
The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
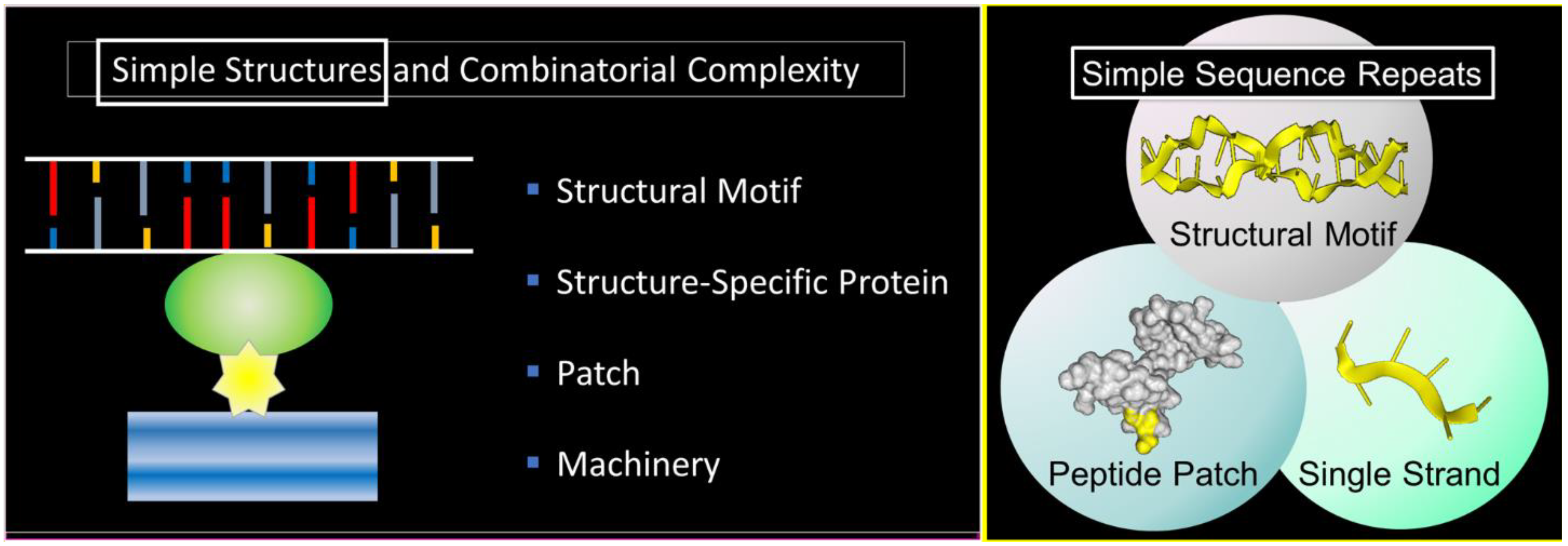
1. Starting Simple
2. Condensates
3. Classical Genetics
4. Simple Sequence Repeats
5. SSR Genomic Spread
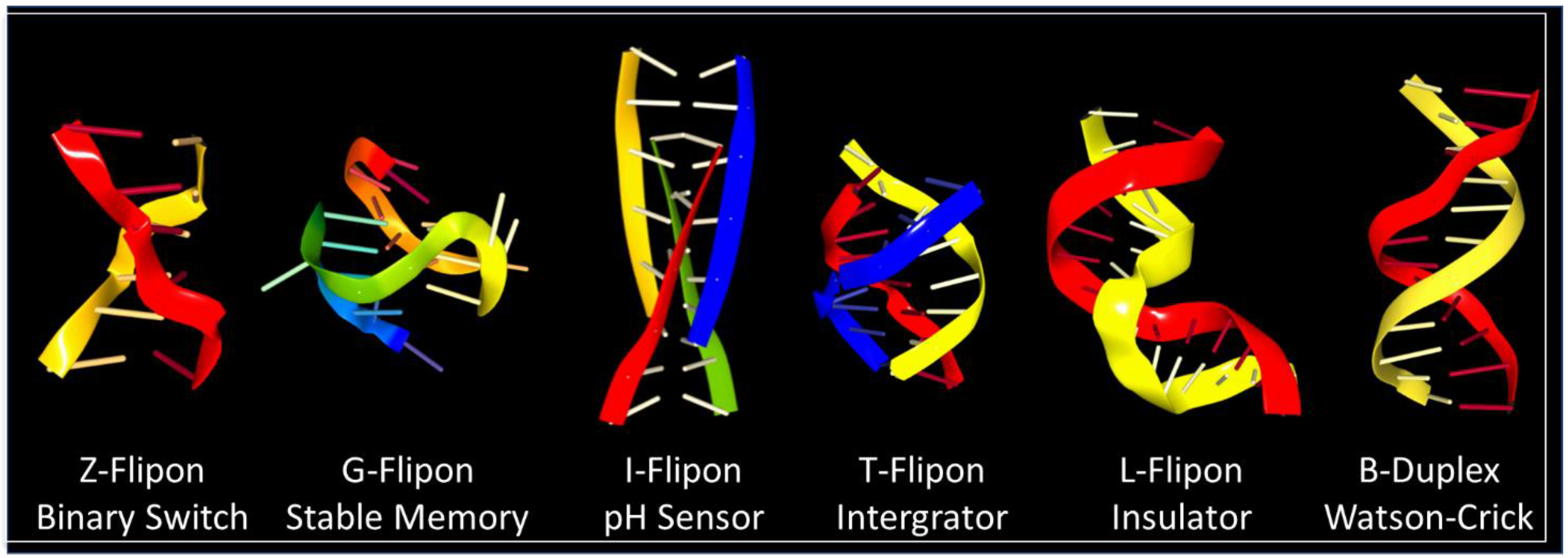
6. Flipon Genetics
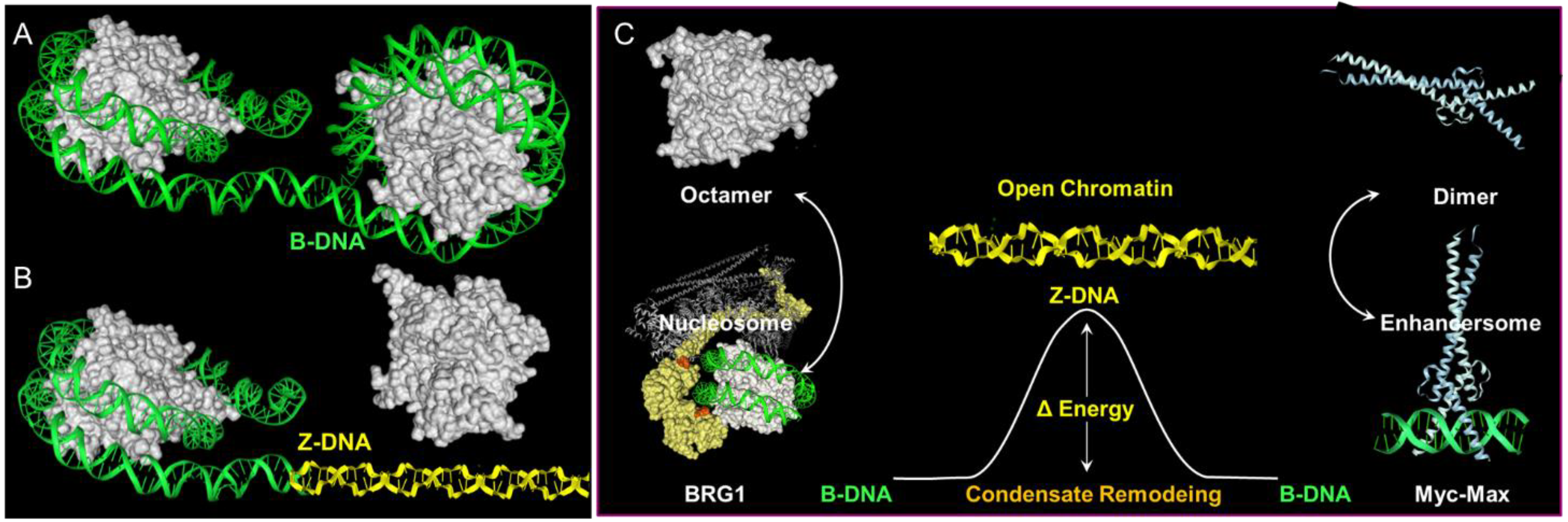
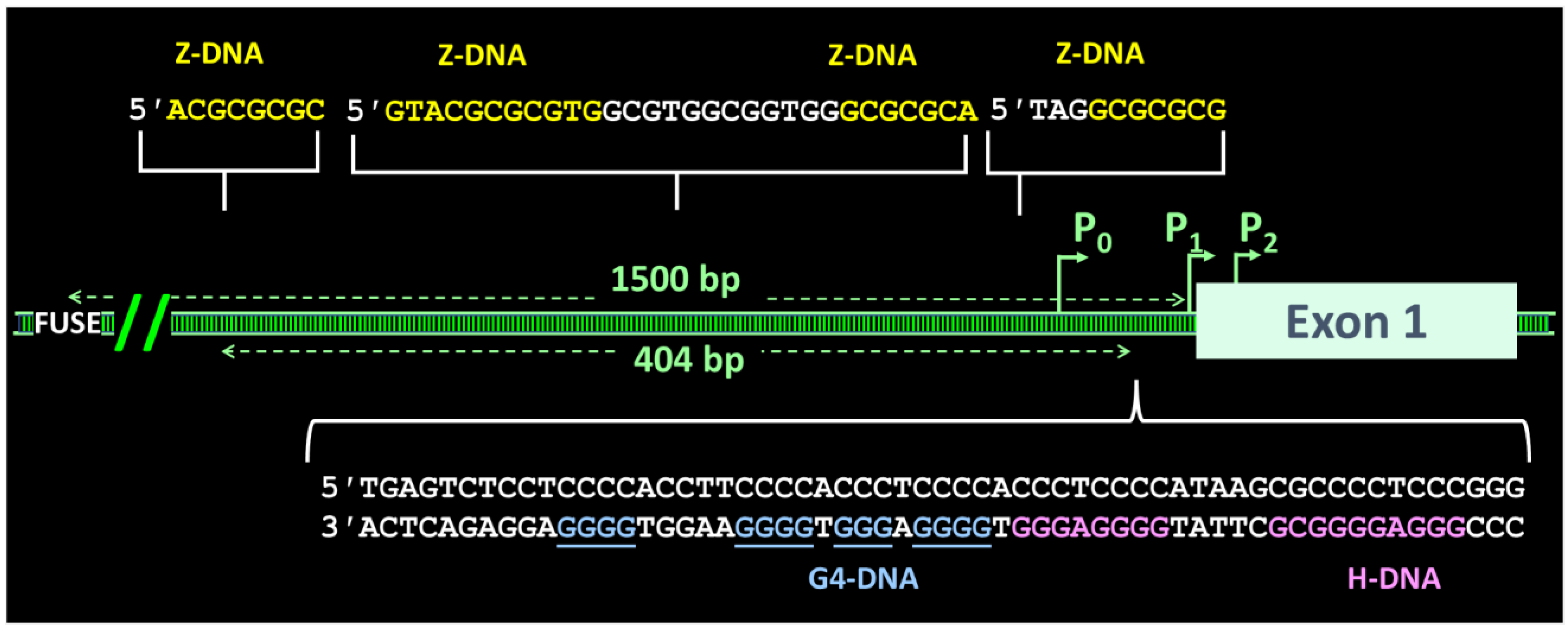
7. Z-Flipons
8. G-Flipons
9. Flipons as Scaffolds for Condensates
10. The Search for Better Outcomes
11. When Flipons and Codons Clash
12. When Flipons Hang out Together
13. Evolutionary Selection at the Cellular Level
14. Missing Heritability
15. Future Directions
16. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; McAtee, C.K.; Su, X. Phase separation in immune signalling. Nat. Rev. Immunol. 2021, 1–12. [Google Scholar] [CrossRef]
- Nott, T.J.; Craggs, T.D.; Baldwin, A.J. Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat. Chem. 2016, 8, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Honson, D.; Guttman, M. Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol. 2021, 1–18. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S. Intrinsically disordered protein regions and phase separation: Sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci. 2020, 4, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Riback, J.A.; Zhu, L.; Ferrolino, M.C.; Tolbert, M.; Mitrea, D.M.; Sanders, D.W.; Wei, M.T.; Kriwacki, R.W.; Brangwynne, C.P. Composition-dependent thermodynamics of intracellular phase separation. Nature 2020, 581, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.; Jana, T.; Mittelman, K.; Chapal, M.; Kumar, D.K.; Carmi, M.; Barkai, N. Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 2020, 79, 459–471.e4. [Google Scholar] [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. J. Mol. Biol. 2018, 430, 4666–4684. [Google Scholar] [CrossRef]
- Fay, M.M.; Anderson, P.J. The Role of RNA in Biological Phase Separations. J. Mol. Biol. 2018, 430, 4685–4701. [Google Scholar] [CrossRef]
- Frank, L.; Rippe, K. Repetitive RNAs as regulators of chromatin-associated subcompartment formation by phase separation. J. Mol. Biol. 2020, 432, 4270–4286. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28. [Google Scholar] [CrossRef]
- Yamazaki, T.; Nakagawa, S.; Hirose, T. Architectural RNAs for Membraneless Nuclear Body Formation. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 227–237. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillen-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef]
