
Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Sulfonamides


Abstract
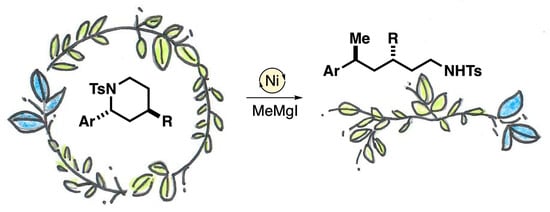
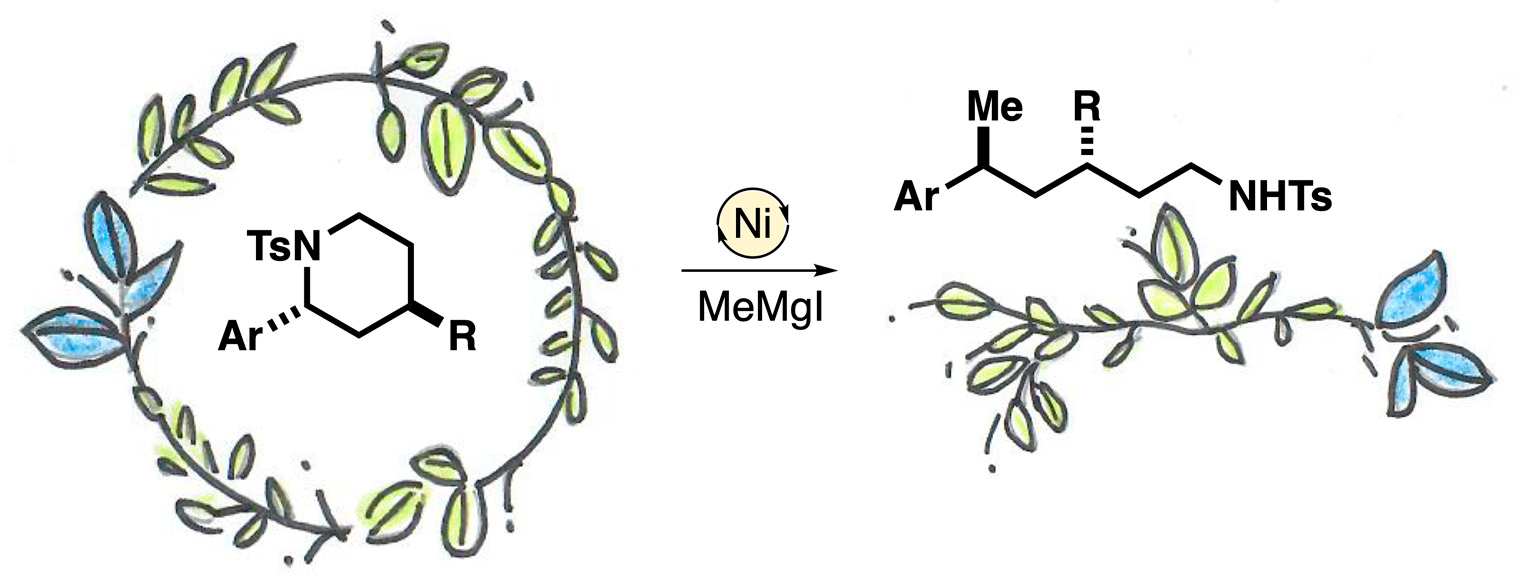
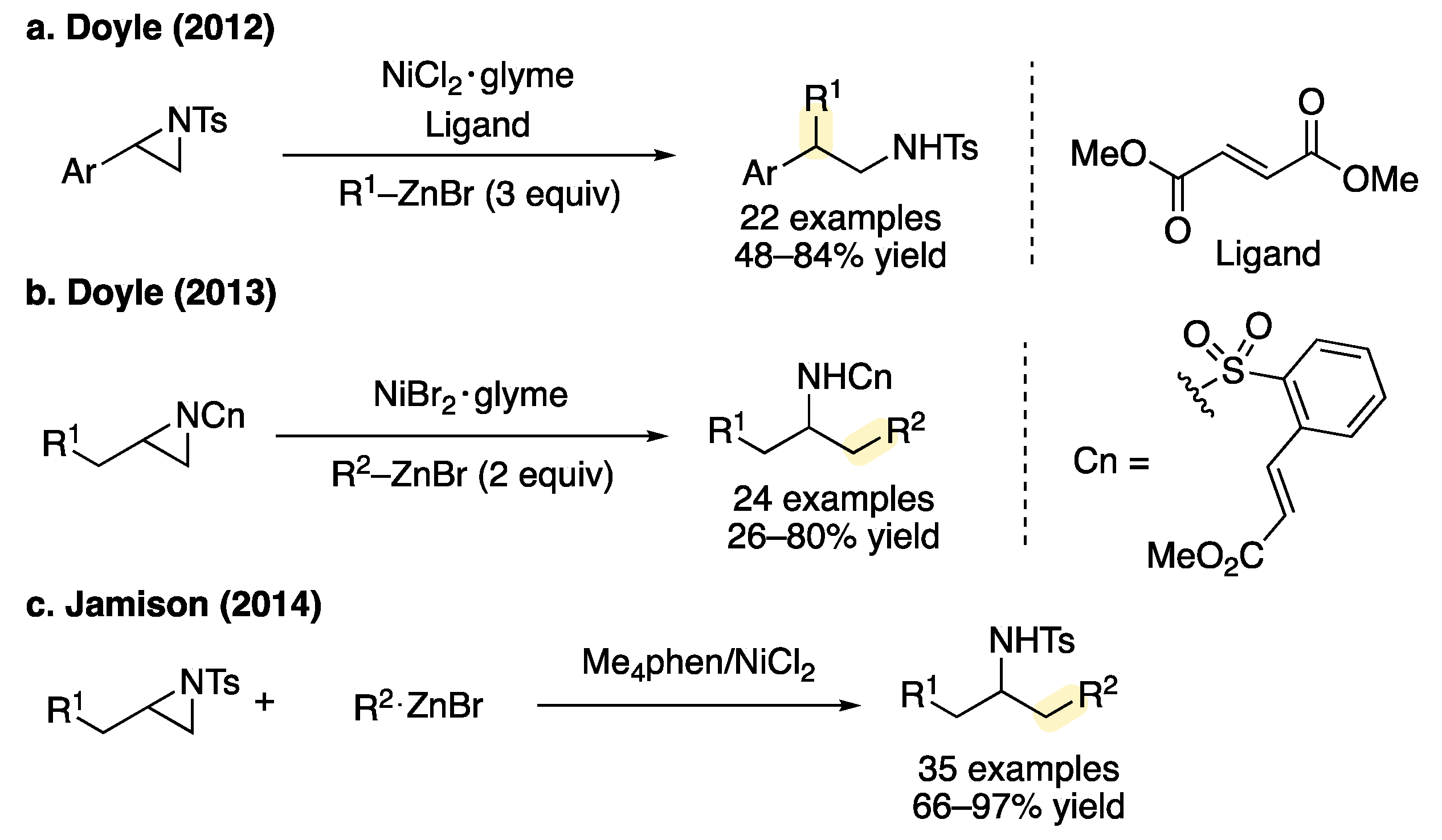
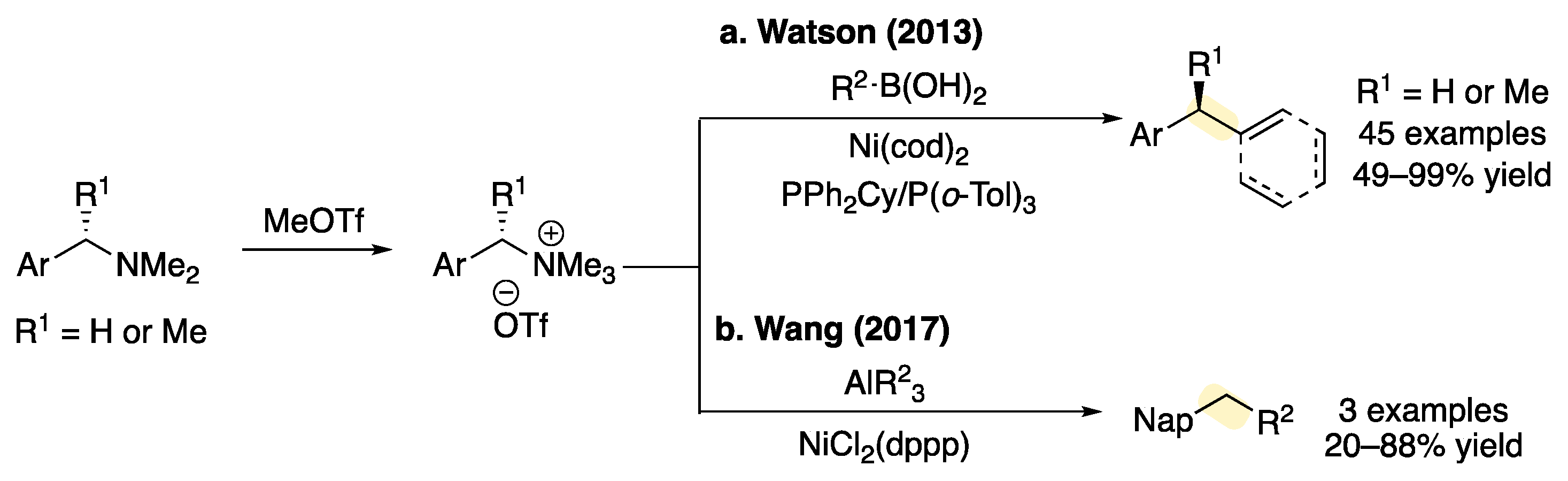
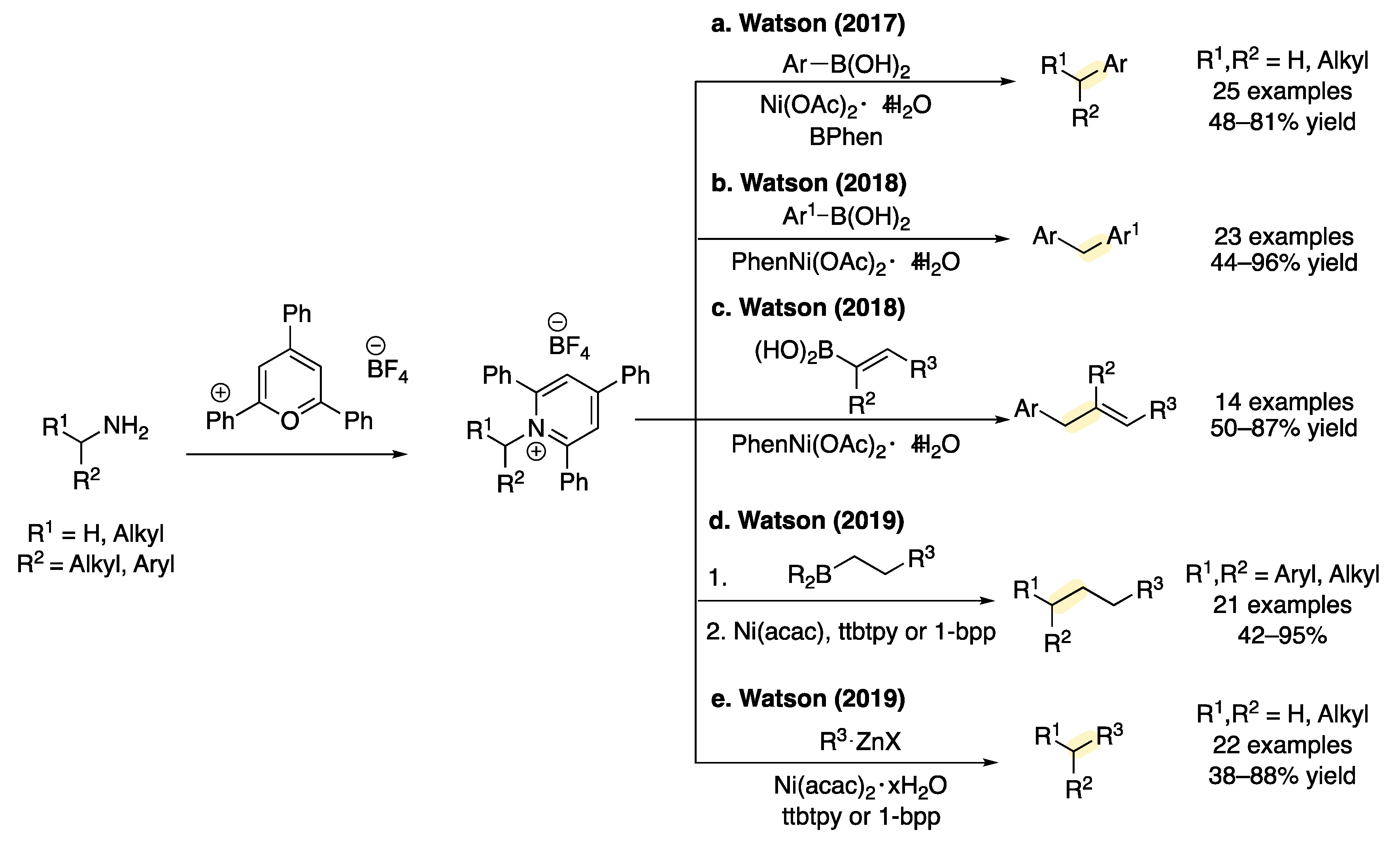
1. Introduction
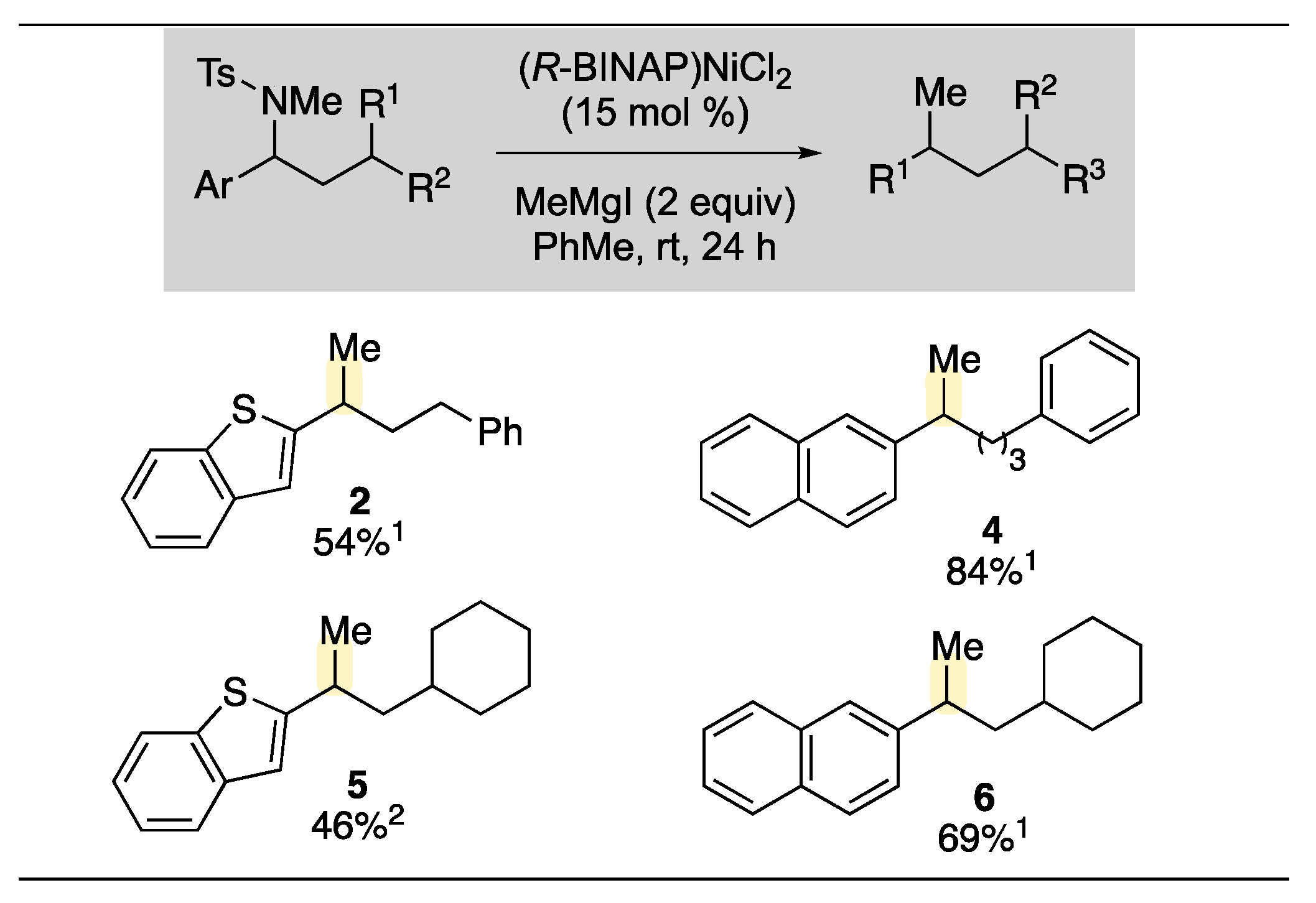
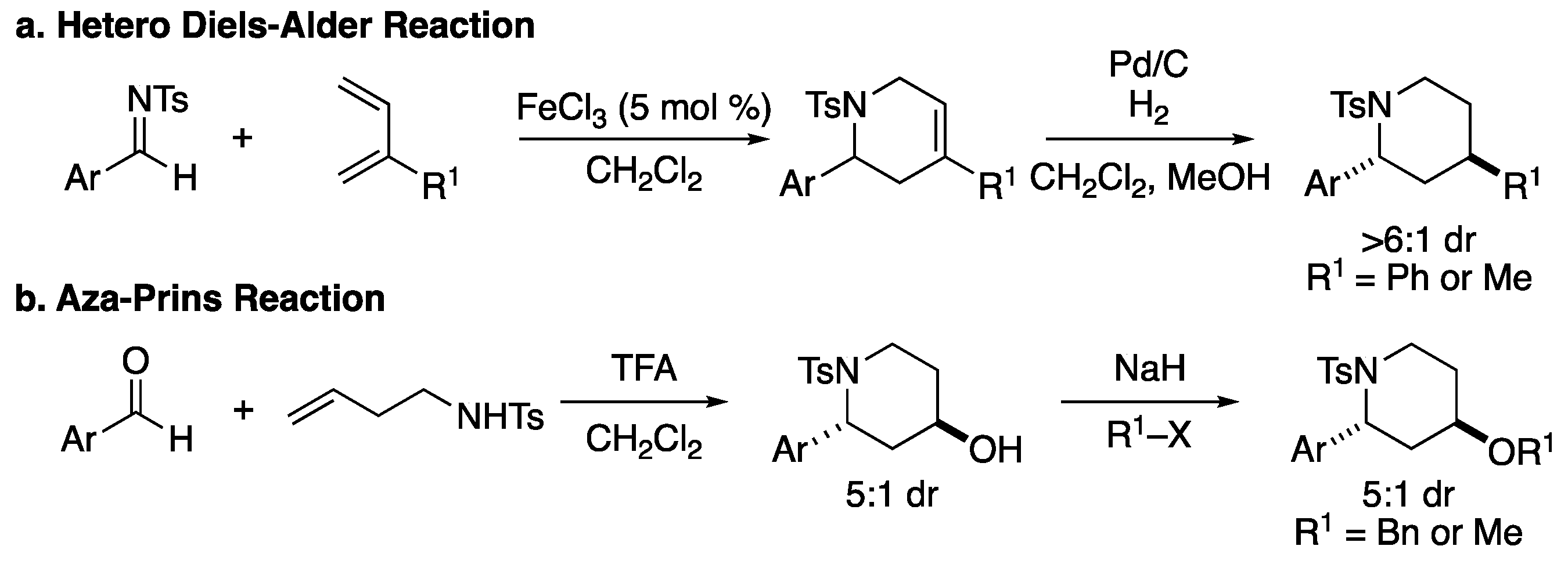
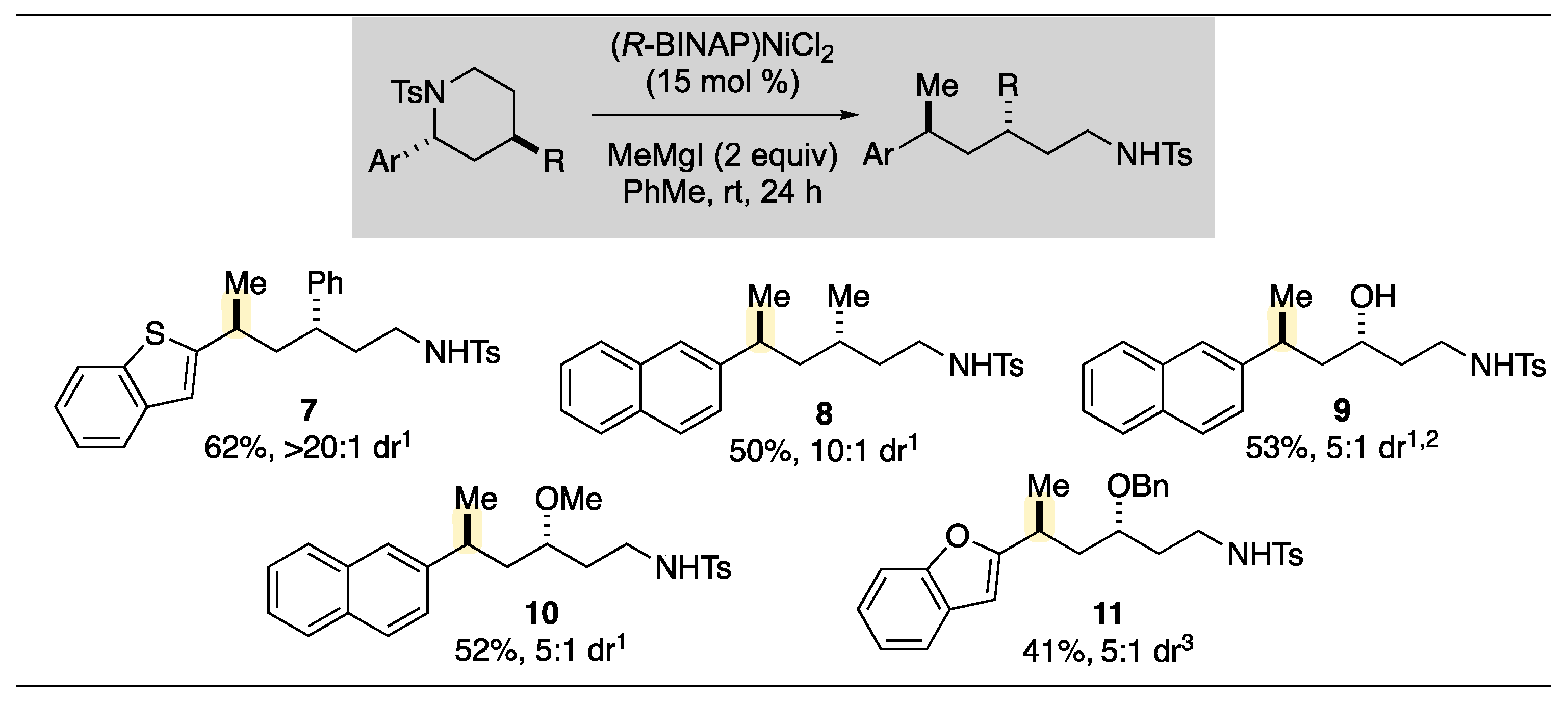
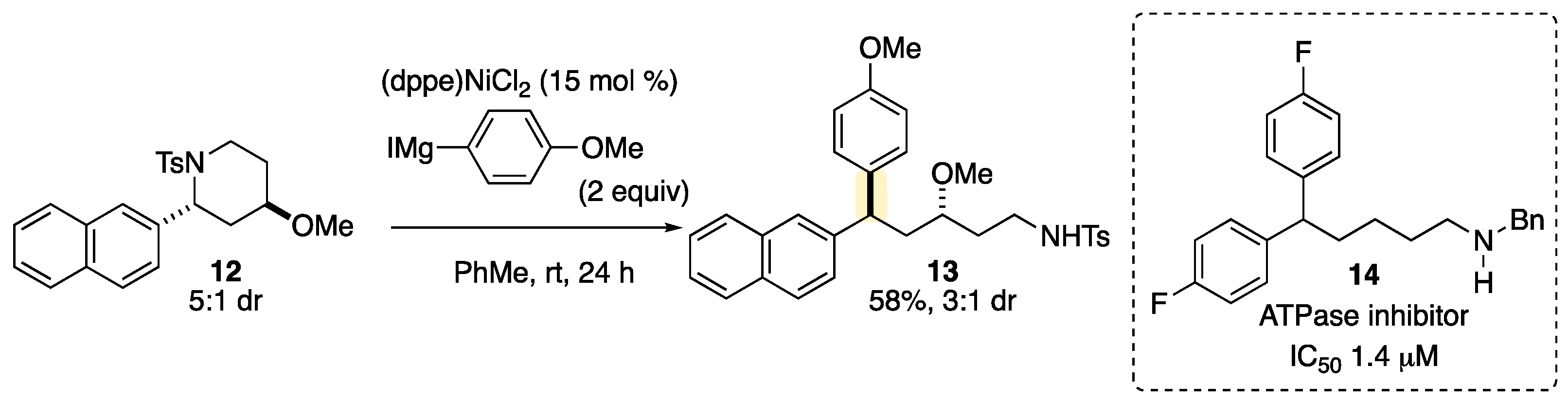
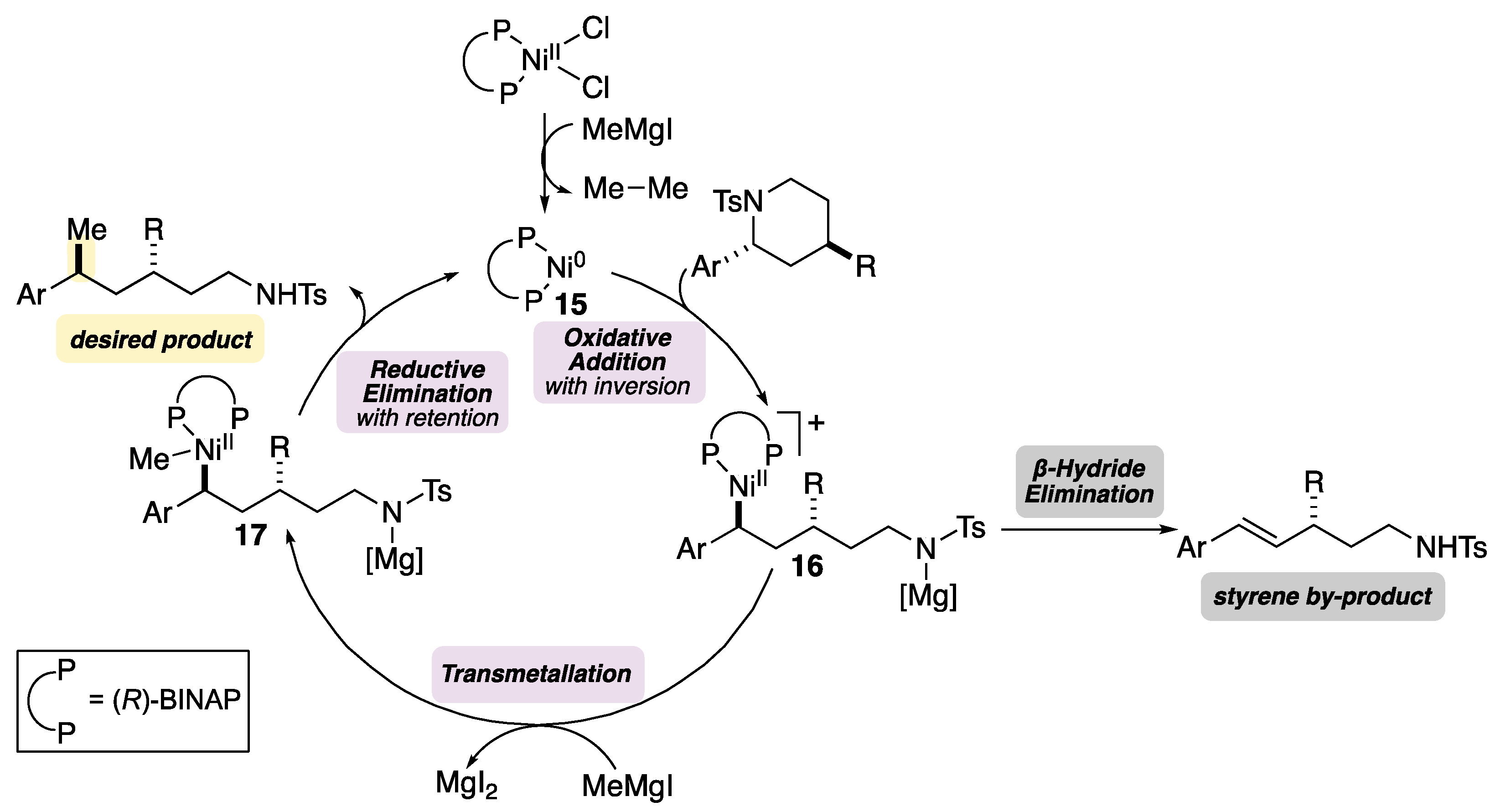
