
Molecular Details on Multiple Cofactor Containing Redox Metalloproteins Revealed by Infrared and Resonance Raman Spectroscopies

 , , , , and
, , , , and
Abstract
1. Introduction
2. Heme Proteins
2.1. Nitrite Reductases (NiR)
2.2. Heme-Containing Respiratory Chain and Analogous Complexes
3. Fe-S Proteins
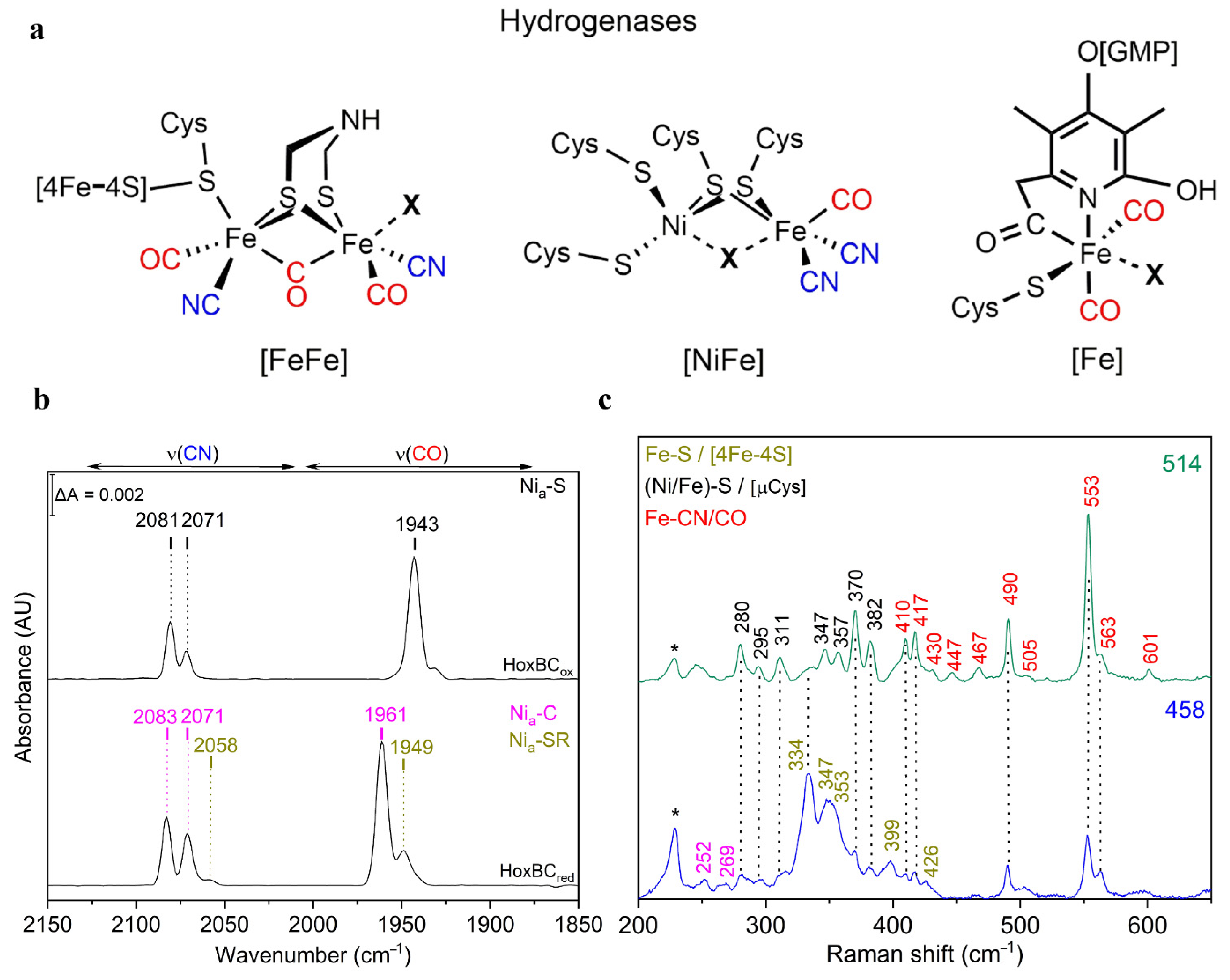
3.1. Hydrogenases
3.2. Multi-Cluster Containing Ferredoxins
4. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siebert, F.; Hildebrandt, P. Vibrational Spectroscopy in Life Science; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Czernuszewicz, R.S.; Fraczkiewicz, G.; Zareba, A.A. A Detailed Resonance Raman Spectrum of Nickel (II)-Substituted Pseudomonas aeruginosa Azurin. Inorg. Chem. 2005, 44, 5745–5752. [Google Scholar] [CrossRef]
- Woodruff, W.H.; Spiro, T.G.; Yonetani, T. Resonance Raman Spectra of Cobalt-Substituted Hemoglobin: Cooperativity and Displacement of the Cobalt Atom upon Oxygenation. Proc. Natl. Acad. Sci. USA 1974, 71, 1065–1069. [Google Scholar] [CrossRef]
- Woodruff, W.H.; Adams, D.H.; Spiro, T.G.; Yonetani, T. Resonance Raman spectra of cobalt myoglobins and cobalt porphyrins. Evaluation of protein effects on porphyrin structure. J. Am. Chem. Soc. 1975, 97, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Garton, S.D.; Garrett, R.M.; Rajagopalan, K.V.; Johnson, M.K. Resonance Raman Characterization of the Molybdenum Center in Sulfite Oxidase: Identification of MoO Stretching Modes. J. Am. Chem. Soc. 1997, 119, 2590–2591. [Google Scholar] [CrossRef]
- Todorovic, S.; Rodrigues, M.L.; Matos, D.; Pereira, I.A.C. Redox Properties of Lysine- and Methionine-Coordinated Hemes Ensure Downhill Electron Transfer in NrfH2A4 Nitrite Reductase. J. Phys. Chem. B 2012, 116, 5637–5643. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, M.; Wang, Y.; Tachikawa, A.H.; Davidson, V.L. Evidence for Redox Cooperativity between c-Type Hemes of MauG Which Is Likely Coupled to Oxygen Activation during Tryptophan Tryptophylquinone Biosynthesis. Biochemistry 2006, 45, 821–828. [Google Scholar] [CrossRef]
- Pauleta, S.R.; Lu, Y.; Goodhew, C.F.; Qiu, Y.; Moura, I.; Pettigrew, G.; Shelnutt, J.A. Structural changes in the calcium-dependent activation of the di-heme cytochrome c peroxidase of Paracoccus pantotrophus. Biophys. J. 2002, 82, 14A. [Google Scholar]
- Wolf, M.W.; Rizzolo, K.; Elliott, S.J.; Lehnert, N. Resonance Raman, Electron Paramagnetic Resonance, and Magnetic Circular Dichroism Spectroscopic Investigation of Diheme Cytochrome c Peroxidases from Nitrosomonas europaea and Shewanella oneidensis. Biochemistry 2018, 57, 6416–6433. [Google Scholar] [CrossRef]
- Hobbs, D.D.; Kriauciunas, A.; Güner, S.; Knaff, D.B.; Ondrias, M.R. Resonance Raman spectroscopy of cytochrome bc1 complexes from Rhodospirillum rubrum: Initial characterization and reductive titrations. Biochim. Biophys. Acta Bioenerg. 1990, 1018, 47–54. [Google Scholar] [CrossRef]
- Todorovic, S.; Teixeira, M. Resonance Raman spectroscopy of Fe-S proteins and their redox properties. JBIC J. Biol. Inorg. Chem. 2018, 23, 647–661. [Google Scholar] [CrossRef]
- Qiu, C.; Cheng, Z.; Lv, C.; Wang, R.; Yu, F. Development of bioorthogonal SERS imaging probe in biological and biomedical applications. Chin. Chem. Lett. 2021. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, R.; Xing, Y.; Zhao, L.; Choo, J.; Yu, F. SERS-based immunoassay using gold-patterned array chips for rapid and sensitive detection of dual cardiac biomarkers. Analyst 2019, 144, 6533–6540. [Google Scholar] [CrossRef]