- Steitz, T.A. Structural studies of protein-nucleic acid interaction: The sources of sequence-specific binding. Q. Rev. Biophys. 1990, 23, 205–280. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef]
- Louka, A.; Zacco, E.; Temussi, P.A.; Tartaglia, G.G.; Pastore, A. RNA as the stone guest of protein aggregation. Nucleic Acids Res. 2020, 48, 11880–11889. [Google Scholar] [CrossRef]
- Fruton, J.S. Proteolytic Enzymes as Specific Agents in the Formation and Breakdown of Proteins. Cold Spring Harb. Symp. Quant. Biol. 1941, 9, 211–217. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Pankert, T.; Seiler, J.; Nemeth, A.; Voit, R.; Grummt, I.; Rippe, K. Alu element-containing RNAs maintain nucleolar structure and function. EMBO J. 2015, 34, 2758–2774. [Google Scholar] [CrossRef]
- Herbert, A. To “Z” or not to “Z”: Z-RNA, self-recognition, and the MDA5 helicase. PLoS Genet. 2021, 17, e1009513. [Google Scholar] [CrossRef]
- Bagshaw, A.T.M. Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 2018, 19, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.A.; Ruggiero, R.P.; Cox, C.; Wacholder, A.C.; Pollock, D.D. Finding and extending ancient simple sequence repeat-derived regions in the human genome. Mob. DNA 2020, 11, 11. [Google Scholar] [CrossRef]
- Herbert, A. Simple Repeats as Building Blocks for Genetic Computers. Trends Genet. 2020, 36, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2015, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Caporale, L.H. Chance favors the prepared genome. Ann. N. Y. Acad. Sci. 1999, 870, 1–21. [Google Scholar] [CrossRef]
- Xie, K.T.; Wang, G.; Thompson, A.C.; Wucherpfennig, J.I.; Reimchen, T.E.; MacColl, A.D.C.; Schluter, D.; Bell, M.A.; Vasquez, K.M.; Kingsley, D.M. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 2019, 363, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Pochet, N.; Pak, T.; Verstrepen, K.J. Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res. 2007, 17, 1787–1796. [Google Scholar] [CrossRef]
- Fazal, S.; Danzi, M.C.; Cintra, V.P.; Bis-Brewer, D.M.; Dolzhenko, E.; Eberle, M.A.; Zuchner, S. Large scale in silico characterization of repeat expansion variation in human genomes. Sci. Data 2020, 7, 294. [Google Scholar] [CrossRef]
