
Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms


Abstract
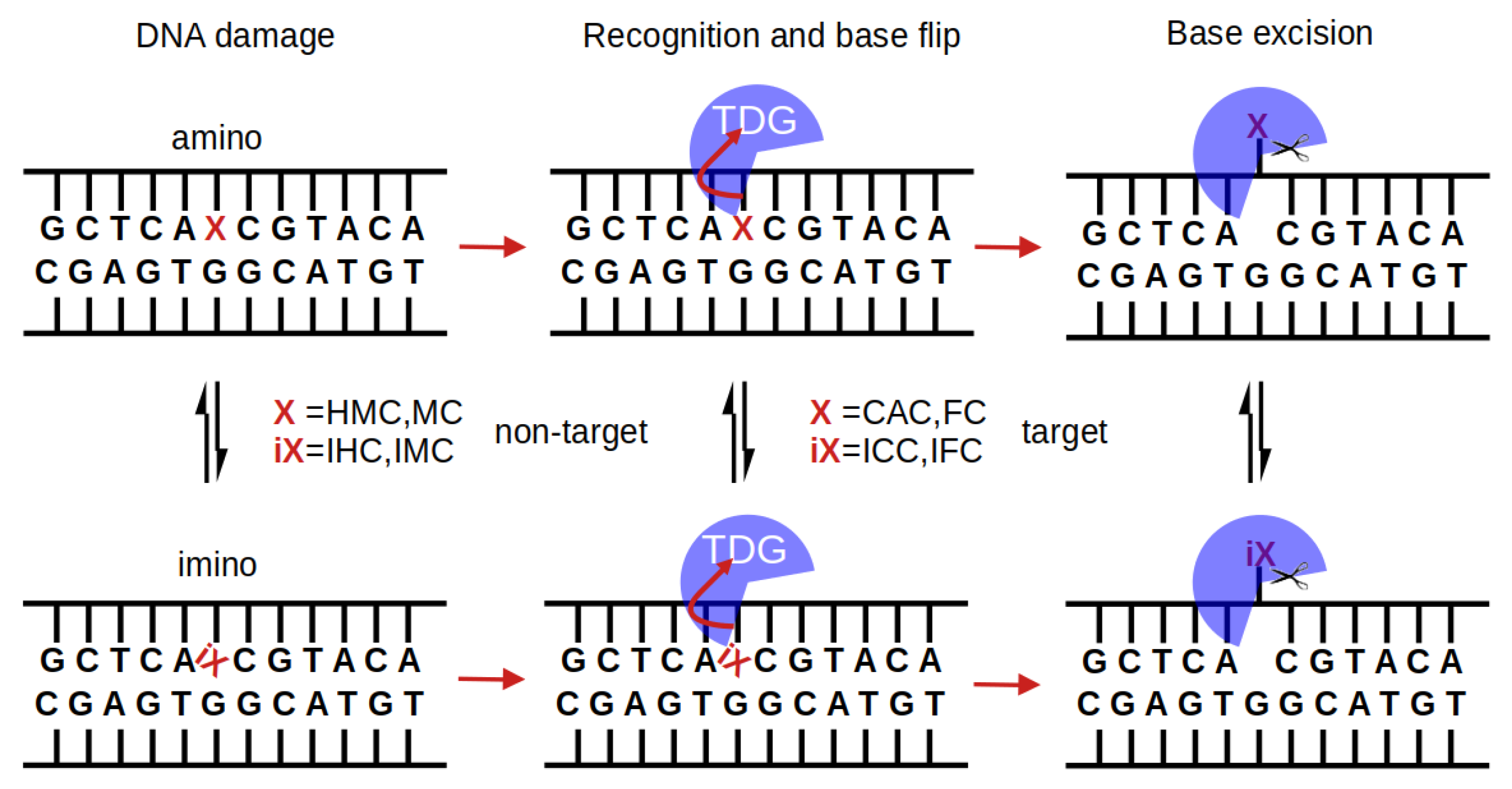
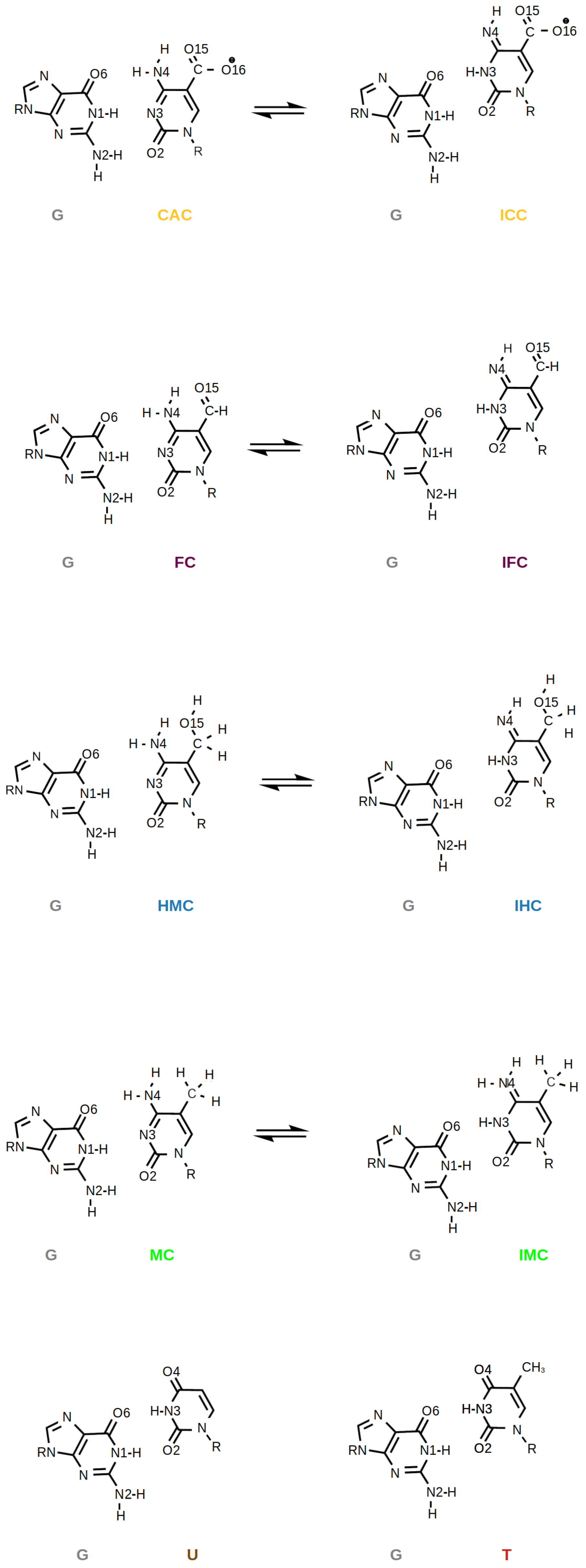
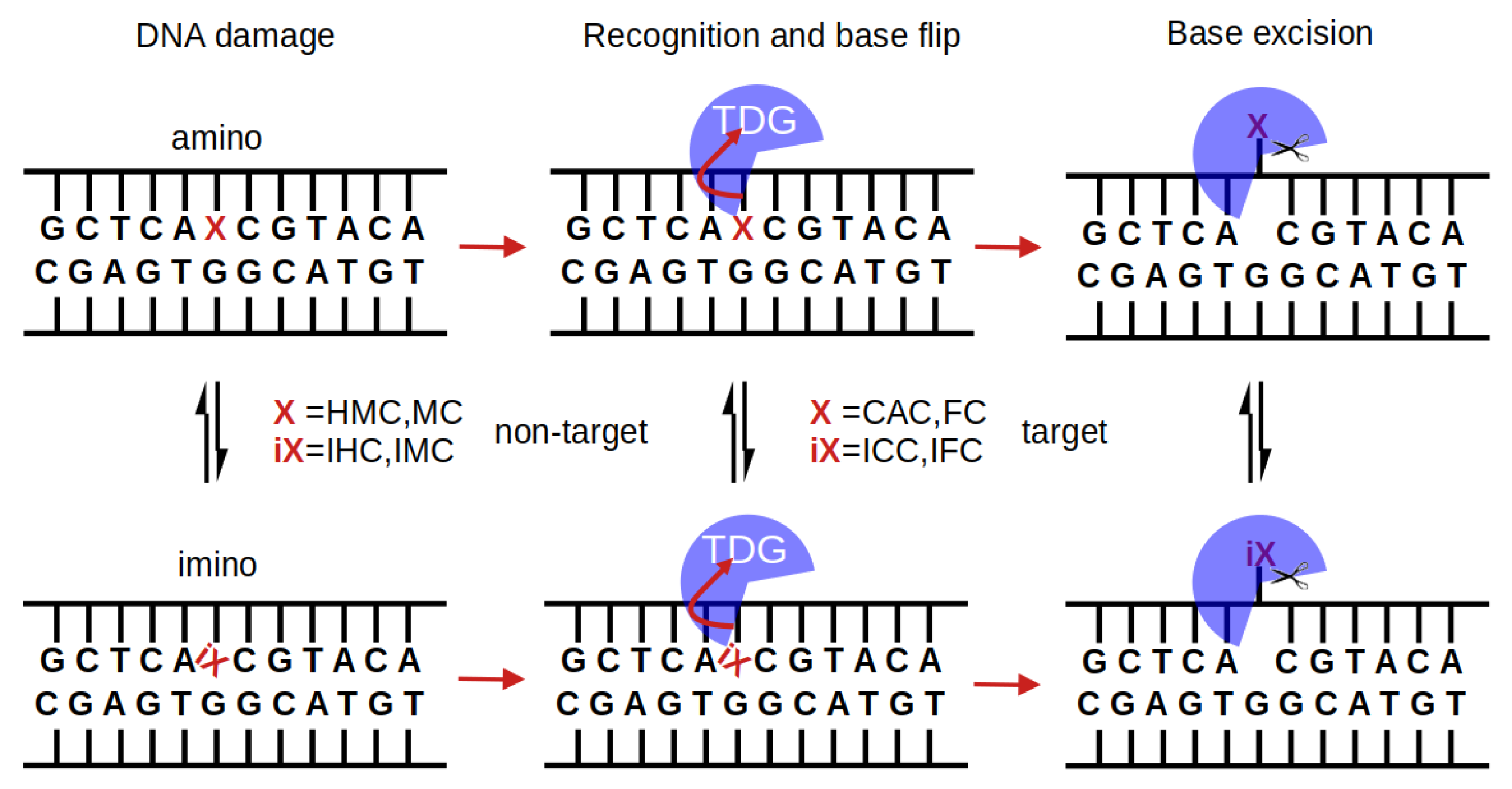
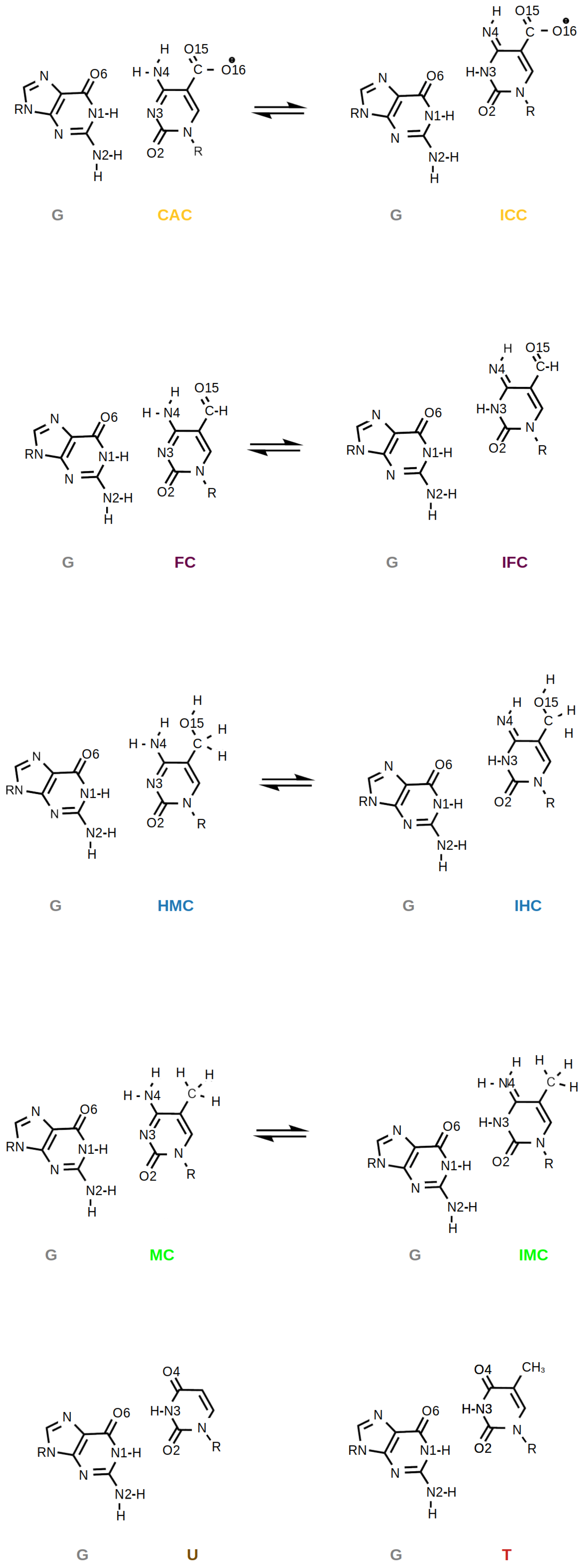
:1. Introduction
2. Methods
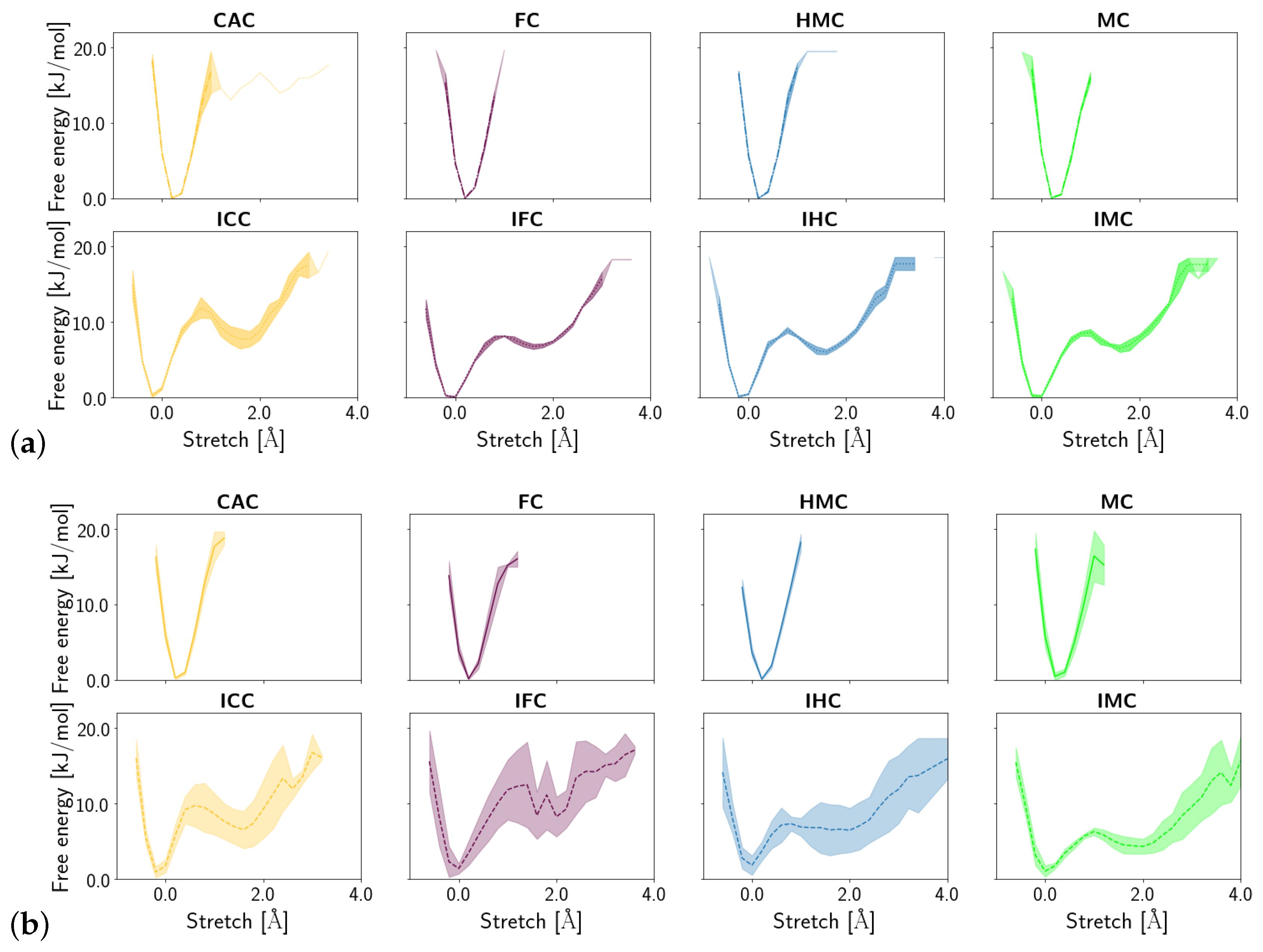
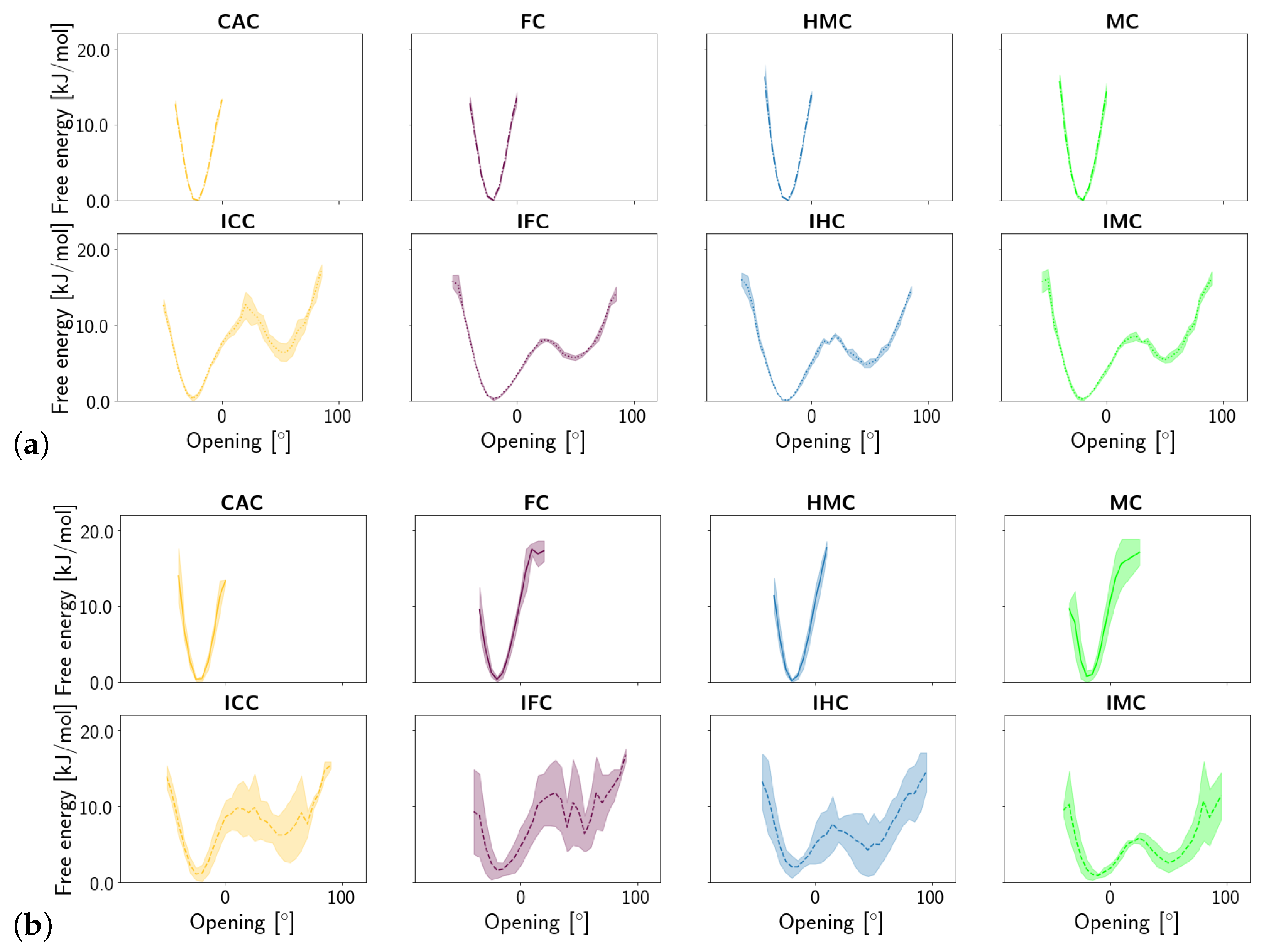
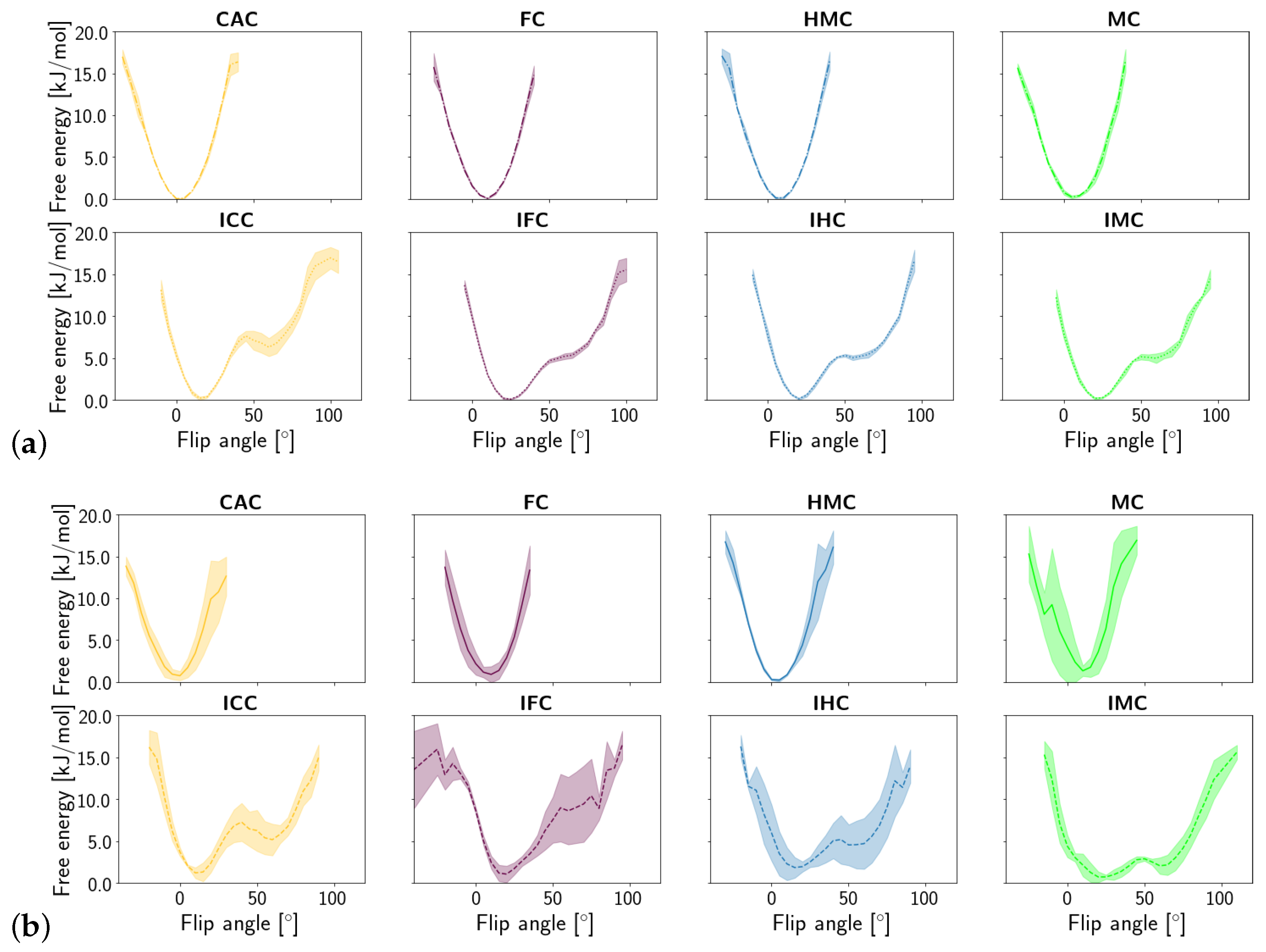
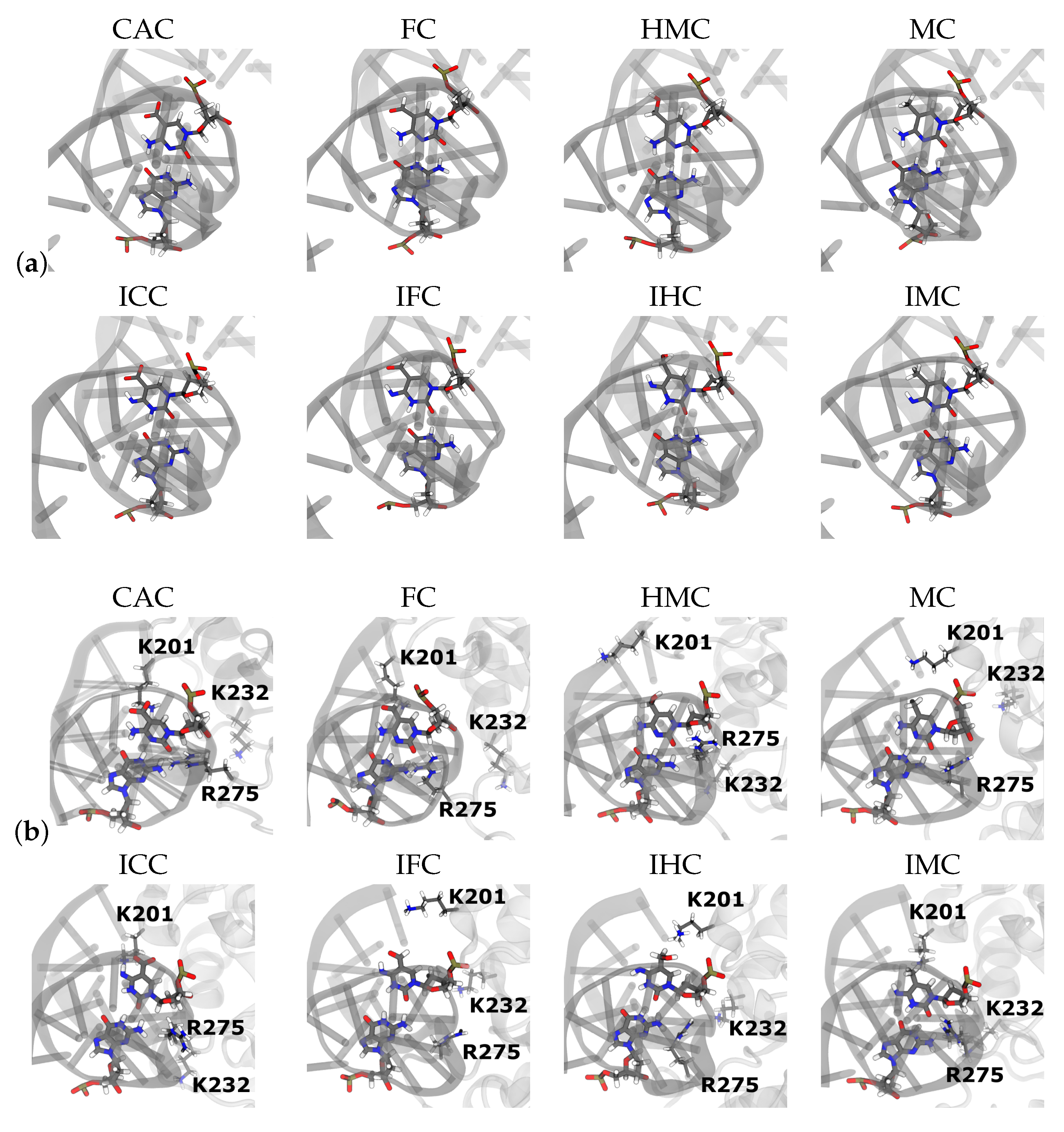
3. Results
3.1. Comparison of Free and Complexed DNA
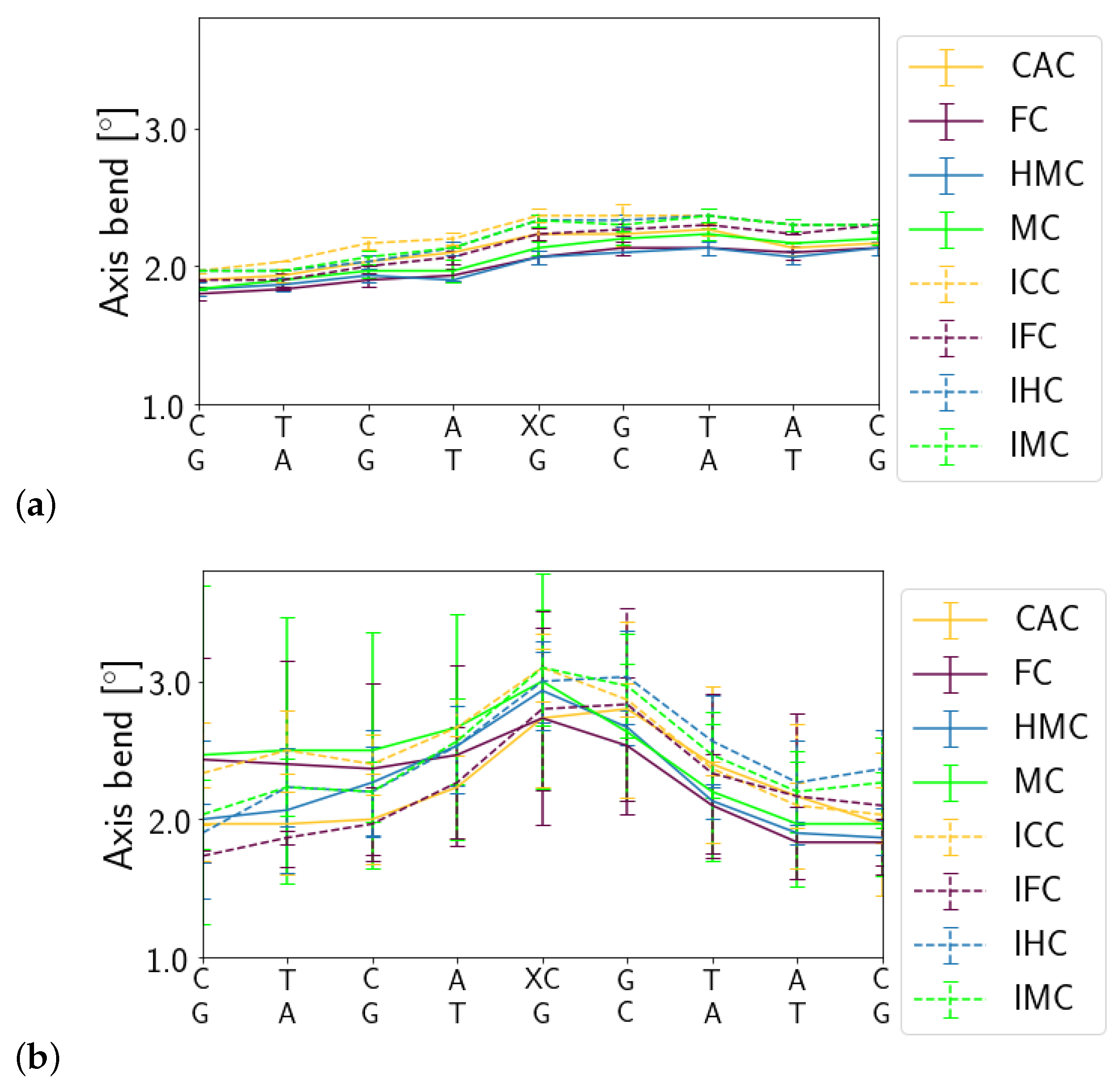
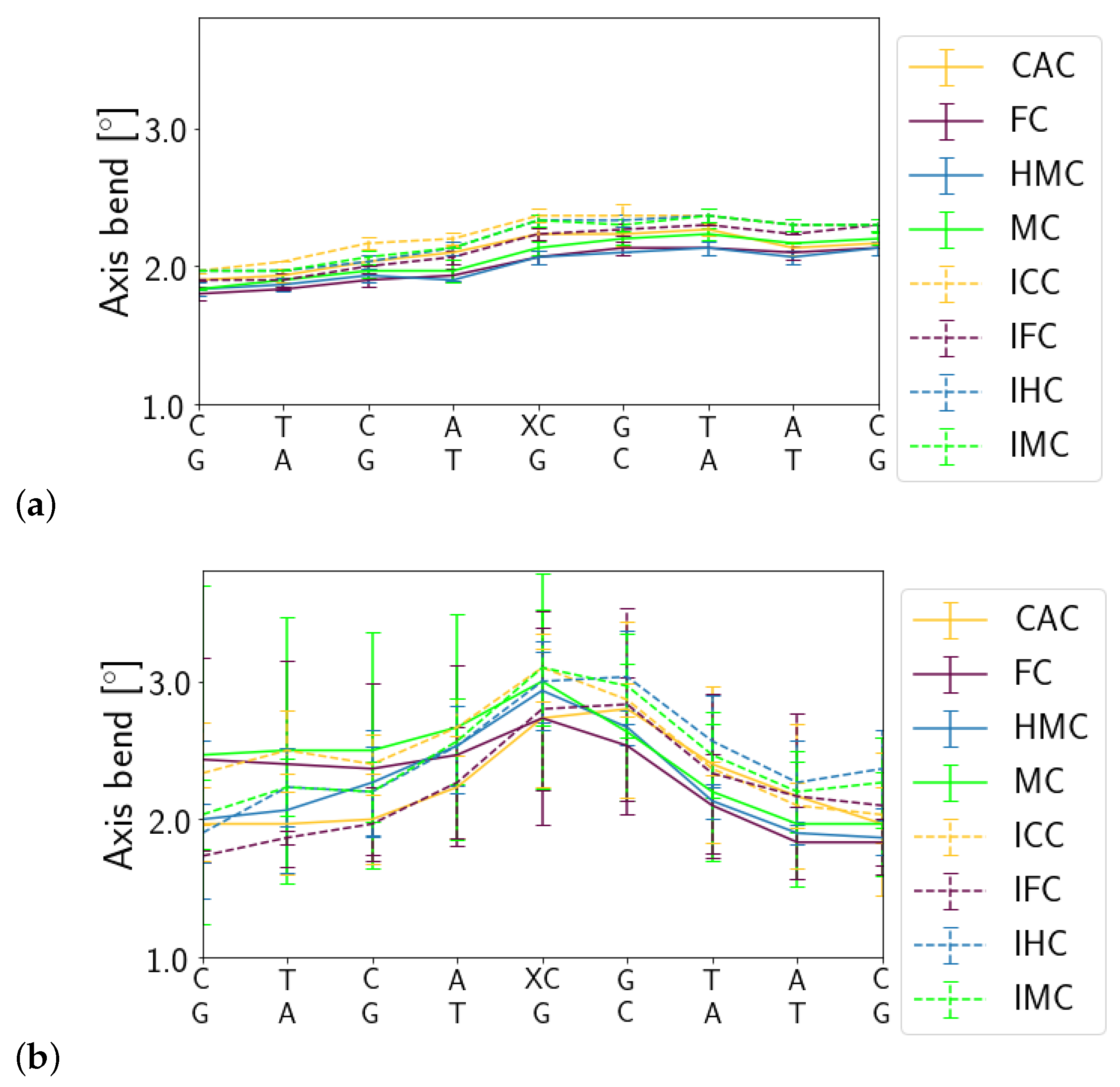
3.1.1. DNA Conformation
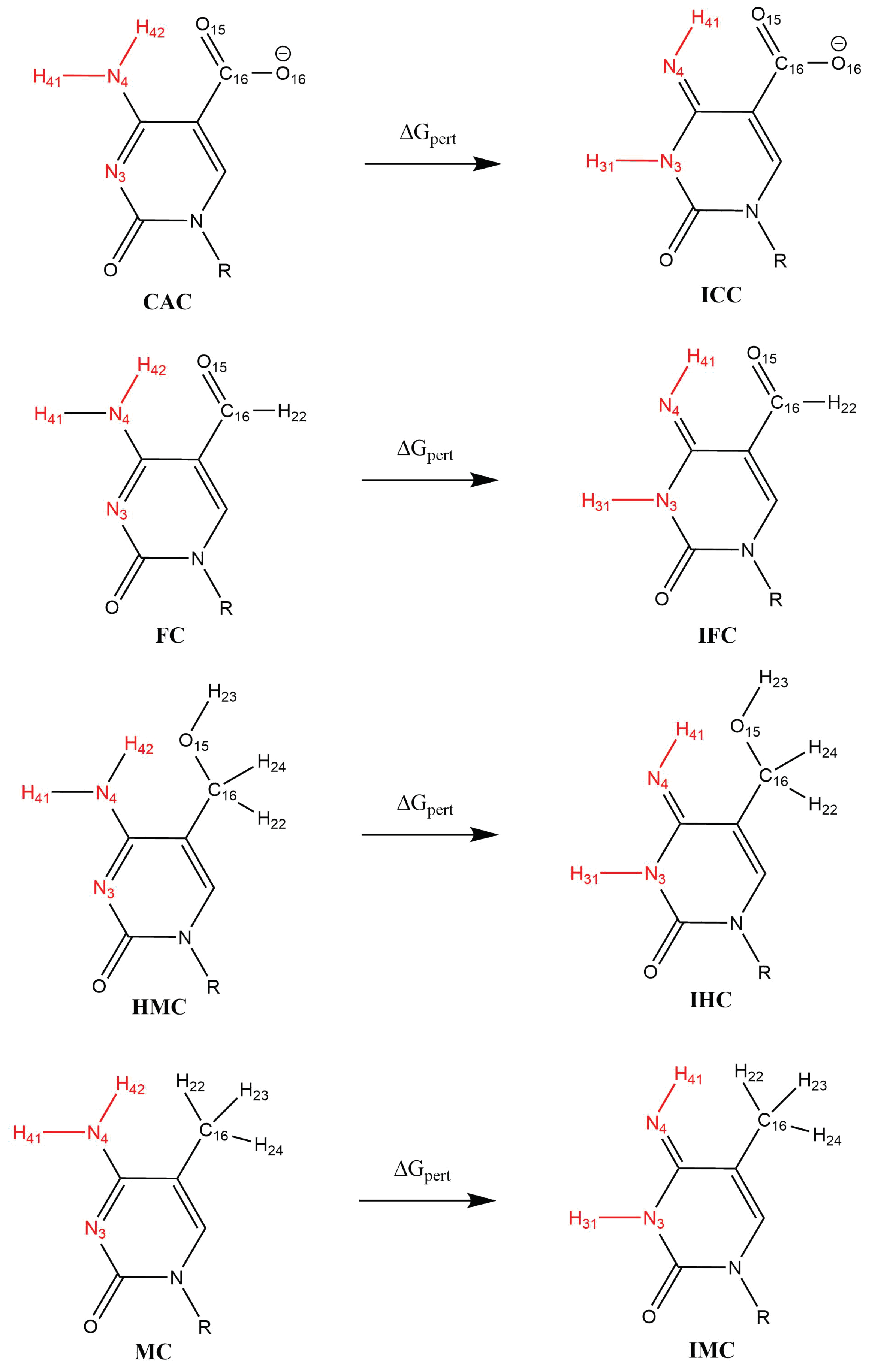
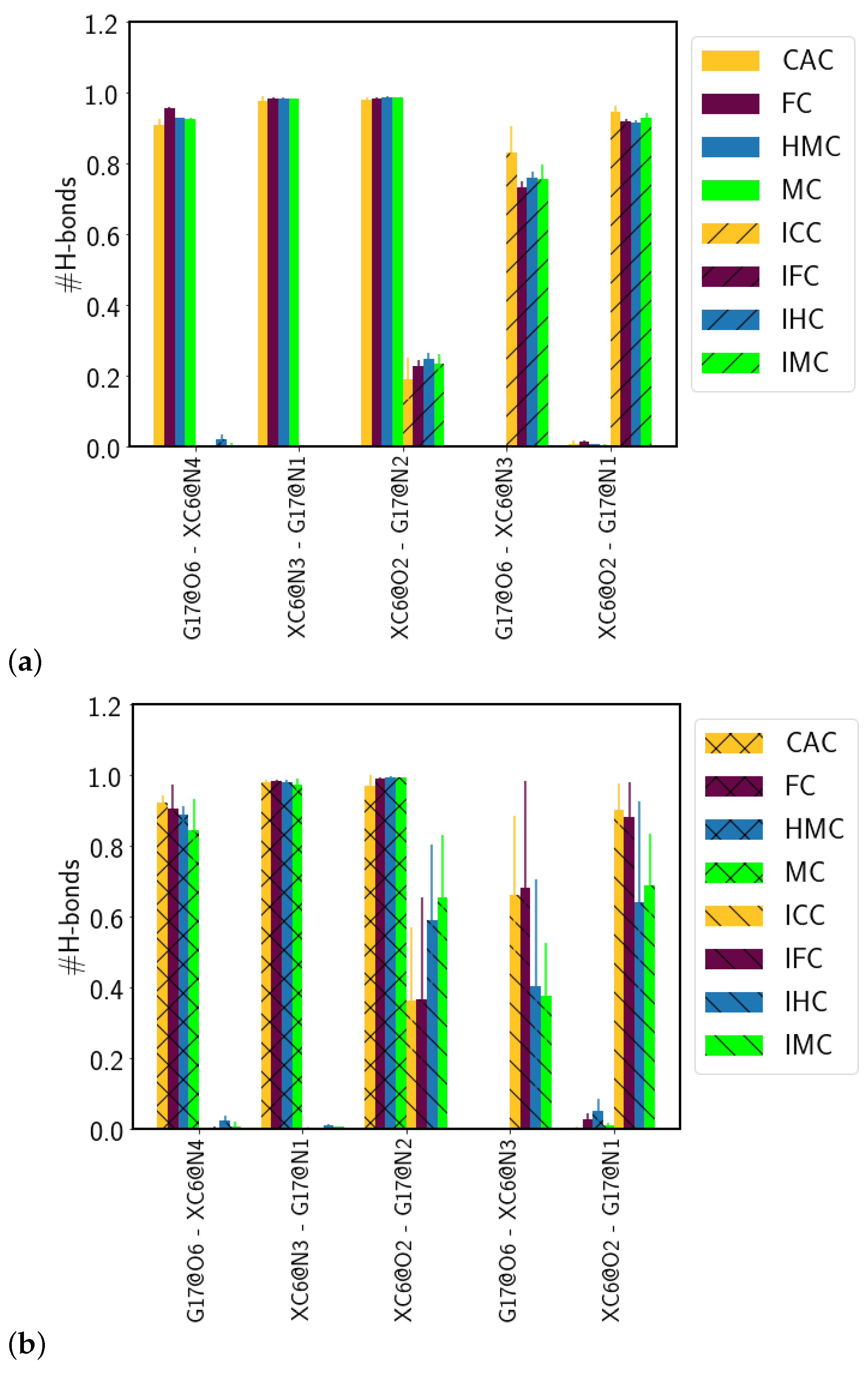
3.1.2. Hydrogen-Bond Interactions
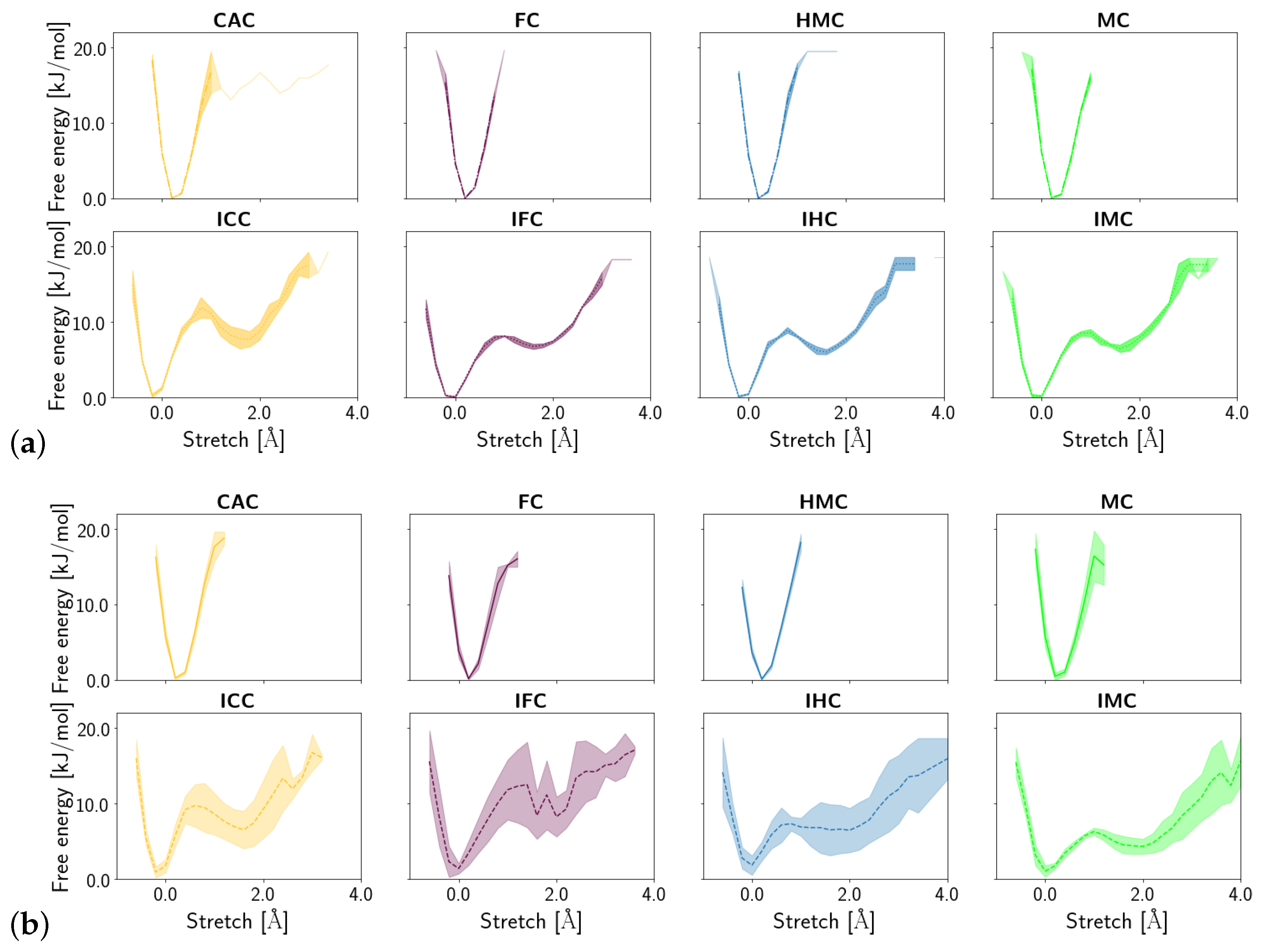
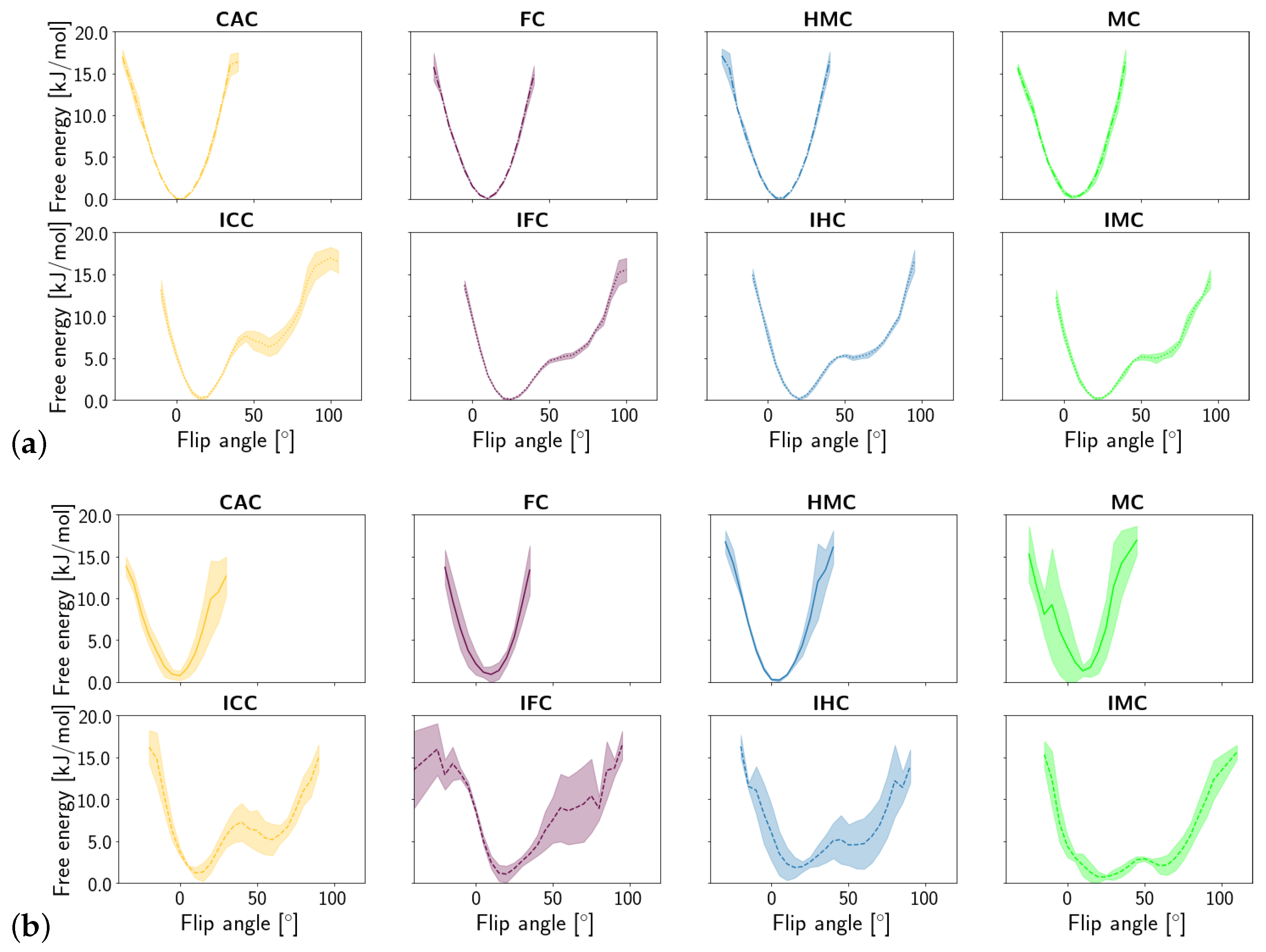
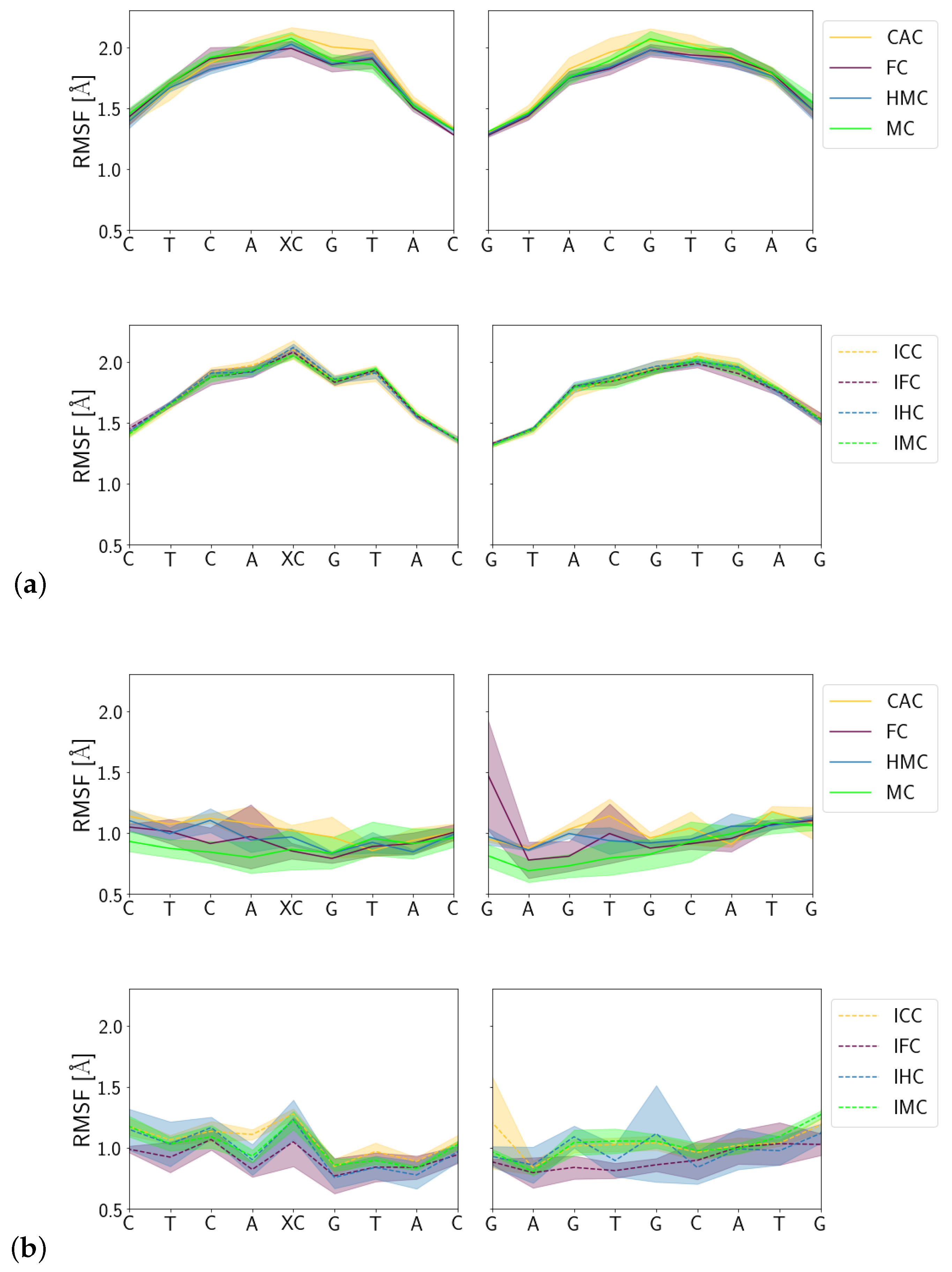
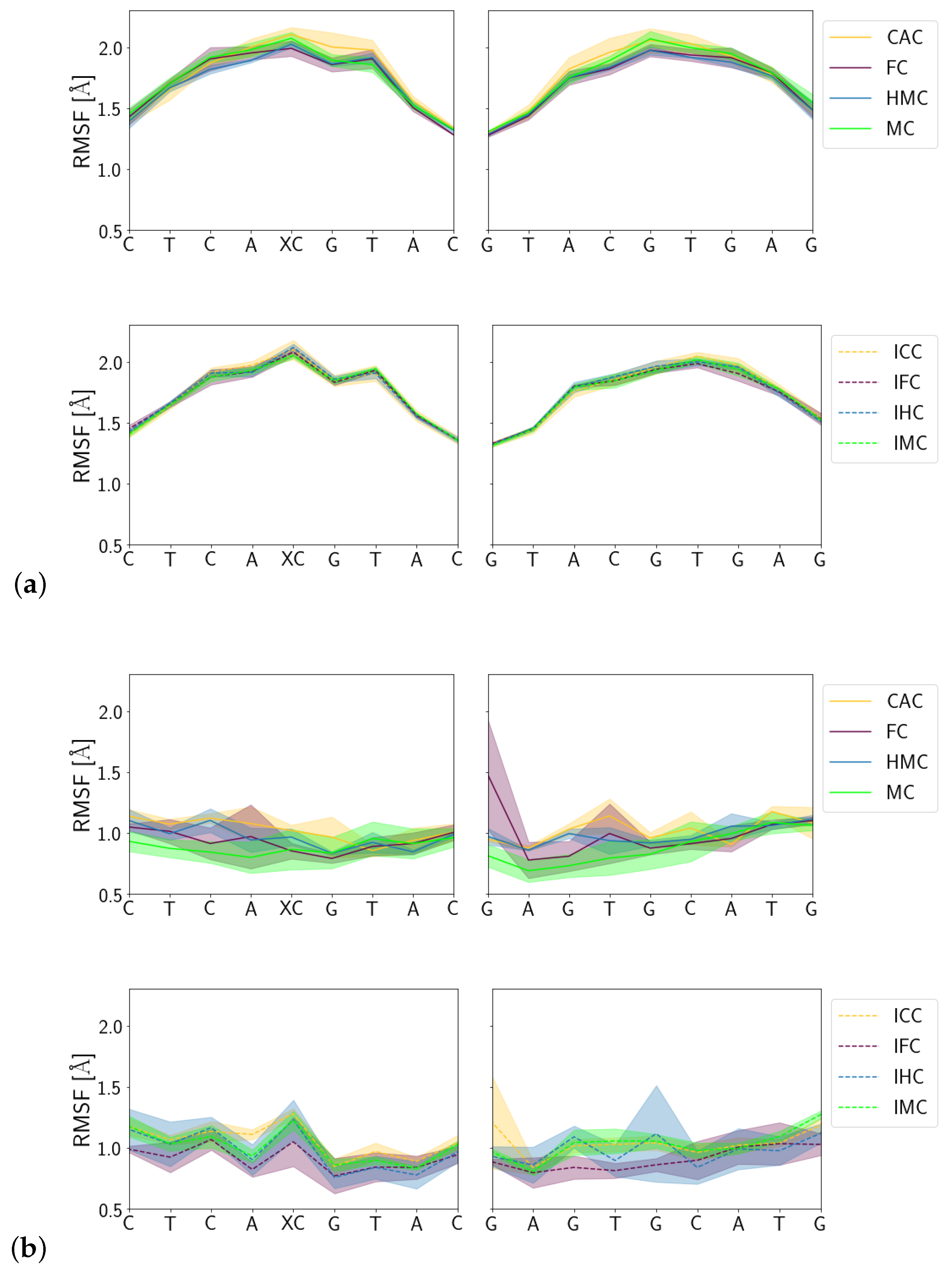
3.1.3. Fluctuations
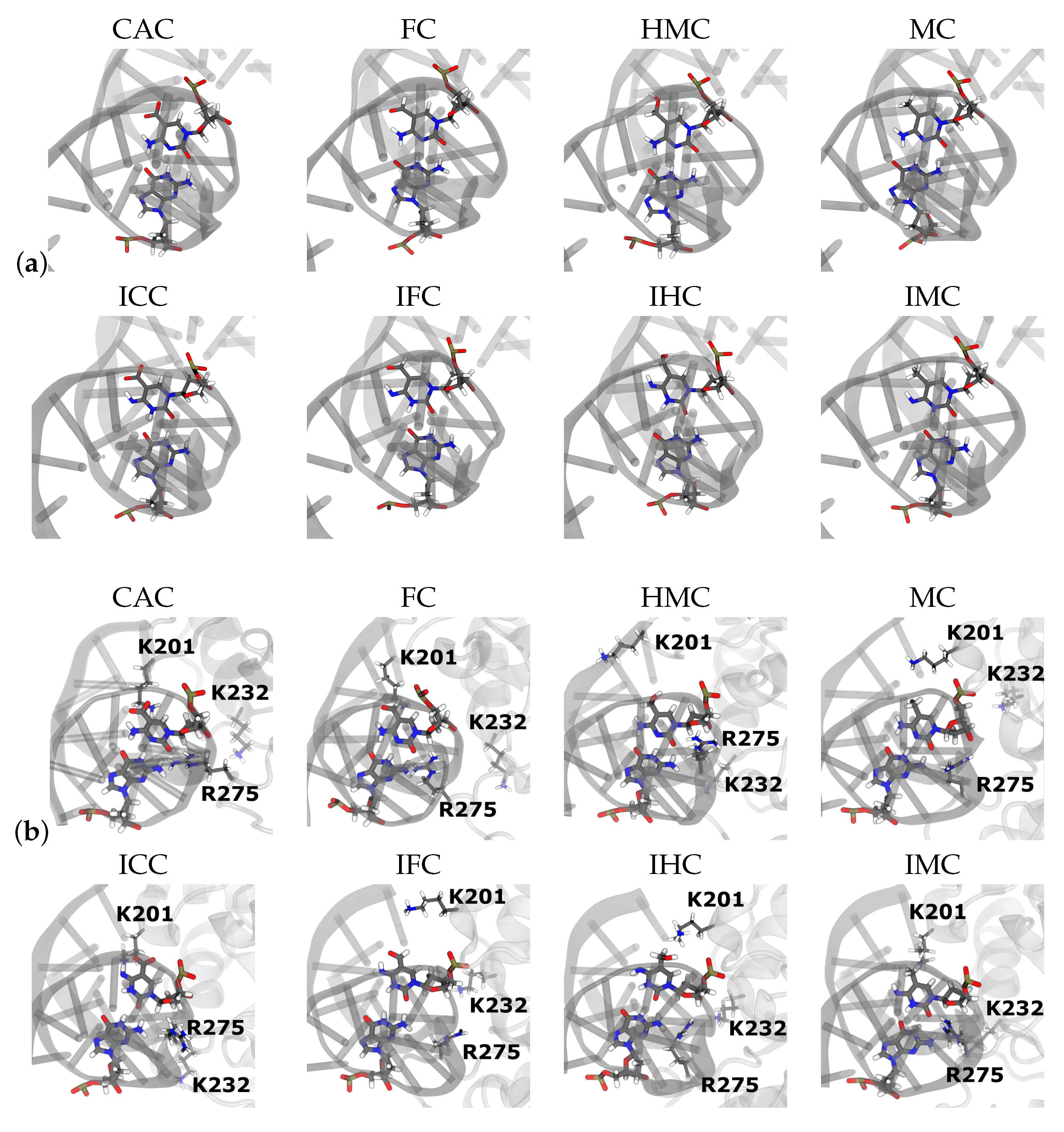
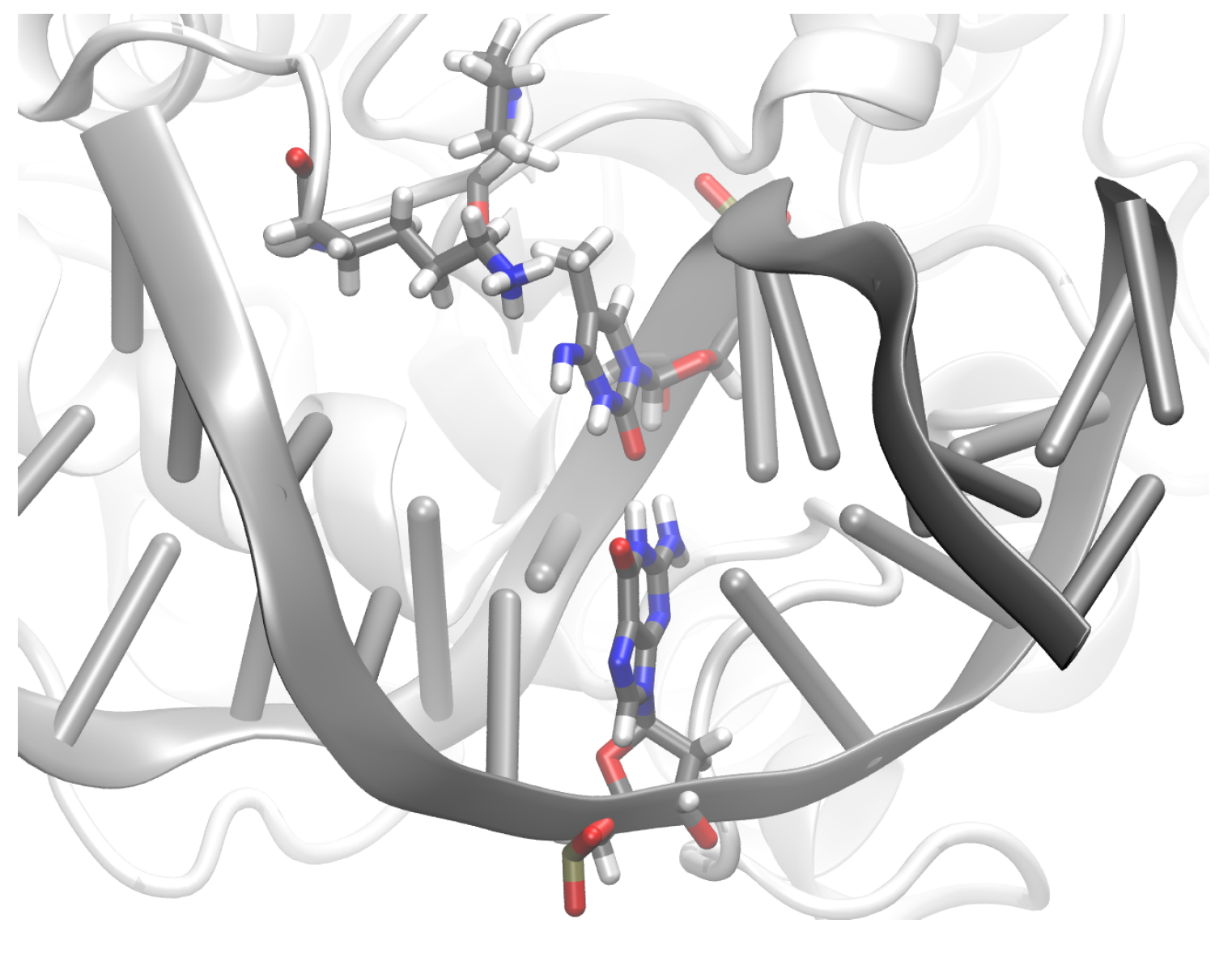
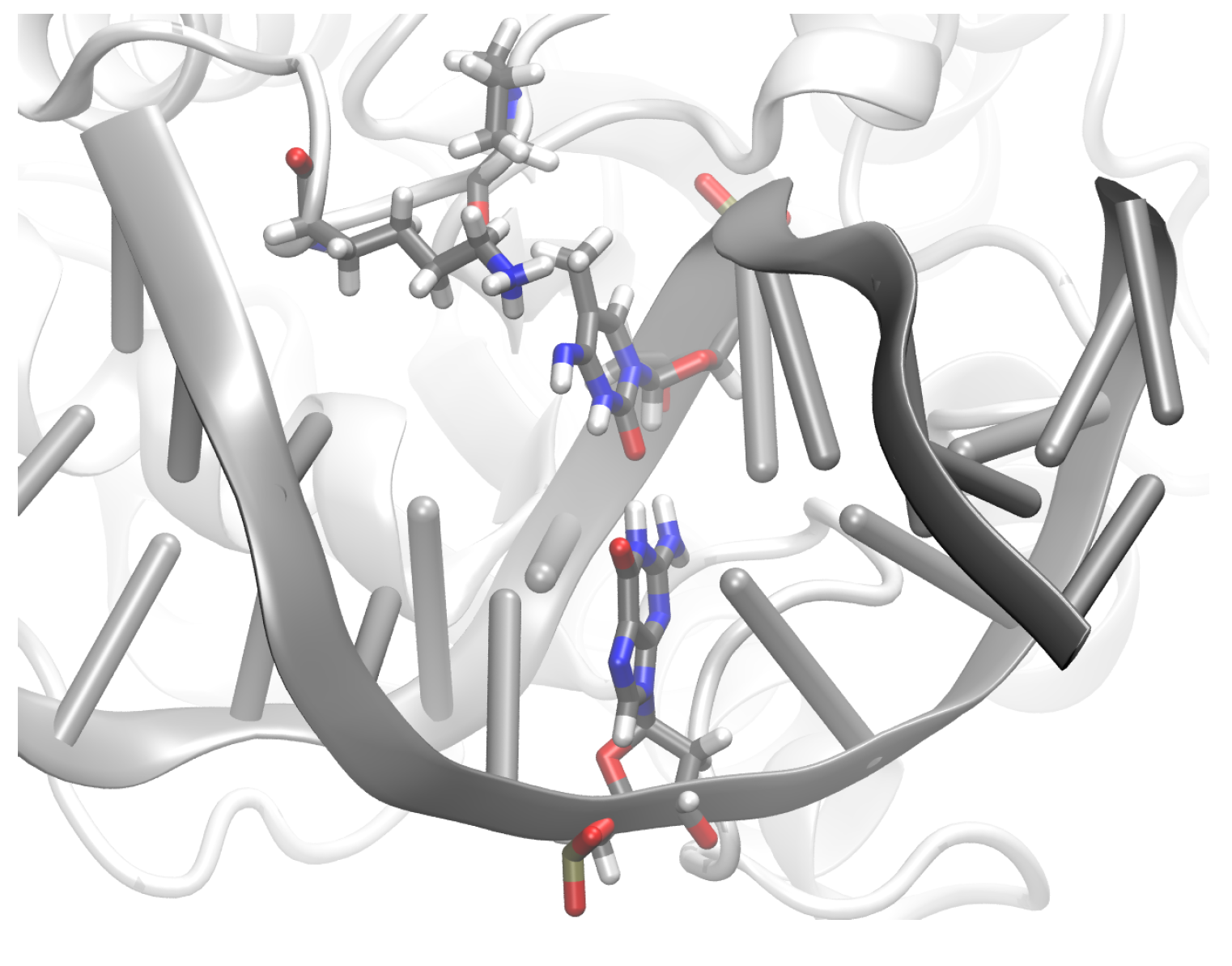
3.2. TDG-DNA Interactions
3.2.1. Hydrogen-Bond Interactions