- Todorovic, S.; Murgida, D.H. Surface-Enhanced Raman Scattering of Biological Materials. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 1–29. [Google Scholar]
- Sezer, M.; Millo, D.; Weidinger, I.; Zebger, I.; Hildebrandt, P. Analyzing the catalytic processes of immobilized redox enzymes by vibrational spectroscopies. IUBMB Life 2012, 64, 455–464. [Google Scholar] [CrossRef]
- Todorovic, S.; Pereira, M.M.; Bandeiras, T.M.; Teixeira, M.; Hildebrandt, P.; Murgida, D.H. Midpoint Potentials of Hemes a and a3 in the Quinol Oxidase from Acidianus ambivalens are Inverted. J. Am. Chem. Soc. 2005, 127, 13561–13566. [Google Scholar] [CrossRef] [PubMed]
- Hrabáková, J.; Ataka, K.; Heberle, J.; Hildebrandt, P.; Murgida, D.H. Long distance electron transfer in cytochrome c oxidase immobilised on electrodes. A surface enhanced resonance Raman spectroscopic study. Phys. Chem. Chem. Phys. 2006, 8, 759–766. [Google Scholar] [CrossRef]
- Behr, J.; Michel, H.; Mäntele, A.W.; Hellwig, P. Functional Properties of the Heme Propionates in Cytochrome c Oxidase from Paracoccus denitrificans. Evidence from FTIR Difference Spectroscopy and Site-Directed Mutagenesis. Biochemistry 2000, 39, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Millo, D.; Pandelia, M.-E.; Utesch, T.; Wisitruangsakul, N.; Mroginski, M.A.; Lubitz, W.; Hildebrandt, P.; Zebger, I. Spectroelectrochemical Study of the [NiFe] Hydrogenase from Desulfovibrio vulgaris Miyazaki F in Solution and Immobilized on Biocompatible Gold Surfaces. J. Phys. Chem. B 2009, 113, 15344–15351. [Google Scholar] [CrossRef] [PubMed]
- Wisitruangsakul, N.; Lenz, O.; Ludwig, M.; Friedrich, B.; Lendzian, F.; Hildebrandt, P.; Zebger, I. Monitoring Catalysis of the Membrane-Bound Hydrogenase from Ralstonia eutropha H16 by Surface-Enhanced IR Absorption Spectroscopy. Angew. Chem. Int. Ed. 2009, 48, 611–613. [Google Scholar] [CrossRef]
- Hidalgo, R.; Ash, P.; Healy, A.J.; Vincent, K.A. Infrared Spectroscopy During Electrocatalytic Turnover Reveals the Ni-L Active Site State During H2 Oxidation by a NiFe Hydrogenase. Angew. Chem. Int. Ed. 2015, 54, 7110–7113. [Google Scholar] [CrossRef]
- Senger, M.; Mebs, S.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Heberle, J.; Haumann, M.; Stripp, S.T. Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 8454–8459. [Google Scholar] [CrossRef]
- Martins, G.; Rodrigues, L.; Cunha, F.M.; Matos, D.; Hildebrandt, P.; Murgida, D.H.; Pereira, I.A.C.; Todorovic, S. Substrate Binding to a Nitrite Reductase Induces a Spin Transition. J. Phys. Chem. B 2010, 114, 5563–5566. [Google Scholar] [CrossRef]
- Rinaldo, S.; Cutruzzolà, F. Chapter 3—Nitrite Reductases in Denitrification. In Biology of the Nitrogen Cycle; Bothe, H., Ferguson, S.J., Newton, W.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 37–55. [Google Scholar]
- Castiglione, N.; Rinaldo, S.; Giardina, G.; Stelitano, V.; Cutruzzolà, F. Nitrite and Nitrite Reductases: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 17, 684–716. [Google Scholar] [CrossRef]
- Cotton, T.M.; Timkovich, R.; Cork, M.S. Resonance Raman and surface-enhanced resonance Raman studies of cytochrome cd1. FEBS Lett. 1981, 133, 39–44. [Google Scholar] [CrossRef]
- Das, T.; Wilson, E.; Cutruzzola, F.; Brunori, M.; Rousseau, D.L. Binding of NO and CO to the d1 Heme of cd1 Nitrite Reductase from Pseudomonas aeruginosa. Biochemistry 2001, 40, 10774–10781. [Google Scholar] [CrossRef]