- Champ, P.C.; Maurice, S.; Vargason, J.M.; Camp, T.; Ho, P.S. Distributions of Z-DNA and nuclear factor I in human chromosome 22: A model for coupled transcriptional regulation. Nucleic Acids Res. 2004, 32, 6501–6510. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Chambers, V.S.; Sahakyan, A.B.; McCauley, P.; Boutell, J.M.; Antonio, M.D.; Balasubramanian, S. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 2019, 47, 3862–3874. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhou, J.; Wallace, S.S.; Burrows, C.J. A Role for the Fifth G-Track in G-Quadruplex Forming Oncogene Promoter Sequences during Oxidative Stress: Do These “Spare Tires” Have an Evolved Function? ACS Cent. Sci. 2015, 1, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, E.A.; Kumar, A.S.; Sowpati, D.T.; Mishra, R.K. Single Amino Acid Repeats in the Proteome World: Structural, Functional, and Evolutionary Insights. PLoS ONE 2016, 11, e0166854. [Google Scholar] [CrossRef]
- Delucchi, M.; Schaper, E.; Sachenkova, O.; Elofsson, A.; Anisimova, M. A New Census of Protein Tandem Repeats and Their Relationship with Intrinsic Disorder. Genes 2020, 11, 407. [Google Scholar] [CrossRef]
- Chong, P.A.; Vernon, R.M.; Forman-Kay, J.D. RGG/RG Motif Regions in RNA Binding and Phase Separation. J. Mol. Biol. 2018, 430, 4650–4665. [Google Scholar] [CrossRef]
- Hsin, J.P.; Manley, J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef]
- Napolitano, G.; Lania, L.; Majello, B. RNA polymerase II CTD modifications: How many tales from a single tail. J. Cell. Physiol. 2014, 229, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. A Genetic Instruction Code Based on DNA Conformation. Trends Genet. 2019, 35, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. ALU non-B-DNA conformations, flipons, binary codes and evolution. R. Soc. Open Sci. 2020, 7, 200222. [Google Scholar] [CrossRef]
- Mao, S.Q.; Ghanbarian, A.T.; Spiegel, J.; Martinez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.; Rould, M.A.; Lowenhaupt, K.; Herbert, A.; Rich, A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 1999, 284, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Placido, D.; Brown, B.A., 2nd; Lowenhaupt, K.; Rich, A.; Athanasiadis, A. A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure 2007, 15, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Nakahama, T.; Yamasaki, R.; Costa Cruz, P.H.; Vongpipatana, T.; Inoue, M.; Kanou, N.; Xing, Y.; Todo, H.; Shibuya, T.; et al. RNA editing at a limited number of sites is sufficient to prevent MDA5 activation in the mouse brain. PLoS Genet. 2021, 17, e1009516. [Google Scholar] [CrossRef]
- Takeuchi, H.; Hanamura, N.; Hayasaka, H.; Harada, I. B-Z transition of poly(dG-m5dC) induced by binding of Lys-containing peptides. FEBS Lett. 1991, 279, 253–255. [Google Scholar] [CrossRef]