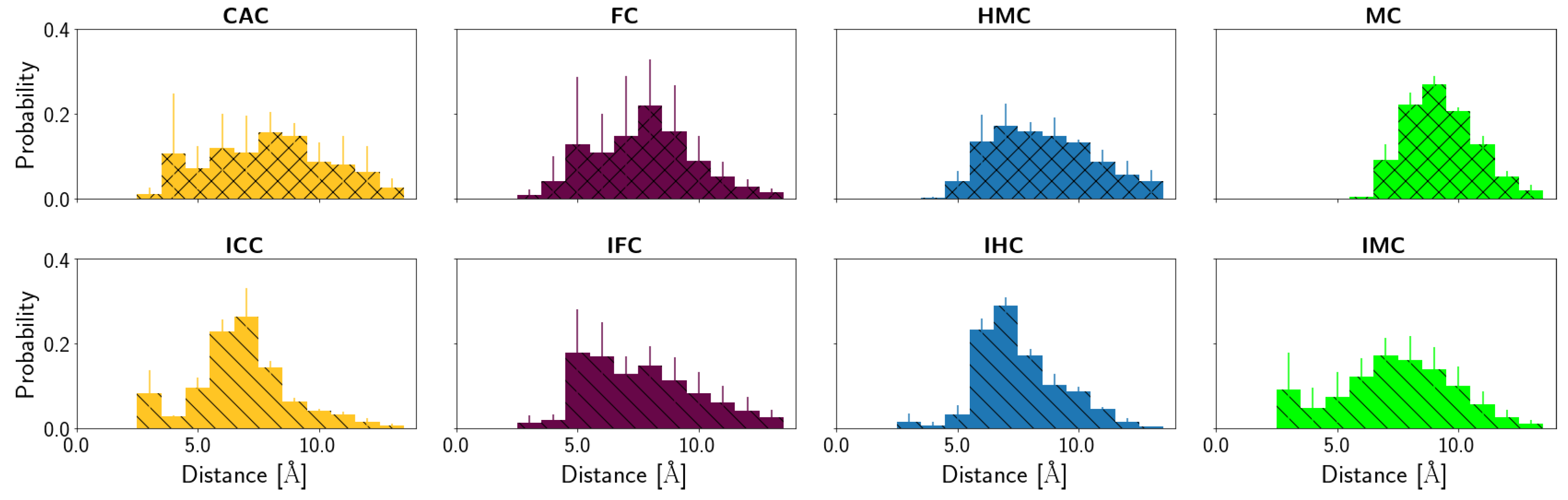
3.2.2. Protein-XC Distances
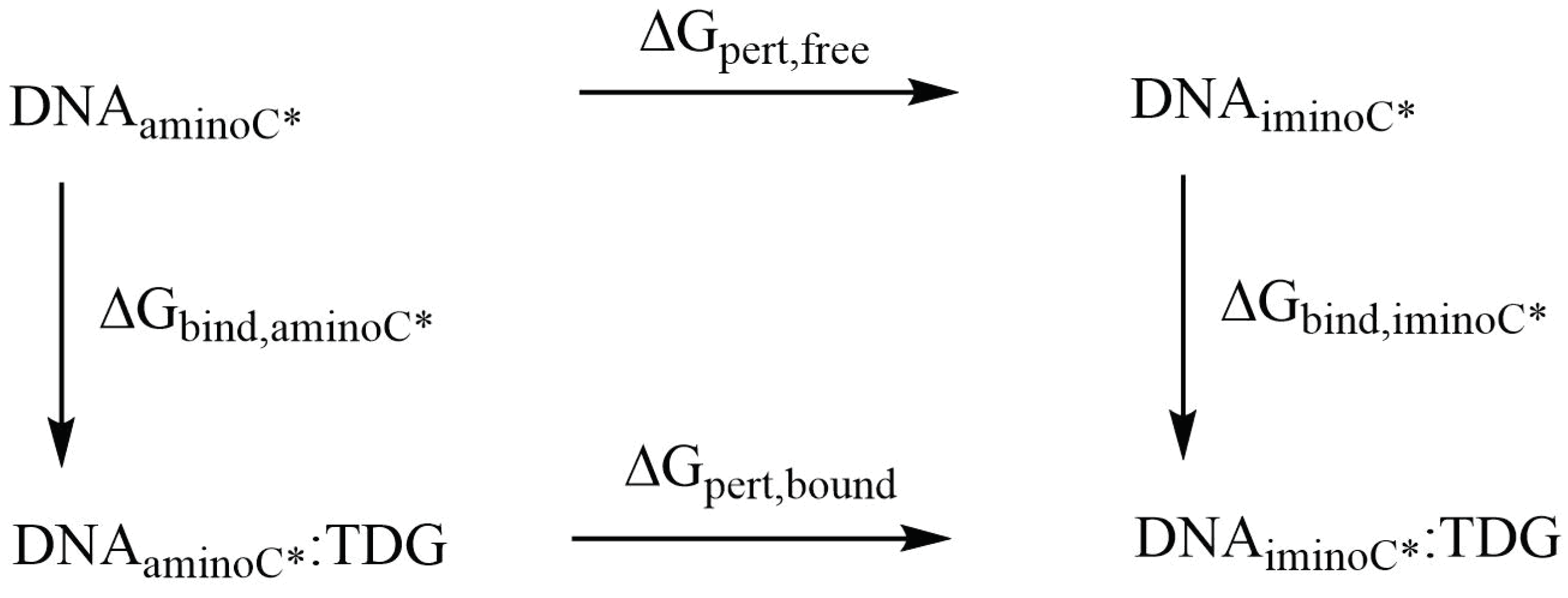
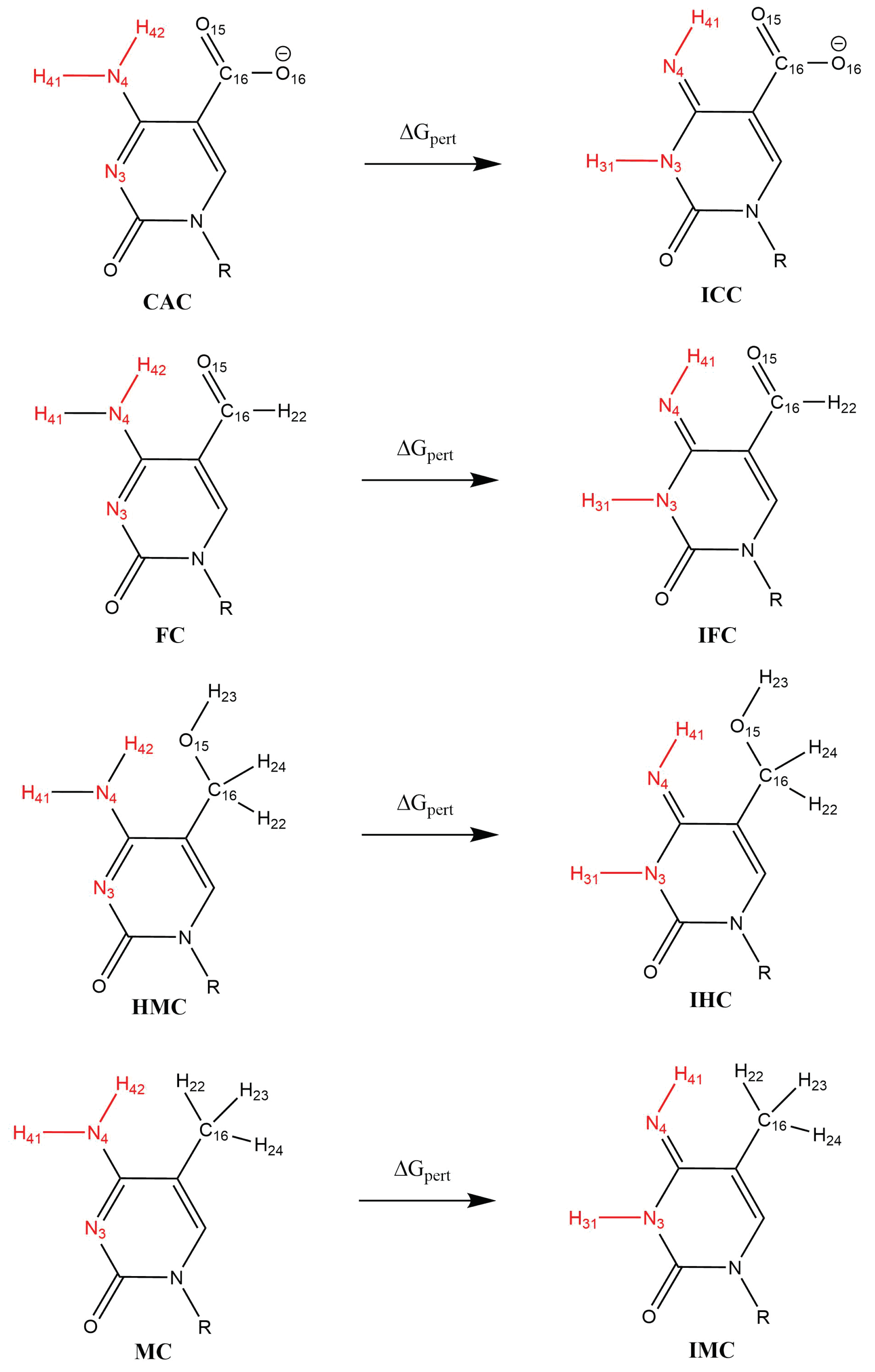
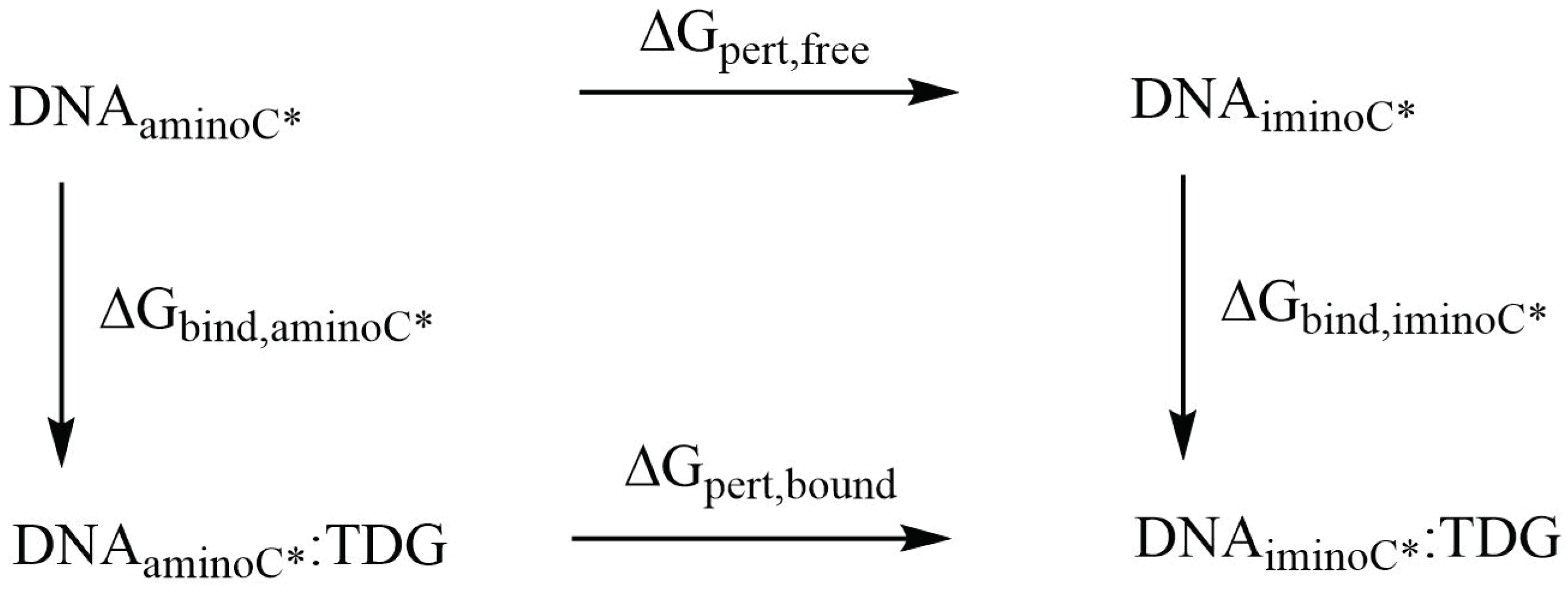
3.2.3. Relative Binding Affinities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- He, Y.; Li, B.Z.; Liu, Z.L.P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; Sun, Y.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.; Collins, L.; Swenberg, J.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA Glycosylase Can Rapidly Excise 5-Formylcytosine and 5-Carboxylcytosine potential implications for active demethylatio of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, J.H.; Hong, S.; Bhagwat, A.S.; Zhang, X.; Cheng, X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: Its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 10203–10214. [Google Scholar] [CrossRef]
- Maiti, A.; Morgan, M.T.; Pozharski, E.; Drohat, A.C. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. USA 2008, 105, 8890–8895. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Zhang, X.; Cheng, X. Activity and crystal structure of human thymine DNA glycosylase mutant N140A with 5-carboxylcytosine DNA at low pH. DNA Repair 2013, 12, 535–540. [Google Scholar] [CrossRef] [Green Version]
- Coey, C.T.; Malik, S.S.; Pidugu, L.S.; Varney, K.M.; Pozharski, E.; Drohat, A.C. Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res. 2016, 44, 10248–10258. [Google Scholar] [CrossRef]
- Stivers, J.T. Extrahelical damaged base recognition by DNA glycosylase enzymes. Chem.-A Eur. J. 2008, 14, 786–793. [Google Scholar] [CrossRef]
- Friedman, J.I.; Stivers, J.T. Detection of Damaged DNA Bases by DNA Glycosylase Enzymes. Biochemistry 2010, 49, 4957–4967. [Google Scholar] [CrossRef] [Green Version]
- Da, L.T.; Yu, J. Base-flipping dynamics from an intrahelical to an extrahelical state exerted by thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 2018, 46, 5410–5425. Available online: https://academic.oup.com/nar/article-pdf/46/11/5410/25067247/gky386.pdf (accessed on 30 July 2021). [CrossRef] [PubMed]
- Maiti, A.; Michelson, A.Z.; Armwood, C.J.; Lee, J.K.; Drohat, A.C. Divergent Mechanisms for Enzymatic Excision of 5-Formylcytosine and 5-Carboxylcytosine from DNA. J. Am. Chem. Soc. 2013, 135, 15813–15822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drohat, A.C.; Maitia, A. Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem. 2014, 12, 8367–8378. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Drohat, A. Dependance of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and provessing ofG:T mispairs. DNA Repair 2011, 10, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.T.; Wang, Y. A Density Functional Theory Study on the Kinetics and Thermodynamics of N-Glycosidic Bond Cleavage in 5-Substituted 2′-Deoxycytidines. Biochemistry 2012, 51, 6458–6462. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.E.R.; Lenz, S.A.P.; Wetmore, S.D. DFT Study on the Deglycosylation of Methylated, Oxidized, and Canonical Pyrimidine Nucleosides in Water: Implications for Epigenetic Regulation and DNA Repair. J. Phys. Chem. B 2020, 124, 2392–2400. [Google Scholar] [CrossRef]