- Silveira, C.M.; Quintas, P.O.; Moura, I.; Moura, J.J.G.; Hildebrandt, P.; Almeida, M.G.; Todorovic, S. SERR Spectroelectrochemical Study of Cytochrome cd1 Nitrite Reductase Co-Immobilized with Physiological Redox Partner Cytochrome c552 on Biocompatible Metal Electrodes. PLoS ONE 2015, 10, e0129940. [Google Scholar] [CrossRef]
- Spiro, T.G.; Jarzecki, A.A. Heme-based sensors: Theoretical modeling of heme-ligand–protein interactions. Curr. Opin. Chem. Biol. 2001, 5, 715–723. [Google Scholar] [CrossRef]
- Andrew, C.R.; George, S.; Lawson, D.M.; Eady, R.R. Six- to Five-Coordinate Heme―Nitrosyl Conversion in Cytochrome c‘ and Its Relevance to Guanylate Cyclase. Biochemistry 2002, 41, 2353–2360. [Google Scholar] [CrossRef]
- Quintas, P.; Catarino, T.; Todorovic, S.; Turner, D. Highly Selective Ligand Binding by Methylophilus methylotrophus Cytochrome c″. Biochemistry 2011, 50, 5624–5632. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Qin, H.; Knaff, D.; Zhang, L.; Yu, L.; Yu, C.; Gray, K.; Daldal, F.; Ondrias, M. Q-Band resonance Raman investigation of turnip cytochrome f and Rhodobacter capsulatus cytochrome c1. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1999, 1430, 203–213. [Google Scholar] [CrossRef]
- De Vitry, C.; Desbois, A.; Redeker, V.; Zito, F.; Wollman, F.-A. Biochemical and Spectroscopic Characterization of the Covalent Binding of Heme to Cytochrome b6. Biochemistry 2004, 43, 3956–3968. [Google Scholar] [CrossRef] [PubMed]
- Picaud, T.; Le Moigne, C.; De Gracia, A.G.; Desbois, A. Soret-Excited Raman Spectroscopy of the Spinach Cytochrome b6f Complex. Structures of the b- and c-Type Hemes, Chlorophylla, and β-Carotene. Biochemistry 2001, 40, 7309–7317. [Google Scholar] [CrossRef] [PubMed]
- Bandeiras, T.M.; Refojo, P.; Todorovic, S.; Murgida, D.H.; Hildebrandt, P.; Bauer, C.; Pereira, M.M.; Kletzin, A.; Teixeira, M. The cytochrome ba complex from the thermoacidophilic crenarchaeote Acidianus ambivalens is an analog of bc1 complexes. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 37–45. [Google Scholar] [CrossRef]
- Hellwig, P.; Grzybek, S.; Behr, J.; Ludwig, B.; Michel, H.; Mäntele, W. Electrochemical and ultraviolet/visible/infrared spectroscopic analysis of heme a and a3 redox reactions in the cytochrome c oxidase from Paracoccus denitrificans: Separation of heme a and a3. Biochemistry 1999, 38, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Giotta, L.; Akinsiku, A.O.; Schägger, H.; Fisher, N.; Breton, J.; Rich, P.R. Redox-Induced Transitions in Bovine Cytochrome bc1 Complex Studied by Perfusion-Induced ATR-FTIR Spectroscopy. Biochemistry 2003, 42, 11109–11119. [Google Scholar] [CrossRef]
- Iwaki, M.; Yakovlev, G.; Hirst, J.; Osyczka, A.; Dutton, P.L.; Marshall, D.; Rich, P.R. Direct Observation of Redox-Linked Histidine Protonation Changes in the Iron−Sulfur Protein of the Cytochrome bc1 Complex by ATR-FTIR Spectroscopy. Biochemistry 2005, 44, 4230–4237. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Miller, S.; Babcock, G.T. Heme/Copper Terminal Oxidases. Chem. Rev. 1996, 96, 2889–2908. [Google Scholar] [CrossRef]
- Uno, T.; Nishimura, Y.; Tsuboi, M.; Kita, K.; Anraku, Y. Resonance Raman study of cytochrome b562-o complex, a terminal oxidase of Escherichia coli in its ferric, ferrous, and CO-ligated states. J. Biol. Chem. 1985, 260, 6755–6760. [Google Scholar] [CrossRef]
- Wang, J.; Rumbley, J.; Ching, Y.-C.; Takahashi, S.; Gennis, R.B.; Rousseau, D.L. Reaction of Cytochrome bo3 with Oxygen: Extra Redox Center(s) are Present in the Protein. Biochemistry 1995, 34, 15504–15511. [Google Scholar] [CrossRef]