- Li, J.; Wang, R.; Jin, J.; Han, M.; Chen, Z.; Gao, Y.; Hu, X.; Zhu, H.; Gao, H.; Lu, K.; et al. USP7 negatively controls global DNA methylation by attenuating ubiquitinated histone-dependent DNMT1 recruitment. Cell Discov. 2020, 6, 58. [Google Scholar] [CrossRef]
- Liu, H.; Mulholland, N.; Fu, H.; Zhao, K. Cooperative activity of BRG1 and Z-DNA formation in chromatin remodeling. Mol. Cell. Biol. 2006, 26, 2550–2559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ohta, T.; Maruyama, A.; Hosoya, T.; Nishikawa, K.; Maher, J.M.; Shibahara, S.; Itoh, K.; Yamamoto, M. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell. Biol. 2006, 26, 7942–7952. [Google Scholar] [CrossRef]
- Mulholland, N.; Xu, Y.; Sugiyama, H.; Zhao, K. SWI/SNF-mediated chromatin remodeling induces Z-DNA formation on a nucleosome. Cell Biosci. 2012, 2, 3. [Google Scholar] [CrossRef]
- McRae, E.K.S.; Booy, E.P.; Padilla-Meier, G.P.; McKenna, S.A. On Characterizing the Interactions between Proteins and Guanine Quadruplex Structures of Nucleic Acids. J. Nucleic Acids 2017, 2017, 9675348. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferre-D’Amare, A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Traczyk, A.; Liew, C.W.; Gill, D.J.; Rhodes, D. Structural basis of G-quadruplex DNA recognition by the yeast telomeric protein Rap1. Nucleic Acids Res. 2020, 48, 4562–4571. [Google Scholar] [CrossRef]
- Masuzawa, T.; Oyoshi, T. Roles of the RGG Domain and RNA Recognition Motif of Nucleolin in G-Quadruplex Stabilization. ACS Omega 2020, 5, 5202–5208. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Lo Sterzo, C.; Mori, M.; Di Matteo, A.; Bertini, I.; Banci, L.; Brunori, M.; Federici, L. Structure of nucleophosmin DNA-binding domain and analysis of its complex with a G-quadruplex sequence from the c-MYC promoter. J. Biol. Chem. 2012, 287, 26539–26548. [Google Scholar] [CrossRef]
- Zanotti, K.J.; Lackey, P.E.; Evans, G.L.; Mihailescu, M.-R. Thermodynamics of the Fragile X Mental Retardation Protein RGG Box Interactions with G Quartet Forming RNA. Biochemistry 2006, 45, 8319–8330. [Google Scholar] [CrossRef]
- Kondo, K.; Mashima, T.; Oyoshi, T.; Yagi, R.; Kurokawa, R.; Kobayashi, N.; Nagata, T.; Katahira, M. Plastic roles of phenylalanine and tyrosine residues of TLS/FUS in complex formation with the G-quadruplexes of telomeric DNA and TERRA. Sci. Rep. 2018, 8, 2864. [Google Scholar] [CrossRef]