- Kaur, R.; Nikkel, D.J.; Wetmore, S.D. Computational studies of DNA repair: Insights into the function of monofunctional DNA glycosylases in the base excision repair pathway. WIREs Comput. Mol. Sci. 2020, 10, e1471. Available online: https://wires.onlinelibrary.wiley.com/doi/pdf/10.1002/wcms.1471 (accessed on 30 July 2021). [CrossRef]
- Bennett, M.; Rodgers, M.; Hebert, A.; Ruslander, L.; Eisele, L.; Drohat, A. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. JACS 2006, 128, 12510–12519. [Google Scholar] [CrossRef] [Green Version]
- Kanaan, N.; Crehuet, R.; Imhof, P. Mechanism of the Glycosidic Bond Cleavage of mismatched Thymine in Human Thymine DNA Glycosylase Revealed by Quantum Mechanical/Molecular Mechanical Calculations. J. Phys. Chem. B 2015, 119, 12365–12380. [Google Scholar] [CrossRef]
- Naydenova, E.; Dietschreit, J.C.B.; Ochsenfeld, C. Reaction Mechanism for the N-Glycosidic Bond Cleavage of 5-Formylcytosine by Thymine DNA Glycosylase. J. Phys. Chem. B 2019, 123, 4173–4179. [Google Scholar] [CrossRef]