- Heibel, G.E.; Hildebrandt, P.; Ludwig, B.; Steinruecke, P.; Soulimane, T.; Buse, G. Comparative resonance Raman study of cytochrome c oxidase from beef heart and Paracoccus denitrificans. Biochemistry 1993, 32, 10866–10877. [Google Scholar] [CrossRef]
- Varotsis, C.; Babcock, G.T.; Garcia-Horsman, J.A.; Gennis, R.B. Resonance Raman Spectroscopy of the Heme Groups of Cytochrome cbb3 in Rhodobacter sphaeroides. J. Phys. Chem. 1995, 99, 16817–16820. [Google Scholar] [CrossRef]
- Pinakoulaki, E.; Pfitzner, U.; Ludwig, B.; Varotsis, C. The Role of the Cross-link His-Tyr in the Functional Properties of the Binuclear Center in Cytochrome c Oxidase. J. Biol. Chem. 2002, 277, 13563–13568. [Google Scholar] [CrossRef] [PubMed]
- Varotsis, C.; Vamvouka, M. Resonance Raman and Fourier Transform Infrared Detection of Azide Binding to the Binuclear Center of Cytochrome bo3 Oxidase from Escherichia coli. J. Phys. Chem. B 1999, 103, 3942–3946. [Google Scholar] [CrossRef]
- Varotsis, C.; Vamvouka, M. Resonance Raman and FTIR Studies of Carbon Monoxide-Bound Cytochrome aa3-600 Oxidase of Bacillus subtilis. J. Phys. Chem. B 1998, 102, 7670–7673. [Google Scholar] [CrossRef]
- Oertling, W.A.; Surerus, K.K.; Einarsdóttir, Ó.; Fee, J.A.; Dyer, R.B.; Woodruff, W.H. Spectroscopic characterization of cytochrome ba3, a terminal oxidase from Thermus thermophilus: Comparison of the a3/CuB site to that of bovine cytochrome aa3. Biochemistry 1994, 33, 3128–3141. [Google Scholar] [CrossRef]
- Varotsis, C.; Ohta, T.; Kitagawa, T.; Soulimane, T.; Pinakoulaki, E. The Structure of the Hyponitrite Species in a Heme Fe-Cu Binuclear Center. Angew. Chem. Int. Ed. 2007, 46, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Pinakoulaki, E.; Vamvouka, M.; Varotsis, C. Resonance Raman Detection of the Fe2+-C-N Modes in Heme−Copper Oxidases: A Probe of the Active Site. Inorg. Chem. 2004, 43, 4907–4910. [Google Scholar] [CrossRef]
- Egawa, T.; Lin, M.T.; Hosler, J.P.; Gennis, R.B.; Yeh, S.-R.; Rousseau, D.L. Communication between R481 and CuB in Cytochrome bo3 Ubiquinol Oxidase from Escherichia coli. Biochemistry 2009, 48, 12113–12124. [Google Scholar] [CrossRef][Green Version]
- Lauraeus, M.; Wikstrom, M.; Varotsis, C.; Tecklenburg, M.M.J.; Babcock, G.T. Optical and resonance Raman spectroscopy of the heme groups of the quinol-oxidizing cytochrome aa3 of Bacillus subtilis. Biochemistry 1992, 31, 10054–10060. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gray, K.A.; Daldal, F.; Rousseau, D.L. The cbb3-Type Cytochrome c Oxidase from Rhodobacter capsulatus Contains a Unique Active Site. J. Am. Chem. Soc. 1995, 117, 9363–9364. [Google Scholar] [CrossRef]
- Ji, H.; Das, T.K.; Puustinen, A.; Wikström, M.; Yeh, S.-R.; Rousseau, D.L. Modulation of the active site conformation by site-directed mutagenesis in cytochrome c oxidase from Paracoccus denitrificans. J. Inorg. Biochemistry 2010, 104, 318–323. [Google Scholar] [CrossRef][Green Version]
- Egawa, T.; Lee, H.J.; Gennis, R.B.; Yeh, S.-R.; Rousseau, D.L. Critical structural role of R481 in cytochrome c oxidase from Rhodobacter sphaeroides. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1272–1275. [Google Scholar] [CrossRef]
- Egawa, T.; Lee, H.J.; Ji, H.; Gennis, R.B.; Yeh, S.-R.; Rousseau, D.L. Identification of heme propionate vibrational modes in the resonance Raman spectra of cytochrome c oxidase. Anal. Biochemistry 2009, 394, 141–143. [Google Scholar] [CrossRef][Green Version]
- Das, T.; Gomes, C.M.; Bandeiras, T.M.; Pereira, M.M.; Teixeira, M.; Rousseau, D.L. Active site structure of the aa3 quinol oxidase of Acidianus ambivalens. Biochim. Biophys. Acta Bioenerg. 2004, 1655, 306–320. [Google Scholar] [CrossRef]