- Vasilyev, N.; Polonskaia, A.; Darnell, J.C.; Darnell, R.B.; Patel, D.J.; Serganov, A. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. USA 2015, 112, E5391–E5400. [Google Scholar] [CrossRef]
- Lerner, L.K.; Sale, J.E. Replication of G Quadruplex DNA. Genes 2019, 10, 95. [Google Scholar] [CrossRef]
- Pipier, A.; Devaux, A.; Lavergne, T.; Adrait, A.; Couté, Y.; Britton, S.; Calsou, P.; Riou, J.F.; Defrancq, E.; Gomez, D. Constrained G4 structures unveil topology specificity of known and new G4 binding proteins. bioRxiv 2021. [Google Scholar] [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Zheng, K.W.; Zhang, J.Y.; He, Y.D.; Gong, J.Y.; Wen, C.J.; Chen, J.N.; Hao, Y.H.; Zhao, Y.; Tan, Z. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 2020, 48, 11706–11720. [Google Scholar] [CrossRef]
- Wang, E.; Thombre, R.; Shah, Y.; Latanich, R.; Wang, J. G-Quadruplexes as pathogenic drivers in neurodegenerative disorders. Nucleic Acids Res. 2021, 49, 4816–4830. [Google Scholar] [CrossRef]
- Maas, S.; Melcher, T.; Herb, A.; Seeburg, P.H.; Keller, W.; Krause, S.; Higuchi, M.; O’Connell, M.A. Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J. Biol. Chem. 1996, 271, 12221–12226. [Google Scholar] [CrossRef]
- Busa, V.F.; Leung, A.K.L. Thrown for a (stem) loop: How RNA structure impacts circular RNA regulation and function. Methods 2021, in press. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Matunis, M.J.; Pinol-Roma, S.; Burd, C.G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Potapova, T.A.; Gerton, J.L. Ribosomal DNA and the nucleolus in the context of genome organization. Chromosome Res. 2019, 27, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Mestre-Fos, S.; Penev, P.I.; Richards, J.C.; Dean, W.L.; Gray, R.D.; Chaires, J.B.; Williams, L.D. Profusion of G-quadruplexes on both subunits of metazoan ribosomes. PLoS ONE 2019, 14, e0226177. [Google Scholar] [CrossRef]
- Von Hacht, A.; Seifert, O.; Menger, M.; Schutze, T.; Arora, A.; Konthur, Z.; Neubauer, P.; Wagner, A.; Weise, C.; Kurreck, J. Identification and characterization of RNA guanine-quadruplex binding proteins. Nucleic Acids Res. 2014, 42, 6630–6644. [Google Scholar] [CrossRef]
- McKinney, J.A.; Wang, G.; Mukherjee, A.; Christensen, L.; Subramanian, S.H.S.; Zhao, J.; Vasquez, K.M. Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun. 2020, 11, 236. [Google Scholar] [CrossRef]