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Experimental
3.2.1. General Kumada Cross-Coupling Reaction Procedures
Method A: Kumada Cross-Coupling Reaction
- (1)
- Preparation of Grignard Reagent Under a N2 atmosphere, a three-necked flask equipped with a stir bar, reflux condenser, and Schlenk filtration apparatus was charged with magnesium turnings (1.1 g, 45 mmol). The flask and magnesium turnings were then flame-dried under vacuum and the flask was back-filled with N2. Anhydrous Et2O (7.0 mL) and a crystal of iodine (ca. 2.0 mg) were added to the flask. Freshly distilled iodomethane (1.9 mL, 31 mmol) or 4-iodoanisole as a solution in Et2O (4.7 g, 20. mmol, 6.7 M in Et2O) was slowly added over 30 min to maintain a gentle reflux. The mixture was stirred for 2 h at room temperature then filtered through the fritted Schlenk filter into a Schlenk flask under N2 atmosphere. The magnesium turnings were washed with Et2O (2 × 1.0 mL) then the Schlenk flask was sealed, removed, and placed under an N2 atmosphere. The resulting methylmagnesium iodide was typically between 2.4 and 3.0 M as titrated by Knochel’s method [72] and could be stored, sealed under N2 atmosphere or in a glovebox, for up to 4 weeks.
- (2)
- Preparation of (R-BINAP)NiCl2 This method was adapted from a procedure reported by Jamison [57]. To a flame-dried 50 mL round bottom flask equipped with a stir bar was added NiCl2·6H2O (0.24 g, 1.0 mmol, 1.0 equiv). The flask was placed under vacuum and flame-dried until nearly all of the nickel compound had turned from emerald green to yellow-orange. Some of the green hexahydrate is necessary for the reaction to proceed. The flask was allowed to cool to room temperature then (R-BINAP) (0.62 g, 1.0 mmol, 1.0 equiv) was added. The flask was then equipped with a reflux condenser and was evacuated and backfilled with N2. Then the solids were dissolved in MeCN (20 mL, 0.05 M) and the reaction mixture was allowed to reflux for 24 h. Upon completion, the reaction was cooled to room temperature and the black crystalline precipitate was filtered under vacuum to yield a fine black powder (0.53 g, 0.71 mmol, 71% yield).