- Rousseau, D.L.; Han, S. 26—Time-Resolved Resonance Raman Spectroscopy of Intermediates in Cytochrome Oxidase. In Enzyme Kinetics and Mechanism; Purich, D.L., Ed.; Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2002; Volume 354, pp. 351–368. [Google Scholar] [CrossRef]
- Ogura, T.; Kitagawa, T. Resonance Raman characterization of the P intermediate in the reaction of bovine cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2004, 1655, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, T. Structures of reaction intermediates of bovine cytochrome c oxidase probed by time-resolved vibrational spectroscopy. J. Inorg. Biochem. 2000, 82, 9–18. [Google Scholar] [CrossRef]
- Han, S.; Takahashi, S.; Rousseau, D.L. Time Dependence of the Catalytic Intermediates in Cytochrome c Oxidase. J. Biol. Chem. 2000, 275, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Proshlyakov, D.A.; Pressler, M.A.; Babcock, G.T. Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 1998, 95, 8020–8025. [Google Scholar] [CrossRef]
- Pinakoulaki, E.; Daskalakis, V.; Varotsis, C. The origin of the FeIV=O intermediates in cytochrome aa3 oxidase. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 552–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nyquist, R.M.; Heitbrink, D.; Bolwien, C.; Gennis, R.B.; Heberle, J. Direct observation of protonation reactions during the catalytic cycle of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 8715–8720. [Google Scholar] [CrossRef]
- Todorovic, S.; Verissimo, A.; Wisitruangsakul, N.; Zebger, I.; Hildebrandt, P.; Pereira, M.M.; Teixeira, M.; Murgida, D.H. SERR-Spectroelectrochemical Study of a cbb3 Oxygen Reductase in a Biomimetic Construct. J. Phys. Chem. B 2008, 112, 16952–16959. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Giebeta, F.; Naumann, R.; Knoll, W.; Ataka, K.; Heberle, J.; Hrabakova, J.; Murgida, D.H.; Hildebrandt, P. Active site structure and redox processes of cytochrome c oxidase immobilised in a novel biomimetic lipid membrane on an electrode. Chem. Commun. 2004, 2376–2377. [Google Scholar] [CrossRef] [PubMed]
- Ataka, K.; Richter, B.; Heberle, J. Orientational Control of the Physiological Reaction of Cytochrome c Oxidase Tethered to a Gold Electrode. J. Phys. Chem. B 2006, 110, 9339–9347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sezer, M.; Woelke, A.-L.; Knapp, E.W.; Schlesinger, R.; Mroginski, M.A.; Weidinger, I. Redox induced protonation of heme propionates in cytochrome c oxidase: Insights from surface enhanced resonance Raman spectroscopy and QM/MM calculations. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, P.; Soulimane, T.; Mäntele, W. Electrochemical, FT-IR and UV/VIS spectroscopic properties of the caa3 oxidase from T. thermophilus. JBIC J. Biol. Inorg. Chem. 2002, 269, 4830–4838. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, P.; Gomes, C.M.; Teixeira, M. FTIR Spectroscopic Characterization of the Cytochrome aa3 from Acidianus ambivalens: Evidence for the Involvement of Acidic Residues in Redox Coupled Proton Translocation. Biochemistry 2003, 42, 6179–6184. [Google Scholar] [CrossRef] [PubMed]
- Stavrakis, S.; Pinakoulaki, E.; Urbani, A.; Varotsis, C. Fourier Transform Infrared Evidence for a Ferric Six-Coordinate Nitrosylheme b3 Complex of Cytochrome cbb3 Oxidase from Pseudomonas stutzeri at Ambient Temperature. J. Phys. Chem. B 2002, 106, 12860–12862. [Google Scholar] [CrossRef]