- Imhof, P.; Zahran, M. The effect of a G:T mispair on the dynamics of DNA. PLoS ONE 2013, 8, e53305. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.; Imhof, P. Interactions of the DNA Repair Enzyme human Thymine DNA Glycosylase with cognate and non-cognate DNA. Biochemistry 2018, 57, 5654–5665. [Google Scholar] [CrossRef] [PubMed]
- Helabad, M.B.; Kanaan, N.; Imhof, P. Base-flip in DNA studied by Molecular Dynamics Simulations of differently oxidised forms of methyl-Cytosine. Int. J. Mol. Sci. 2014, 15, 11799–11816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, T.T.M.; Yoo, J.; Dai, Q.; Zhang, Q.; He, C.; Aksimentiev, A.; Ha, T. Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat. Commun. 2016, 7, 10813. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; Liu, L.; Yang, Q.L.; Wang, Y.; Xu, P.; Zhang, L.; Liu, S.; Dai, Q.; Ji, Q.; Xu, G.L.; et al. Thymine DNA glycosylase recognizes the geometry alteration of minor grooves induced by 5-formylcytosine and 5-carboxylcytosine. Chem. Sci. 2019, 10, 7407–7417. [Google Scholar] [CrossRef] [Green Version]
- Sanstead, P.J.; Ashwood, B.; Dai, Q.; He, C.; Tokmakoff, A. Oxidized Derivatives of 5-Methylcytosine Alter the Stability and Dehybridization Dynamics of Duplex DNA. J. Phys. Chem. B 2020, 124, 1160–1174. [Google Scholar] [CrossRef]
- Szulik, M.W.; Pallan, P.S.; Nocek, B.; Voehler, M.; Banerjee, S.; Brooks, S.; Joachimiak, A.; Egli, M.; Eichman, B.F.; Stone, M.P. Differential Stabilities and Sequence-Dependent Base Pair Opening Dynamics of Watson–Crick Base Pairs with 5-Hydroxymethylcytosine, 5-Formylcytosine, or 5-Carboxylcytosine. Biochemistry 2015, 54, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Sanstead, P.J.; Peng, C.S.; Han, D.; He, C.; Tokmakoff, A. Weakened N3 Hydrogen Bonding by 5-Formylcytosine and 5-Carboxylcytosine Reduces Their Base-Pairing Stability. ACS Chem. Biol. 2016, 11, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Pidugu, L.S.; Dai, Q.; Malik, S.S.; Pozharski, E.; Drohat, A.C. Excision of 5-Carboxylcytosine by Thymine DNA Glycosylase. J. Am. Chem. Soc. 2019, 141, 18851–18861. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018; Available online: http://ambermd.org (accessed on 30 July 2021).