3.2.2. Characterization Data for Kumada Cross-Coupled Products




3.2.3. Characterization Data for Ring Opening Kumada Cross-Coupled Products






3.2.4. General Procedures for Synthesis of Starting Materials
Method B: Condensation Reaction

Method C: Methylation of Sulfonamide with Methyl Iodide

Method D: Fe-Catalyzed Formal [4+2] Cycloaddition

Method E: Pd/C Reduction of Alkenes

Method F: TFA Mediated Aza-Prins Cyclization

Method G: Alkylation of Secondary Alcohol

3.2.5. Synthesis and Characterization Data of Sulfonamide Starting Materials
















4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References and Note
- De Meijere, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Winheim, Germany, 2004. [Google Scholar]
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-Organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [PubMed]
- Cherney, A.H.; Kadunce, N.T.; Reisman, S.E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C bonds. Chem. Rev. 2015, 115, 9587–9652. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Fu, G.C. Transition Metal-Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [Google Scholar] [CrossRef] [PubMed]
- Campeau, L.-C.; Hazari, N. Cross-Coupling and Related Reactions: Connecting Past Successes to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Tamaru, Y. (Ed.) Modern Organonickel Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent Advances in Homogeneous Nickel Catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, S. (Ed.) Nickel Catalysis in Organic Synthesis: Methods and Reactions; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Singer, R.A.; Monfette, S.; Bernhardson, D.; Tcyrulnikov, S.; Hubbell, A.K.; Hansen, E.C. Recent Advances in Nonprecious Metal Catalysis. Org. Process. Res. Dev. 2021, 25, 1802–1815. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-Catalyzed Cross-Couplings Involving Carbon-Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef]
- Tollefson, E.J.; Hanna, L.E.; Jarvo, E.R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Benzylic Ethers and Esters. Acc. Chem. Res. 2015, 48, 2344–2353. [Google Scholar] [CrossRef]
- Yu, D.-G.; Li, B.-J.; Shi, Z.-J. Exploration of New C–O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res. 2010, 43, 1486–1495. [Google Scholar] [CrossRef]
- Ouyang, K.; Hao, W.; Zhang, W.-X.; Xi, Z. Transition-Metal-Catalyzed Cleavage of C–N Single Bonds. Chem. Rev. 2015, 115, 12045–12090. [Google Scholar] [CrossRef]
- Wang, W.; Su, Y.; Li, L.; Huang, H. Transition-Metal Catalysed C–N Bond Activation. Chem. Soc. Rev. 2016, 45, 1257–1272. [Google Scholar] [CrossRef]
- Pound, S.M.; Watson, M.P. Asymmetric Synthesis via Stereospecific C–N and C–O Bond Activation of Alkyl Amine and Alcohol Derivatives. Chem. Commun. 2018, 54, 12286–12301. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Garg, N.K. Breaking Amides using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, S.; Szostak, M. Amide Bond Activation: The Power of Resonance. Trends Chem. 2020, 2, 914–928. [Google Scholar] [CrossRef]
- Li, M.-B.; Tang, X.-L.; Tian, S.-K. Cross-Coupling of Grignard Reagents with Sulfonyl-Activated sp3 Carbon-Nitrogen Bonds. Adv. Synth. Catal. 2011, 353, 1980–1984. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Doyle, A.G. The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev. 2014, 114, 8153–8198. [Google Scholar] [CrossRef]
- Lin, B.L.; Clough, C.R.; Hillhouse, G.L. Interactions of Aziridines with Nickel Complexes: Oxidative-Addition and Reductive-Elimination Reactions that Break and Make C−N Bonds. J. Am. Chem. Soc. 2002, 124, 2890–2891. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Doyle, A.G. Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J. Am. Chem. Soc. 2012, 134, 9541–9544. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Doyle, A.G. Electron-Deficient Olefin Ligands Enable Generation of Quaternary Carbons by Ni-Catalyzed Cross Coupling. J. Am. Chem. Soc. 2015, 137, 5638–5641. [Google Scholar] [CrossRef]
- Nielsen, D.K.; Huang, C.-Y.; Doyle, A.G. Directed Nickel-Catalyzed Negishi Cross Coupling of Alkyl Aziridines. J. Am. Chem. Soc. 2013, 135, 13605–13609. [Google Scholar] [CrossRef]
- Jensen, K.L.; Standley, E.A.; Jamison, T.F. Highly Regioselective Nickel-Catalyzed Cross-Coupling of N-Tosylaziridines and Alkylzinc Reagents. J. Am. Chem. Soc. 2014, 136, 11145–11152. [Google Scholar] [CrossRef] [PubMed]
- Wenkert, E.; Han, A.-L.; Jenny, C.-J. Nickel-Induced Conversion of Carbon-Nitrogen into Carbon-Carbon Bonds. One-Step Transformations of Aryl, Quaternary Ammonium Salts into Alkylarenes and Biaryls. J. Chem. Soc. Chem. Commun. 1988, 975–976. [Google Scholar] [CrossRef]
- Blakey, S.B.; MacMillan, D.W.C. The First Suzuki Cross-Couplings of Aryltrimethylammonium Salts. J. Am. Chem. Soc. 2003, 125, 6046–6047. [Google Scholar] [CrossRef] [PubMed]
- Maity, P.; Shacklady-McAtee, D.M.; Yap, G.P.A.; Sirianni, E.R.; Watson, M.P. Nickel-Catalyzed Cross-Couplings of Benzylic Ammonium Salts and Boronic Acids: Stereospecific Formation of Diarylethanes via C–N Bond Activation. J. Am. Chem. Soc. 2013, 135, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Shacklady-McAtee, D.M.; Roberts, K.M.; Basch, C.H.; Song, Y.-G.; Watson, M.P. A General, Simple Catalyst for Enantiospecific Cross Couplings of Benzylic Ammonium Triflates and Boronic Acids: No Phosphine Ligand Required. Tetrahedron 2014, 70, 4257–4263. [Google Scholar] [CrossRef][Green Version]
- He, F.; Wang, Z.-X. Nickel-Catalyzed Cross-Coupling of Aryl or -2-Menaphthyl Quaternary Ammonium Triflates with Organoaluminum Reagents. Tetrahedron 2017, 73, 4450–4457. [Google Scholar] [CrossRef]