- Koutsoupakis, C.; Kolaj-Robin, O.; Soulimane, T.; Varotsis, C. Probing Protonation/Deprotonation of Tyrosine Residues in Cytochrome ba3 Oxidase from Thermus thermophilus by Time-resolved Step-scan Fourier Transform Infrared Spectroscopy. J. Biol. Chem. 2011, 286, 30600–30605. [Google Scholar] [CrossRef]
- Gorbikova, E.A.; Vuorilehto, K.; Wikström, M.; Verkhovsky, M.I. Redox Titration of All Electron Carriers of Cytochrome c Oxidase by Fourier Transform Infrared Spectroscopy. Biochemistry 2006, 45, 5641–5649. [Google Scholar] [CrossRef]
- Melin, F.; Xie, H.; Meyer, T.; Ahn, Y.O.; Gennis, R.B.; Michel, H.; Hellwig, P. The unusual redox properties of c-type oxidases. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1892–1899. [Google Scholar] [CrossRef]
- Pinakoulaki, E.; Varotsis, C. Resonance Raman Spectroscopy of Nitric Oxide Reductase and cbb3 Heme-Copper Oxidase. J. Phys. Chem. B 2008, 112, 1851–1857. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Ruediger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.-P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef]
- Ash, P.A.; Hidalgo, R.; Vincent, K.A. Proton Transfer in the Catalytic Cycle of [NiFe] Hydrogenases: Insight from Vibrational Spectroscopy. ACS Catal. 2017, 7, 2471–2485. [Google Scholar] [CrossRef]
- Caserta, G.; Pelmenschikov, V.; Lorent, C.; Waffo, A.F.T.; Katz, S.; Lauterbach, L.; Schoknecht, J.; Wang, H.; Yoda, Y.; Tamasaku, K.; et al. Hydroxy-bridged resting states of a [NiFe]-hydrogenase unraveled by cryogenic vibrational spectroscopy and DFT computations. Chem. Sci. 2021, 12, 2189–2197. [Google Scholar] [CrossRef]
- Lorent, C.; Katz, S.; Duan, J.; Kulka, C.J.; Caserta, G.; Teutloff, C.; Yadav, S.; Apfel, U.-P.; Winkler, M.; Happe, T.; et al. Shedding Light on Proton and Electron Dynamics in [FeFe] Hydrogenases. J. Am. Chem. Soc. 2020, 142, 5493–5497. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, P.S.; Tirsch, J.L.; Silakov, A. Investigation of the Unusual Ability of the [FeFe] Hydrogenase from Clostridium beijerinckii to Access an O2-Protected State. J. Am. Chem. Soc. 2020, 142, 12409–12419. [Google Scholar] [CrossRef]
- Rodríguez-Maciá, P.; Breuer, N.; Debeer, S.; Birrell, J.A. Insight into the Redox Behavior of the [4Fe–4S] Subcluster in [FeFe] Hydrogenases. ACS Catal. 2020, 10, 13084–13095. [Google Scholar] [CrossRef]
- Land, H.; Senger, M.; Berggren, G.; Stripp, S.T. Current State of [FeFe]-Hydrogenase Research: Biodiversity and Spectroscopic Investigations. ACS Catal. 2020, 10, 7069–7086. [Google Scholar] [CrossRef]
- Greene, B.L.; Van Such, G.E.; Chica, B.C.; Adams, M.W.W.; Dyer, R.B. Applications of Photogating and Time Resolved Spectroscopy to Mechanistic Studies of Hydrogenases. Acc. Chem. Res. 2017, 50, 2718–2726. [Google Scholar] [CrossRef] [PubMed]
- Horch, M.; Schoknecht, J.; Wrathall, S.L.D.; Greetham, G.M.; Lenz, O.; Hunt, N.T. Understanding the structure and dynamics of hydrogenases by ultrafast and two-dimensional infrared spectroscopy. Chem. Sci. 2019, 10, 8981–8989. [Google Scholar] [CrossRef]
- Tai, H.; Hirota, S.; Stripp, S.T. Proton Transfer Mechanisms in Bimetallic Hydrogenases. Acc. Chem. Res. 2021, 54, 232–241. [Google Scholar] [CrossRef]
- Ilina, Y.; Lorent, C.; Katz, S.; Jeoung, J.H.; Shima, S.; Horch, M.; Zebger, I.; Dobbek, H. X-ray Crystallography and Vibrational Spectroscopy Reveal the Key Determinants of Biocatalytic Dihydrogen Cycling by [NiFe] Hydrogenases. Angew. Chem. Int. Ed. 2019, 58, 18710–18714. [Google Scholar] [CrossRef]
- Katz, S.; Noth, J.; Horch, M.; Shafaat, H.S.; Happe, T.; Hildebrandt, P.; Zebger, I. Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem. Sci. 2016, 7, 6746–6752. [Google Scholar] [CrossRef]
