med. Chem. 2016, 59, 7389–7409. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Tagami, S.; Yanagida, K.; Takami, M.; Kodama, T.S.; Mori, K.; Nakayama, T.; Ihara, Y.; Takeda, M. γ-Secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013, 3, 42–51. [Google Scholar] [CrossRef]
- Golde, T.E.; Schneider, L.S.; Koo, E.H. Anti-Aβ therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 2011, 69, 203–213. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Eng. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Comm. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the βamyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfe, M.S. Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease. Molecules 2021, 26, 388. https://doi.org/10.3390/molecules26020388
Wolfe MS. Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease. Molecules. 2021; 26(2):388. https://doi.org/10.3390/molecules26020388
Chicago/Turabian StyleWolfe, Michael S. 2021. "Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease" Molecules 26, no. 2: 388. https://doi.org/10.3390/molecules26020388
APA StyleWolfe, M. S. (2021). Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease. Molecules, 26(2), 388. https://doi.org/10.3390/molecules26020388