- Bapat, J.B.; Blade, R.J.; Boulton, A.J.; Epsztajn, J.; Katritzky, A.R.; Lewis, J.; Molina-Buendia, P.; Nie, P.-L.; Ramsden, C.A. Pyridines as Leaving Groups in Synthetic Transformations: Nucleophilic Displacements of Amino Groups, and Novel Preparations of Nitriles and Isocyanate. Tetrahedron Lett. 1976, 17, 2691–2694. [Google Scholar] [CrossRef]
- Katritzky, A.R.; De Ville, G.; Patel, R.C. Carbon-Alkylation of Simple Nitronate Anions by N-Substituted Pyridiniums. Tetrahedron 1981, 37, 25–30. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Marson, C.M. Pyrylium Mediated Transformations of Primary Amino Groups into Other Functional Groups. New Synthetic Methods (41). Angew. Chem. Int. Ed. 1984, 23, 420–429. [Google Scholar] [CrossRef]
- Said, S.A.; Fiksdahl, A. Stereoselective Transformation of Amines via Chiral 2,4,6-Triphenylpyridinium Intermediates. Tetrahedron Asymmetry 2001, 12, 1947–1951. [Google Scholar] [CrossRef]
- Klauck, F.J.; James, M.J.; Glorius, F. Deaminative Strategy for the Visible-Light-Mediated Generation of Alkyl Radicals. Angew. Chem. Int. Ed. 2017, 56, 12336–12339. [Google Scholar] [CrossRef] [PubMed]
- He, F.-S.; Ye, S.; Wu, J. Recent Advances in Pyridinium Salts as Radical Reservoirs in Organic Synthesis. ACS Catal. 2019, 9, 8943–8960. [Google Scholar] [CrossRef]
- Pang, Y.; Moser, D.; Cornella, J. Pyrylium Salts: Selective Reagents for the Activation of Primary Amino Groups in Organic Synthesis. Synthesis 2020, 52, 489–503. [Google Scholar]
- Rössler, S.L.; Jelier, B.J.; Magnier, E.; Dagousset, G.; Carreira, E.M.; Togni, A. Pyridinium Salts as Redox-Active Functional Group Transfer Reagents. Angew. Chem. Int. Ed. 2020, 59, 9264–9280. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-N.; Xiao, F.; Guo, Y.; Zeng, Y.-F. Recent Developments in Deaminative Functionalization of Alkyl Amines. Eur. J. Org. Chem. 2021, 2021, 1215–1228. [Google Scholar] [CrossRef]
- Kong, D.; Moon, P.J.; Lundgren, R.J. Radical Coupling from Alkyl Amines. Nat. Catal. 2019, 2, 473–476. [Google Scholar] [CrossRef]
- Basch, C.H.; Liao, J.; Xu, J.; Piane, J.J.; Watson, M.P. Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C–N Bond Activation. J. Am. Chem. Soc. 2017, 139, 5313–5316. [Google Scholar] [CrossRef] [PubMed]
- Hoerner, M.E.; Baker, K.M.; Basch, C.H.; Bampo, E.M.; Watson, M.P. Deaminative Arylation of Amino Acid-Derived Pyridinium Salts. Org. Lett. 2019, 21, 7356–7360. [Google Scholar] [CrossRef]
- Liao, J.; Guan, W.; Boscoe, B.P.; Tucker, J.W.; Tomlin, J.W.; Garnsey, M.R.; Watson, M.P. Transforming Benzylic Amines into Diarylmethanes: Cross-Couplings of Benzylic Pyridinium Salts via C–N Bond Activation. Org. Lett. 2018, 20, 3030–3033. [Google Scholar] [CrossRef]
- Baker, K.M.; Baca, D.L.; Plunkett, S.; Daneker, M.E.; Watson, M.P. Engaging Alkenes and Alkynes in Deaminative Alkyl–Alkyl and Alkyl–Vinyl Cross-Couplings of Alkylpyridinium Salts. Org. Lett. 2019, 21, 9738–9741. [Google Scholar] [CrossRef]
- Guan, W.; Liao, J.; Watson, M.P. Vinylation of Benzylic Amines via C–N Bond Functionalization of Benzylic Pyridinium Salts. Synthesis 2018, 50, 3231–3237. [Google Scholar]
- Plunkett, S.; Basch, C.H.; Santana, S.O.; Watson, M.P. Harnessing Alkylpyridinium Salts as Electrophiles in Deaminative Alkyl–Alkyl Cross-Couplings. J. Am. Chem. Soc. 2019, 141, 2257–2262. [Google Scholar] [CrossRef]
- Taylor, B.L.H.; Swift, E.C.; Waetzig, J.D.; Jarvo, E.R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Ethers: Enantioselective Synthesis of Diarylethanes. J. Am. Chem. Soc. 2011, 133, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, E.J.; Dawson, D.D.; Osborne, C.A.; Jarvo, E.R. Stereospecific Cross-Coupling Reactions of Aryl-Substituted Tetrahydrofurans, Tetrahydropyrans, and Lactones. J. Am. Chem. Soc. 2014, 136, 14951–14958. [Google Scholar] [CrossRef]
- Marshall, D.R.; Thomas, P.J.; Stirling, C.J.M. Leaving Group Ability in Base-Promoted Alkene-Forming 1,2-Eliminations. J. Chem. Soc. Chem. Commun. 1975, 23, 940–941. [Google Scholar] [CrossRef]
- Lucas, E.L.; Hewitt, K.A.; Chen, P.-P.; Castro, A.J.; Hong, X.; Jarvo, E.R. Engaging Sulfonamides: Intramolecular Cross-Electrophile Coupling Reaction of Sulfonamides with Alkyl Chlorides. J. Org. Chem. 2020, 85, 1775–1793. [Google Scholar] [CrossRef] [PubMed]
- Steinman, T.J.; Liu, J.; Mengiste, A.; Doyle, A.G. Synthesis of β-Phenethylmines via Ni/Photoredox Cross-Electrophile Coupling of Aliphatic Aziridines and Aryl Iodides. J. Am. Chem. Soc. 2020, 142, 7598–7605. [Google Scholar] [CrossRef]
- Tcyrulnikov, S.; Cai, Q.; Twitty, J.C.; Xu, J.; Atifi, A.; Bercher, O.P.; Yap, G.P.A.; Rosenthal, J.; Watson, M.P.; Kozlowski, M.C. Dissection of Alkylpyridinium Structures to Understand Deamination Reactions. ACS Catal. 2021, 11, 8456–8466. [Google Scholar] [CrossRef]
- Xu, J.; Bercher, O.P.; Talley, M.R.; Watson, M.P. Nickel-Catalyzed, Stereospecific C−C and C−B Cross-Couplings via C−N and C−O Bond Activation. ACS Catal. 2021, 11, 1604–1612. [Google Scholar] [CrossRef]
- Moragas, T.; Gaydou, M.; Martin, R. Nickel-Catalyzed Carboxylation of Benzylic C–N Bonds with CO2. Angew. Chem. Int. Ed. 2016, 55, 5053–5057. [Google Scholar] [CrossRef]
- Hanessian, S.; Giroux, S.; Mascitti, V. The Iterative Synthesis of Acyclic Deoxypropionate Units and Their Implication in Polyketide-Derived Natural Products. Synthesis 2006, 7, 1057–1076. [Google Scholar] [CrossRef]
- Chen, R.; Shen, Y.; Yang, S.; Zhang, Y. Conformational Design Principles in Total Synthesis. Angew. Chem. Int. Ed. 2020, 59, 14198–14210. [Google Scholar] [CrossRef]
- Standley, E.A.; Smith, S.J.; Müller, P.; Jamison, T.F. A Broadly Applicable Strategy for Entry into Homogenous Nickel(0) Catalysts from Air-Stable Nickel(II) Complexes. Organometallics 2014, 33, 2012–2018. [Google Scholar] [CrossRef]