- Horch, M.; Schoknecht, J.; Mroginski, M.A.; Lenz, O.; Hildebrandt, P.; Zebger, I. Resonance Raman Spectroscopy on [NiFe] Hydrogenase Provides Structural Insights into Catalytic Intermediates and Reactions. J. Am. Chem. Soc. 2014, 136, 9870–9873. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Maciá, P.; Galle, L.M.; Bjornsson, R.; Lorent, C.; Zebger, I.; Yoda, Y.; Cramer, S.P.; Debeer, S.; Span, I.; Birrell, J.A. Caught in the H inact: Crystal Structure and Spectroscopy Reveal a Sulfur Bound to the Active Site of an O2− stable State of [FeFe] Hydrogenase. Angew. Chem. Int. Ed. 2020, 59, 16786–16794. [Google Scholar] [CrossRef]
- Ash, P.A.; Carr, S.B.; Reeve, H.A.; Skorupskaitė, A.; Rowbotham, J.S.; Shutt, R.; Frogley, M.D.; Evans, R.M.; Cinque, G.; Armstrong, F.A.; et al. Generating single metalloprotein crystals in well-defined redox states: Electrochemical control combined with infrared imaging of a NiFe hydrogenase crystal. Chem. Commun. 2017, 53, 5858–5861. [Google Scholar] [CrossRef] [PubMed]
- Morra, S.; Duan, J.; Winkler, M.; Ash, P.A.; Happe, T.; Vincent, K.A. Electrochemical control of [FeFe]-hydrogenase single crystals reveals complex redox populations at the catalytic site. Dalton Trans. 2021. [Google Scholar] [CrossRef]
- Ash, P.A.; Kendall-Price, S.; Evans, R.M.; Carr, S.B.; Brasnett, A.; Morra, S.; Hidalgo, R.; Healy, A.J.; Cinque, G.; Frogley, M.; et al. The crystalline state as a dynamic system: IR microspectroscopy under electrochemical control for a [NiFe] hydrogenase. Chem. Sci. 2021. [Google Scholar] [CrossRef]
- Lorent, C.; Pelmenschikov, V.; Frielingsdorf, S.; Schoknecht, J.; Caserta, G.; Yoda, Y.; Wang, H.; Tamasaku, K.; Lenz, O.; Cramer, S.P.; et al. Exploring Structure and Function of Redox Intermediates in [NiFe]-Hydrogenases by an Advanced Experimental Approach for Solvated, Lyophilized and Crystallized Metalloenzymes. Angew. Chem. Int. Ed. 2021, 60, 15854–15862. [Google Scholar] [CrossRef]
- Siebert, E.; Rippers, Y.; Frielingsdorf, S.; Fritsch, J.; Schmidt, A.; Kalms, J.; Katz, S.; Lenz, O.; Scheerer, P.; Paasche, L.; et al. Resonance Raman Spectroscopic Analysis of the [NiFe] Active Site and the Proximal [4Fe-3S] Cluster of an O2-Tolerant Membrane-Bound Hydrogenase in the Crystalline State. J. Phys. Chem. B 2015, 119, 13785–13796. [Google Scholar] [CrossRef]
- Frielingsdorf, S.; Fritsch, J.; Schmidt, A.; Hammer, M.; Löwenstein, J.; Siebert, E.; Pelmenschikov, V.; Jaenicke, T.; Kalms, J.; Rippers, Y.; et al. Reversible [4Fe-3S] cluster morphing in an O2-tolerant [NiFe] hydrogenase. Nat. Chem. Biol. 2014, 10, 378–385. [Google Scholar] [CrossRef]
- Siebert, E.; Schmidt, A.; Frielingsdorf, S.; Kalms, J.; Kuhlmann, U.; Lenz, O.; Scheerer, P.; Zebger, I.; Hildebrandt, P. Resonance Raman spectroscopic analysis of the iron–sulfur cluster redox chain of the Ralstonia eutropha membrane-bound [NiFe]-hydrogenase. J. Raman Spectrosc. 2021. [Google Scholar] [CrossRef]
- Sezer, M.; Frielingsdorf, S.; Millo, D.; Heidary, N.; Utesch, T.; Mroginski, M.-A.; Friedrich, B.; Hildebrandt, P.; Zebger, I.; Weidinger, I.M. Role of the HoxZ Subunit in the Electron Transfer Pathway of the Membrane-Bound [NiFe]-Hydrogenase fromRalstonia eutropha Immobilized on Electrodes. J. Phys. Chem. B 2011, 115, 10368–10374. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, S.; Leal, S.S.; Salgueiro, C.; Zebger, I.; Hildebrandt, P.; Murgida, D.H.; Gomes, C.M. A Spectroscopic Study of the Temperature Induced Modifications on Ferredoxin Folding and Iron−Sulfur Moieties. Biochemistry 2007, 46, 10733–10738. [Google Scholar] [CrossRef] [PubMed]