- Schrank, B.; Gautier, J. Assembling nuclear domains: Lessons from DNA repair. J. Cell Biol. 2019, 218, 2444–2455. [Google Scholar] [CrossRef]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.; Srinivasan, B.; Kus, K.; Mata, J.F.; Joao Amorim, M.; Jansen, L.E.T.; Athanasiadis, A. Enrichment of Zalpha domains at cytoplasmic stress granules is due to their innate ability to bind to nucleic acids. J. Cell Sci. 2021, 134, jcs258446. [Google Scholar] [CrossRef]
- Mompean, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253.e10. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K.R.; Benham, C.J.; Casellas, R.; Przytycka, T.M.; et al. Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 2017, 4, 344–356. [Google Scholar] [CrossRef]
- Khristich, A.N.; Mirkin, S.M. On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar] [CrossRef] [PubMed]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 2016, 5, e17820. [Google Scholar] [CrossRef] [PubMed]
- Kovanda, A.; Zalar, M.; Sket, P.; Plavec, J.; Rogelj, B. Anti-sense DNA d(GGCCCC)n expansions in C9ORF72 form i-motifs and protonated hairpins. Sci. Rep. 2015, 5, 17944. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Babić Leko, M.; Župunski, V.; Kirincich, J.; Smilović, D.; Hortobágyi, T.; Hof, P.R.; Šimić, G. Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion. Behav. Neurol. 2019, 2019, 1–18. [Google Scholar] [CrossRef]
- Asamitsu, S.; Yabuki, Y.; Ikenoshita, S.; Kawakubo, K.; Kawasaki, M.; Usuki, S.; Nakayama, Y.; Adachi, K.; Kugoh, H.; Ishii, K.; et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv. 2021, 7, eabd9440. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Kamikubo, H.; Onitsuka, M.; Kataoka, M.; Shortle, D. Conformational Preference of Polyglycine in Solution to Elongated Structure. J. Am. Chem. Soc. 2006, 128, 16338–16344. [Google Scholar] [CrossRef]
- Ellison, M.J.; Fenton, M.J.; Ho, P.S.; Rich, A. Long-range interactions of multiple DNA structural transitions within a common topological domain. EMBO J. 1987, 6, 1513–1522. [Google Scholar] [CrossRef]
- Ioshikhes, I.; Zhabinskaya, D.; Benham, C.J. Theoretical Analysis of Competing Conformational Transitions in Superhelical DNA. PLoS Comput. Biol. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb. Perspect. Med. 2013, 3, a014233. [Google Scholar] [CrossRef]
- Wittig, B.; Wolfl, S.; Dorbic, T.; Vahrson, W.; Rich, A. Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 1992, 11, 4653–4663. [Google Scholar] [CrossRef]