- Dawson, D.D.; Oswald, V.F.; Borovik, A.S.; Jarvo, E.R. Identification of the Active Catalyst for Nickel-Catalyzed Stereospecific Kumada Coupling Reactions of Ethers. Chem. Eur. J. 2020, 26, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Tomifuji, R.; Maeda, K.; Takahashi, T.; Kurahashi, T.; Mastubara, S. FeCl3 as an Ion-Pairing Lewis Acid Catalyst. Formation of Highly Lewis Acidic FeCl2+ and Thermodynamically Stable FeCl4– To Catalyze the Aza-Diels–Alder Reaction with High Turnover Frequency. Org. Lett. 2018, 20, 7474–7477. [Google Scholar] [CrossRef]
- Sabitha, G.; Reddy, N.M.; Prasad, M.N.; Yadav, J.S. Stereoselective Routes for the Total Synthesis of (+)-Crytocarya Diacetate. Helv. Chim. Acta 2009, 92, 967–976. [Google Scholar] [CrossRef]
- Tollefson, E.J.; Erickson, L.W.; Jarvo, E.R. Stereospecific Intramolecular Reductive Cross-Electrophile Coupling Reactions for Cyclopropane Synthesis. J. Am. Chem. Soc. 2015, 137, 9760–9763. [Google Scholar] [CrossRef]
- Kawana, M. The Reaction of Benzylated Pyrrole and Adenine Ribonucleosides with Grignard Reagents. Chem. Lett. 1981, 10, 1541–1542. [Google Scholar] [CrossRef]
- Pöhler, R.; Krahn, J.H.; van den Boom, J.; Dobrynin, G.; Kaschani, F.; Eggenweiler, H.-M.; Zenke, F.T.; Kaiser, M.; Meyer, H. A Non-Competitive Inhibitor of VCP/p97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases. Angew. Chem. Int. Ed. 2018, 57, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Wetterau, J.R.; Gregg, R.E.; Harrity, T.W.; Arbeeny, C.; Cap, M.; Connolly, F.; Chu, C.-H.; George, R.J.; Gordon, D.A.; Jamil, H.; et al. An MTP Inhibitor That Normalizes Atherogenic Lipoprotein Levels in WHHL Rabbits. Science 1998, 282, 751. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Masuda, T.; Yamada, K.; Mitani, M.; Kubota, N.; Kawakatsu, N.; Kishii, K.; Inazu, M.; Kiuchi, Y.; Oguchi, K.; et al. Syntheses of Novel Diphenyl Piperazine Derivatives and Their Activities as Inhibitors of Dopamine Uptake in the Central Nervous System. Bioorg. Med. Chem. 2003, 11, 1621–1630. [Google Scholar] [CrossRef]
- Dei, S.; Coronnello, M.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Udomtanakunchai, C.; Salerno, M.; Teodori, E. Design and synthesis of new potent N,N-is(arylalkyl)piperazine Derivatives as Multidrug Resistance (MDR) Reversing agents. Eur. J. Med. Chem. 2018, 147, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ameen, D.; Snape, T.J. Chiral 1,1-Diaryl Compounds as Important Pharmacophores. MedChemComm 2013, 4, 893–907. [Google Scholar] [CrossRef]
- Yonova, I.M.; Johnson, A.G.; Osborne, C.A.; Moore, C.E.; Morrissette, N.S.; Jarvo, E.R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Grignard Reagents and Identification of Selective Anti-Breast-Cancer Agents. Angew. Chem. Int. Ed. 2014, 53, 2422–2427. [Google Scholar] [CrossRef]
- When (R-BINAP)NiCl2 was employed as the precatalyst, the desired product was not observed and 44% of starting material was recovered.
- Chen, P.-P.; Lucas, E.L.; Greene, M.A.; Zhang, S.; Tollefson, E.J.; Erickson, L.W.; Taylor, B.L.; Jarvo, E.R.; Hong, X. A Unified Explanation for Chemoselectivity and Stereospecificity of Ni-Catalyzed Kumada and Cross-Electrophile Coupling Reactions of Benzylic Ethers: A Combined Computational and Experimental Study. J. Am. Chem. Soc. 2019, 141, 5835–5855. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Krasoviskiy, A.; Knochel, P. Convenient Titration Method for Organometallic Zinc, Magnesium, and Lanthanide Reagents. Synthesis 2006, 5, 890. [Google Scholar]
- García Ruano, J.L.; Alemán, J.; Belén Cid, M.; Parra, A. A General Method for the Preparation of N-Sulfonyl Aldimines and Ketimines. Org. Lett. 2005, 7, 179–182. [Google Scholar] [CrossRef]
- Fu, M.; Chen, L.; Jiang, Y.; Jiang, Z.-X.; Yang, Z. Copper-Catalyzed Intermolecular Chloro- and Bromotrifluoromethylation of Alkenes. Org. Lett. 2016, 18, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Syu, S.; Lee, Y.-T.; Jang, Y.-J.; Lin, W. Organocatalytic Tandem Three-Component Reaction of Imine, Alkyl Vinyl Ketone, and Imide via aza-Baylis−Hillman Reaction. J. Org. Chem. 2011, 76, 2888–2891. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, D.; Folliet, S.; Liu, Y.; Mazet, C. A General Nickel-Catalyzed Kumada Vinylation for the Preparation of 2-Substituted 1,3-Dienes. ACS Catal. 2018, 8, 1392–1398. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, J.; Wu, W.; Li, J.; Ma, J.; Ren, Y.; Jiang, H. Palladium-Catalyzed Intermolecular Oxidative Cyclization of Allyltosylamides with AcOH: Assembly of 3-Pyrrolin-2-ones. J. Org. Chem. 2017, 82, 8191–8198. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Nickel Catalyst | Ligand | Yield 2 (%) 1 | Yield 3 (%) 1 | RSM 1 (%) 1 |
|---|---|---|---|---|---|
| 1 2 | Ni(cod)2 | rac-BINAP | 25 | 10 | 19 |
| 2 | Ni(cod)2 | rac-BINAP | 34 | 30 | 7 |
| 3 | Ni(cod)2 | DPEPhos | 42 | 20 | 0 |
| 4 | Ni(cod)2 | XantPhos | 0 | <5 | 37 |
| 5 | Ni(cod)2 | dppe | 0 | <5 | 65 |
| 6 | Ni(cod)2 | SiMes·BF4 | 12 | 0 | 86 |
| 7 | Ni(cod)2 | BPhen | 0 | 0 | 61 |
| 8 | (R-BINAP)NiCl2 | – | 54 | 40 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hewitt, K.A.; Herbert, C.A.; Matus, A.C.; Jarvo, E.R. Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Sulfonamides. Molecules 2021, 26, 5947. https://doi.org/10.3390/molecules26195947
Hewitt KA, Herbert CA, Matus AC, Jarvo ER. Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Sulfonamides. Molecules. 2021; 26(19):5947. https://doi.org/10.3390/molecules26195947
Chicago/Turabian StyleHewitt, Kirsten A., Claire A. Herbert, Alissa C. Matus, and Elizabeth R. Jarvo. 2021. "Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Sulfonamides" Molecules 26, no. 19: 5947. https://doi.org/10.3390/molecules26195947
APA StyleHewitt, K. A., Herbert, C. A., Matus, A. C., & Jarvo, E. R. (2021). Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Sulfonamides. Molecules, 26(19), 5947. https://doi.org/10.3390/molecules26195947