- Caserta, G.; Lorent, C.; Pelmenschikov, V.; Schoknecht, J.; Yoda, Y.; Hildebrandt, P.; Cramer, S.P.; Zebger, I.; Lenz, O. In Vitro Assembly as a Tool to Investigate Catalytic Intermediates of [NiFe]-Hydrogenase. ACS Catal. 2020, 10, 13890–13894. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Wilson, R.; Siebel, J.F.; Adamska-Venkatesh, A.; Pham, C.C.; Reijerse, E.; Wang, H.; Cramer, S.P.; Lubitz, W.; Rauchfuss, T.B. Spectroscopic Investigations of [FeFe] Hydrogenase Maturated with [57 Fe2 (adt) (CN)2 (CO)4 ]2−. J. Am. Chem. Soc. 2015, 137, 8998–9005. [Google Scholar] [CrossRef]
- Brasseur, G.; Sariba, A.S.; Daldal, F. A compilation of mutation located in cytochrome b subunit of the bacterial and mito-chondrial bc1 complex. Biochim. Biophys. Acta 1996, 1275, 61–69. [Google Scholar] [CrossRef][Green Version]
- Crofts, A.R.; Lhee, S.; Crofts, S.B.; Cheng, J.; Rose, S. Proton pumping in the bc1 complex: A new gating mechanism that prevents short circuits. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1019–1034. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
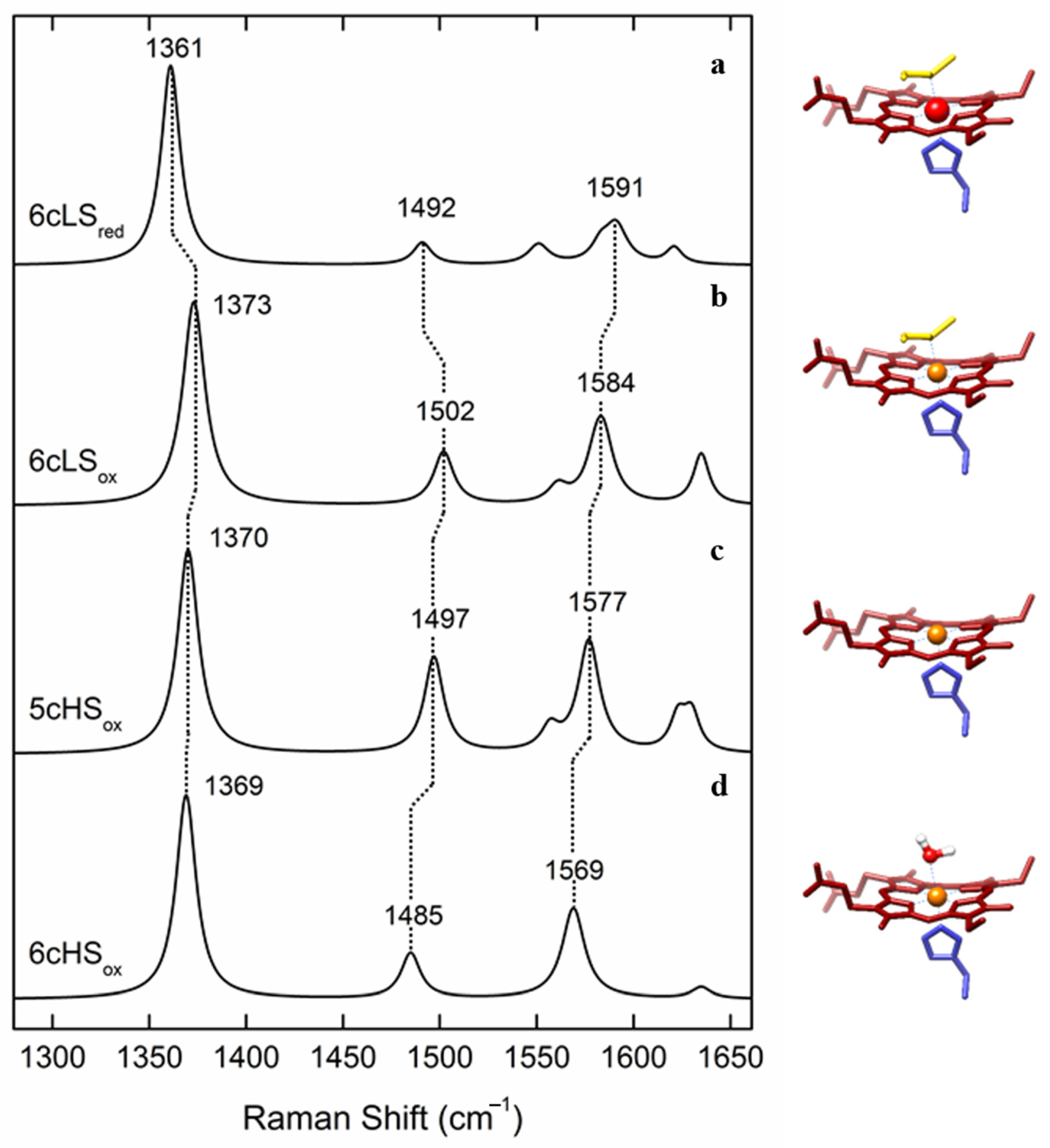
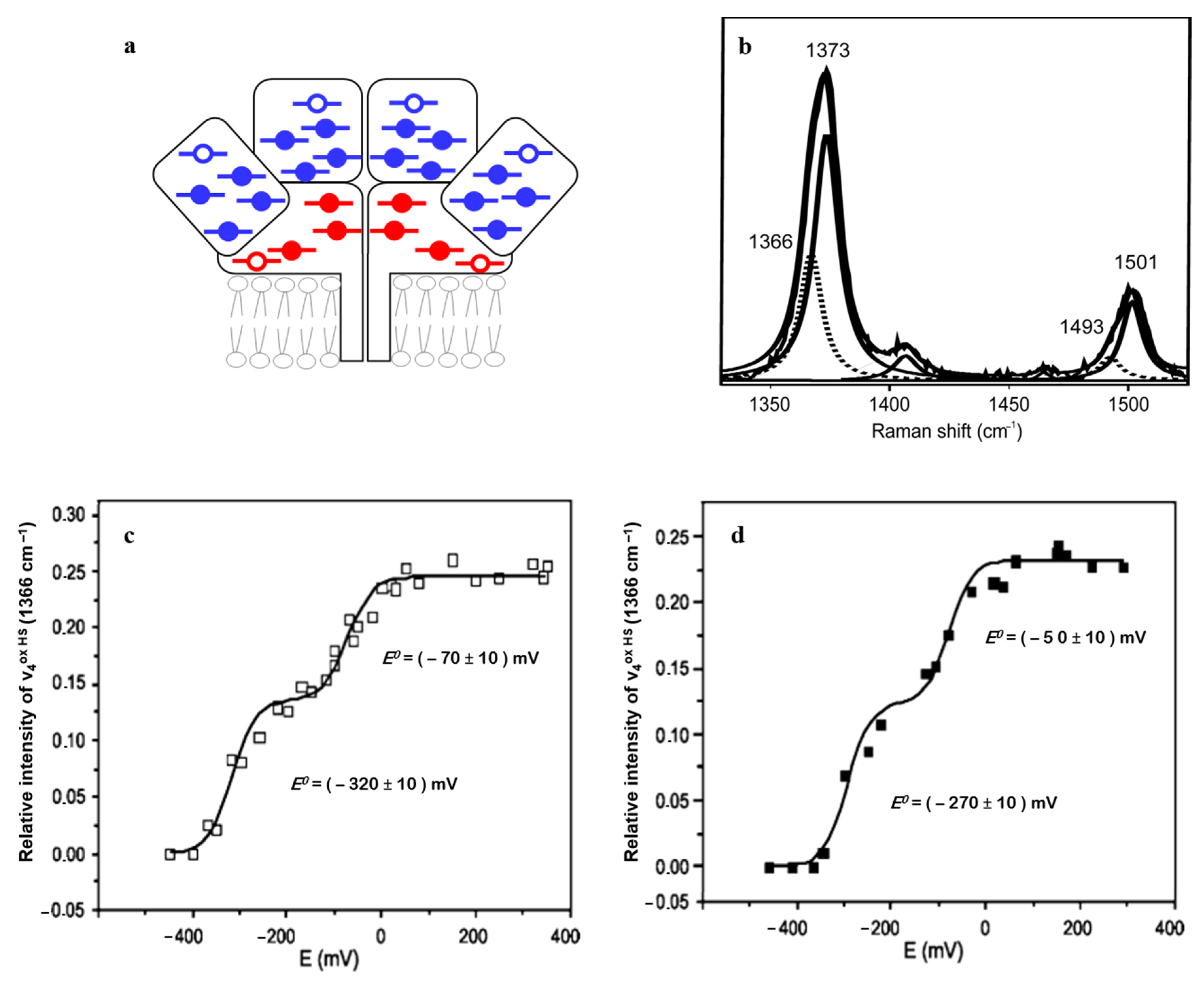
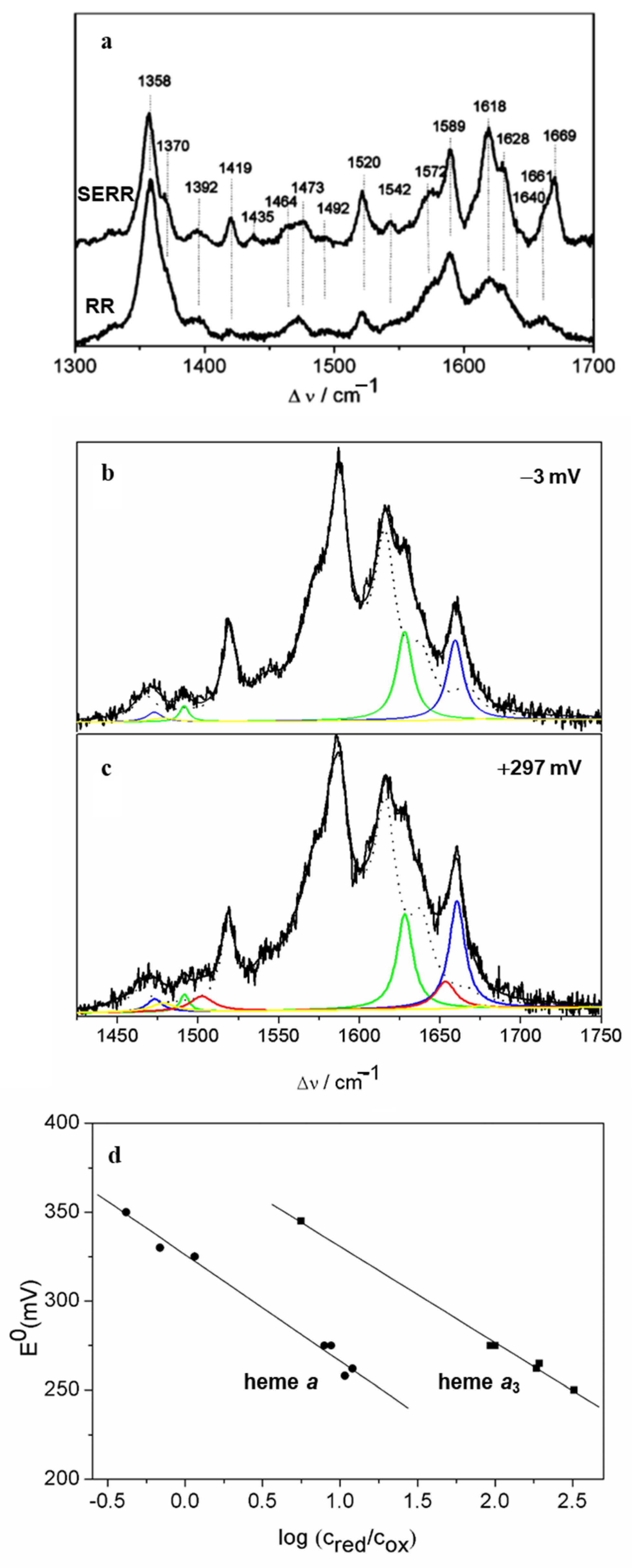
| Heme Group (1300–1700 cm−1) | ||||||
|---|---|---|---|---|---|---|
| Coordination and Spin State | Redox State | ν4 | ν3 | ν2 | ν10 | |
| His/Met | 6cLS | 3+ 2+ | 1374 ± 2 1363 ± 2 | 1505 ± 3 1495 ± 3 | 1586 ± 2 1592 ± 3 | 1638 ± 3 1624 ± 3 |
| His/- | 5cHS | 3+ 2+ | 1371 ± 2 1357 ± 3 | 1496 ± 2 1472 ± 1 | 1576 ± 2 1573 ± 3 | 1627 ± 4 1606 ± 2 |
| Fe-S Clusters (200–450 cm−1) | ||||||
| Cluster | RR Active Redox State | Frequency Range (cm−1) | ||||
| 1Fe-4S | 3+ | 314–318 | ||||
| 2Fe-2S | 2+ | 281–290 (T) | ||||
| 3Fe-4S | 1+ | 346–348 (B) | ||||
| 4Fe-4S | 2+/3+ | 333–339 (B)/341–344 (B) | ||||
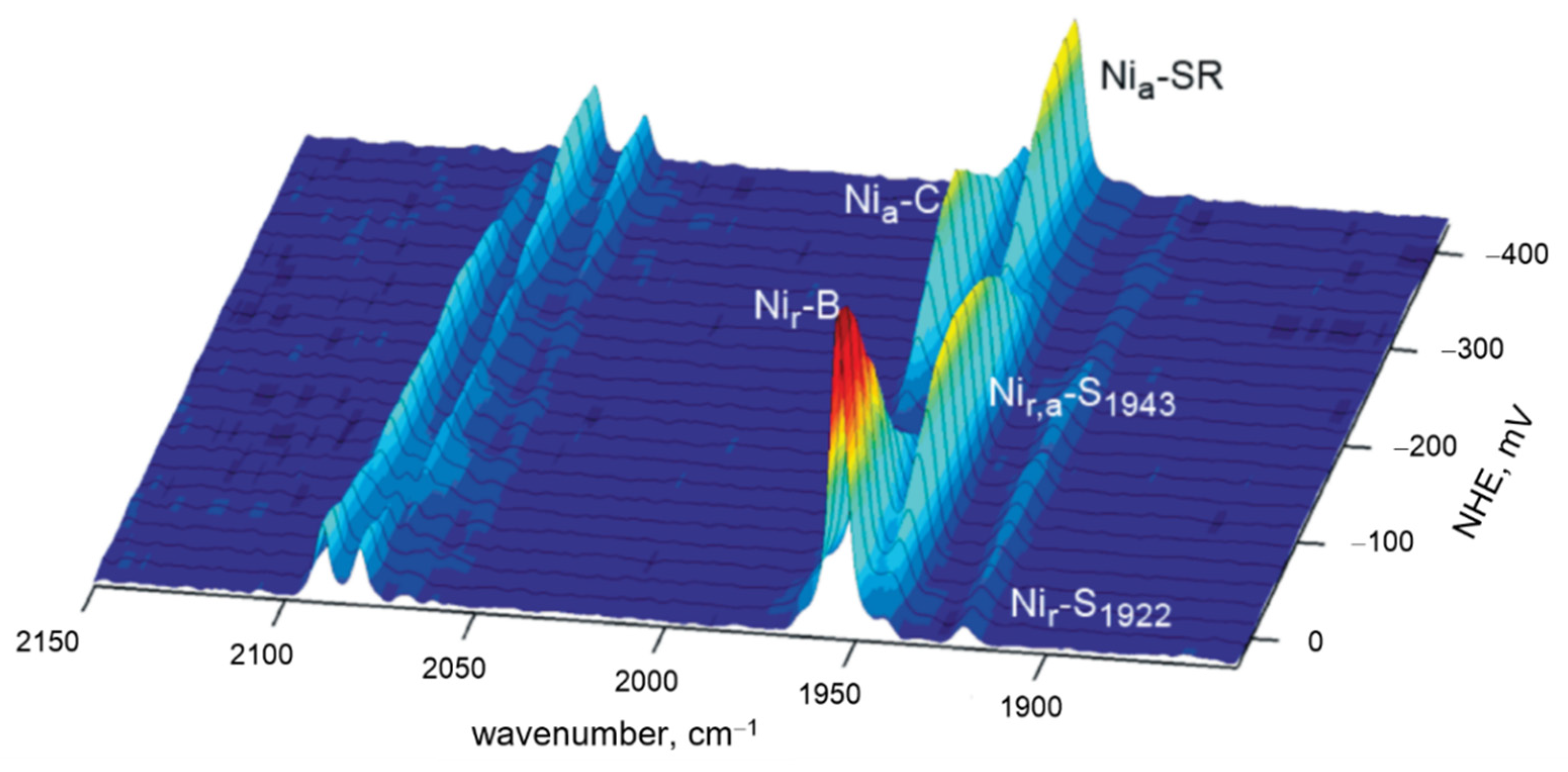
| [NiFe] and [FeFe] Hydrogenase Active Sites | ||||||
| Hydrogenase | Bands | Frequency Range (cm−1) | ||||
| NiFe/FeFe | ν(CO) | IR | 1780–2030 | |||
| NiFe/FeFe | ν(CN) | IR | 2030–2150 | |||
| NiFe/FeFe | Fe-CN/CO | RR | 400–650 | |||
| FeFe | Fe-S [adt] | RR | 300–400 | |||
| NiFe | (Ni/Fe)-S [µCys] | RR | 280–400 | |||
| NiFe | Fe-OH [4Fe-3S] | RR | 500–600 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silveira, C.M.; Zuccarello, L.; Barbosa, C.; Caserta, G.; Zebger, I.; Hildebrandt, P.; Todorovic, S. Molecular Details on Multiple Cofactor Containing Redox Metalloproteins Revealed by Infrared and Resonance Raman Spectroscopies. Molecules 2021, 26, 4852. https://doi.org/10.3390/molecules26164852
Silveira CM, Zuccarello L, Barbosa C, Caserta G, Zebger I, Hildebrandt P, Todorovic S. Molecular Details on Multiple Cofactor Containing Redox Metalloproteins Revealed by Infrared and Resonance Raman Spectroscopies. Molecules. 2021; 26(16):4852. https://doi.org/10.3390/molecules26164852
Chicago/Turabian StyleSilveira, Célia M., Lidia Zuccarello, Catarina Barbosa, Giorgio Caserta, Ingo Zebger, Peter Hildebrandt, and Smilja Todorovic. 2021. "Molecular Details on Multiple Cofactor Containing Redox Metalloproteins Revealed by Infrared and Resonance Raman Spectroscopies" Molecules 26, no. 16: 4852. https://doi.org/10.3390/molecules26164852
APA StyleSilveira, C. M., Zuccarello, L., Barbosa, C., Caserta, G., Zebger, I., Hildebrandt, P., & Todorovic, S. (2021). Molecular Details on Multiple Cofactor Containing Redox Metalloproteins Revealed by Infrared and Resonance Raman Spectroscopies. Molecules, 26(16), 4852. https://doi.org/10.3390/molecules26164852