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020; Available online: https://ambermd.org/doc12/Amber20.pdf (accessed on 30 July 2021).
- Beierlein, F.R.; Clark, T.; Braunschweig, B.; Engelhardt, K.; Glas, L.; Peukert, W. Carboxylate Ion Pairing with Alkali-Metal Ions for β-Lactoglobulin and Its Role on Aggregation and Interfacial Adsorption. J. Phys. Chem. B 2015, 119, 5505–5517. [Google Scholar] [CrossRef] [Green Version]
- Beierlein, F.R.; Paradas Palomo, M.; Sharapa, D.I.; Zozulia, O.; Mokhir, A.; Clark, T. DNA-Dye-Conjugates: Conformations and Spectra of Fluorescence Probes. PLoS ONE 2016, 11, e0160229. [Google Scholar] [CrossRef]
- Hardwick, J.S.; Haugland, M.M.; El-Sagheer, A.H.; Ptchelkine, D.; Beierlein, F.R.; Lane, A.N.; Brown, T.; Lovett, J.E.; Anderson, E.A. 2′-Alkynyl spin-labelling is a minimally perturbing tool for DNA structural analysis. Nucleic Acids Res. 2020, 48, 2830–2840. [Google Scholar] [CrossRef]
- Zozulia, O.; Bachmann, T.; Deussner-Helfmann, N.S.; Beierlein, F.; Heilemann, M.; Mokhir, A. Red light-triggered nucleic acid-templated reaction based on cyclic oligonucleotide substrates. Chem. Commun. 2019, 55, 10713–10716. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Eberlein, L.; Beierlein, F.R.; van Eikema Hommes, N.J.R.; Radadiya, A.; Heil, J.; Benner, S.A.; Clark, T.; Kast, S.M.; Richards, N.G.J. Tautomeric Equilibria of Nucleobases in the Hachimoji Expanded Genetic Alphabet. J. Chem. Theory Comput. 2020, 16, 2766–2777. [Google Scholar] [CrossRef]
- Lankaš, F.; Cheatham, T.E.; Špačáková, N.; Hobza, P.; Langowski, J.; Šponer, J. Critical Effect of the N2 Amino Group on Structure, Dynamics, and Elasticity of DNA Polypurine Tracts. Biophys. J. 2002, 82, 2592–2609. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Tutorial A4. AMBER Web Site. Available online: http://ambermd.org/tutorials/advanced/tutorial4/ (accessed on 30 July 2021).
- Zacharias, M. Atomic Resolution Insight into Sac7d Protein Binding to DNA and Associated Global Changes by Molecular Dynamics Simulations. Angew. Chem. (Int. Ed. Engl.) 2019, 58, 5967–5972. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Blanchet, C.; Pasi, M.; Zakrzewska, K.; Lavery, R. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 2011, 39, W68–W73. [Google Scholar] [CrossRef] [Green Version]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef] [Green Version]
- Tutorial A9. AMBER Web Site. Available online: http://ambermd.org/tutorials/advanced/tutorial9/ (accessed on 30 July 2021).
- Beierlein, F.R.; Kneale, G.G.; Clark, T. Predicting the Effects of Basepair Mutations in DNA-Protein Complexes by Thermodynamic Integration. Biophys. J. 2011, 101, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Mobley, D.L.; Case, D.A. Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations. J. Chem. Phys. 2007, 127, 214108. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
|---|---|---|---|---|---|---|---|---|
| G17:O6-XC6:N4 | 0.91 ± 0.02 | 0.96 ± 0.00 | 0.93 ± 0.00 | 0.93 ± 0.00 | – | – | – | – |
| XC6:N3-G17:N1 | 0.98 ± 0.01 | 0.99 ± 0.00 | 0.98 ± 0.00 | 0.98 ± 0.00 | – | – | – | – |
| XC6:O2-G17:N2 | 0.98 ± 0.01 | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | – | – | – | – |
| G17:O6-XC6:N3 | – | – | – | – | 0.83 ± 0.08 | 0.73 ± 0.02 | 0.76 ± 0.02 | 0.76 ± 0.04 |
| XC6:O2-G17:N1 | – | – | – | – | 0.95 ± 0.02 | 0.92 ± 0.01 | 0.92 ± 0.01 | 0.93 ± 0.01 |
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
| G17:O6-XC6:N4 | 0.92 ± 0.02 | 0.91 ± 0.07 | 0.89 ± 0.02 | 0.85 ± 0.09 | – | – | – | – |
| XC6:N3-G17:N1 | 0.98 ± 0.01 | 0.98 ± 0.01 | 0.98 ± 0.01 | 0.97 ± 0.02 | – | – | – | – |
| XC6:O2-G17:N2 | 0.97 ± 0.03 | 0.99 ± 0.00 | 1.00 ± 0.00 | 0.99 ± 0.00 | – | – | 0.59 ± 0.22 | 0.66 ± 0.18 |
| G17:O6-XC6:N3 | – | – | – | – | 0.66 ± 0.22 | 0.68 ± 0.30 | – | – |
| XC6:O2-G17:N1 | – | – | – | – | 0.90 ± 0.07 | 0.88 ± 0.10 | 0.64 ± 0.28 | 0.69 ± 0.15 |
| Free DNA | Complex | |
|---|---|---|
| CAC | 0.00 ± 0.00 | 0.01 ± 0.01 |
| FC | 0.00 ± 0.00 | 0.00 ± 0.00 |
| HMC | 0.00 ± 0.00 | 0.00 ± 0.00 |
| MC | 0.00 ± 0.00 | 0.00 ± 0.00 |
| ICC | 0.61 ± 0.02 | 0.35 ± 0.20 |
| IFC | 0.64 ± 0.00 | 0.50 ± 0.19 |
| IHC | 0.62 ± 0.01 | 0.48 ± 0.09 |
| IMC | 0.64 ± 0.01 | 0.52 ± 0.12 |
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
|---|---|---|---|---|---|---|---|---|
| G17:N3-W | 0.65 ± 0.01 | 0.62 ± 0.01 | 0.62 ± 0.01 | 0.61 ± 0.02 | 0.66 ± 0.01 | 0.61 ± 0.01 | 0.63 ± 0.01 | 0.62 ± 0.01 |
| G17:N7-W | 0.81 ± 0.00 | 0.81 ± 0.01 | 0.81 ± 0.01 | 0.82 ± 0.00 | 0.85 ± 0.00 | 0.81 ± 0.00 | 0.76 ± 0.02 | 0.80 ± 0.01 |
| G17:O6-W | 0.97 ± 0.02 | 0.88 ± 0.01 | 0.94 ± 0.02 | 0.95 ± 0.01 | – | 0.73 ± 0.03 | 0.80 ± 0.09 | 0.68 ± 0.03 |
| XC6:O15-W | 1.79 ± 0.05 | 0.95 ± 0.01 | 0.76 ± 0.01 | – | 1.89 ± 0.04 | 0.95 ± 0.01 | 0.68 ± 0.01 | – |
| XC6:O16-W | 1.83 ± 0.06 | – | – | – | 2.09 ± 0.04 | – | – | – |
| XC6:O2-W | 0.86 ± 0.02 | 0.79 ± 0.01 | 0.86 ± 0.00 | 0.88 ± 0.01 | 0.72 ± 0.07 | – | – | – |
| XC6:N4-W | – | – | – | – | – | 0.89 ± 0.01 | 1.06 ± 0.15 | 0.94 ± 0.07 |
| W-XC6:O15 | – | – | 0.84 ± 0.01 | – | – | – | 0.81 ± 0.00 | – |
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
| G17:N3-W | 0.63 ± 0.11 | 0.52 ± 0.30 | 0.68 ± 0.13 | 0.53 ± 0.20 | – | 0.52 ± 0.19 | – | 0.55 ± 0.32 |
| G17:N7-W | 0.77 ± 0.02 | 0.80 ± 0.01 | 0.82 ± 0.04 | 0.86 ± 0.11 | 0.81 ± 0.01 | 0.79 ± 0.03 | 0.82 ± 0.05 | 0.80 ± 0.02 |
| G17:O6-W | 0.91 ± 0.06 | 0.89 ± 0.04 | 0.92 ± 0.05 | 1.02 ± 0.16 | – | 0.74 ± 0.23 | 1.06 ± 0.34 | 1.18 ± 0.37 |
| XC6:O15-W | 1.68 ± 0.28 | 0.94 ± 0.10 | 0.59 ± 0.10 | – | 1.67 ± 0.18 | 0.75 ± 0.18 | – | – |
| XC6:O16-W | 1.57 ± 0.20 | – | – | – | 1.79 ± 0.09 | – | – | – |
| XC6:O2-W | 0.68 ± 0.27 | – | – | – | – | – | – | – |
| XC6:N4-W | – | – | – | – | – | 0.87 ± 0.13 | 1.14 ± 0.16 | 1.08 ± 0.14 |
| W-XC6:O15 | – | – | 0.86 ± 0.03 | – | – | – | 0.84 ± 0.04 | – |
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
|---|---|---|---|---|---|---|---|---|
| XC6:O15-LYS201:NZ | 0.24 ± 0.27 | – | – | – | 0.14 ± 0.17 | – | 0.17 ± 0.14 | – |
| XC6:O3’-ARG275:NH | 0.21 ± 0.19 | – | – | – | – | – | – | – |
| XC6:OP-SER200:N | – | 0.47 ± 0.43 | 0.27 ± 0.45 | 0.82 ± 0.06 | – | 0.28 ± 0.49 | 0.54 ± 0.47 | 0.32 ± 0.37 |
| XC6:OP-SER200:OG | – | 0.32 ± 0.50 | 0.19 ± 0.32 | 0.87 ± 0.11 | – | 0.17 ± 0.30 | 0.38 ± 0.39 | 0.22 ± 0.33 |
| XC6:OP-LYS201:N | – | 0.24 ± 0.42 | – | 0.40 ± 0.30 | – | – | 0.34 ± 0.30 | – |
| XC6:OP-LEU143:N | – | – | 0.43 ± 0.37 | – | – | 0.53 ± 0.46 | 0.12 ± 0.22 | 0.29 ± 0.38 |
| XC6:OP-MET144:N | – | – | 0.25 ± 0.40 | – | – | – | – | – |
| XC6:OP-GLY199:N | – | – | 0.22 ± 0.38 | – | – | – | 0.13 ± 0.22 | – |
| Acc-Don | CAC | FC | HMC | MC | ICC | IFC | IHC | IMC |
|---|---|---|---|---|---|---|---|---|
| G7:OP-SER273:OG | – | – | 0.60 ± 0.24 | – | – | – | – | – |
| T8:OP-LYS232:N | – | – | 0.54 ± 0.46 | – | 0.91 ± 0.06 | – | 0.79 ± 0.15 | 0.87 ± 0.04 |
| T8:OP-CYS233:N | – | – | 0.62 ± 0.54 | – | 0.88 ± 0.07 | – | 0.61 ± 0.53 | 0.92 ± 0.01 |
| T8:OP-SER271:N | – | – | 0.56 ± 0.35 | – | – | – | 0.73 ± 0.37 | – |
| A9:OP-LYS232:NZ | – | – | 0.85 ± 0.01 | – | 0.59 ± 0.23 | 0.57 ± 0.36 | 0.86 ± 0.03 | 0.75 ± 0.14 |
| T18:O2-ARG275:NH | 0.60 ± 0.71 | 1.13 ± 0.75 | 1.29 ± 0.58 | 0.96 ± 0.89 | – | 1.05 ± 0.74 | 1.07 ± 0.58 | 0.58 ± 0.39 |
| G19:N3-ARG275:NH | – | – | – | – | – | 0.70 ± 0.35 | – | – |
| XC | |
|---|---|
| CAC→ICC | 0.17 ± 1.10 |
| FC→IFC | 2.26 ± 0.23 |
| HMC→IHC | −0.65 ± 0.43 |
| MC→IMC | 2.37 ± 0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkenandt, S.; Beierlein, F.; Imhof, P. Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms. Molecules 2021, 26, 5728. https://doi.org/10.3390/molecules26195728
Volkenandt S, Beierlein F, Imhof P. Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms. Molecules. 2021; 26(19):5728. https://doi.org/10.3390/molecules26195728
Chicago/Turabian StyleVolkenandt, Senta, Frank Beierlein, and Petra Imhof. 2021. "Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms" Molecules 26, no. 19: 5728. https://doi.org/10.3390/molecules26195728
APA StyleVolkenandt, S., Beierlein, F., & Imhof, P. (2021). Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms. Molecules, 26(19), 5728. https://doi.org/10.3390/molecules26195728