- Wang, W.; Hu, S.; Gu, Y.; Yan, Y.; Stovall, D.B.; Li, D.; Sui, G. Human MYC G-quadruplex: From discovery to a cancer therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188410. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef]
- Shepherd, J.W.; Greenall, R.J.; Probert, M.I.J.; Noy, A.; Leake, M.C. The emergence of sequence-dependent structural motifs in stretched, torsionally constrained DNA. Nucleic Acids Res. 2020, 48, 1748–1763. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Dubois, W.; Benham, C.; Batchelor, E.; Levens, D. FUBP1 and FUBP2 enforce distinct epigenetic setpoints for MYC expression in primary single murine cells. Commun. Biol. 2020, 3, 545. [Google Scholar] [CrossRef]
- Avigan, M.I.; Strober, B.; Levens, D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J. Biol. Chem. 1990, 265, 18538–18545. [Google Scholar] [CrossRef]
- Wiktor-Brown, D.M.; Hendricks, C.A.; Olipitz, W.; Engelward, B.P. Age-dependent accumulation of recombinant cells in the mouse pancreas revealed by in situ fluorescence imaging. Proc. Natl. Acad. Sci. USA 2006, 103, 11862–11867. [Google Scholar] [CrossRef] [PubMed]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; et al. Poltheta reverse transcribes RNA and promotes RNA-templated DNA repair. Sci. Adv. 2021, 7, eabf1771. [Google Scholar] [CrossRef]
- Fayomi, A.P.; Orwig, K.E. Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res. 2018, 29, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Kotaja, N. Small RNAs in spermatogenesis. Mol. Cell. Endocrinol. 2014, 382, 498–508. [Google Scholar] [CrossRef]
- Gapp, K.; Bohacek, J. Epigenetic germline inheritance in mammals: Looking to the past to understand the future. Genes Brain Behav. 2018, 17, e12407. [Google Scholar] [CrossRef] [PubMed]
- Vavouri, T.; Lehner, B. Chromatin organization in sperm may be the major functional consequence of base composition variation in the human genome. PLoS Genet. 2011, 7, e1002036. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbert, A. The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules 2021, 26, 4881. https://doi.org/10.3390/molecules26164881
Herbert A. The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules. 2021; 26(16):4881. https://doi.org/10.3390/molecules26164881
Chicago/Turabian StyleHerbert, Alan. 2021. "The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity" Molecules 26, no. 16: 4881. https://doi.org/10.3390/molecules26164881
APA StyleHerbert, A. (2021). The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules, 26(16), 4881. https://doi.org/10.3390/molecules26164881