
Moving toward a Handheld “Plasma” Spectrometer for Elemental Analysis, Putting the Power of the Atom (Ion) in the Palm of Your Hand


Abstract
:1. Introduction
2. A History of Plasma Spectroscopic Components
3. Advantages of Miniaturization
4. Requirements for Excitation Ionization Sources
4.1. Potential Ionization Sources
4.1.1. Breakdown Plasmas
4.1.2. Arc/Spark
4.1.3. Cold Plasmas, Corona Discharges, and Other Breakdown Plasma
4.2. Voltage over Current: Making the Most of Battery Power
4.3. Plasma Gases
5. Sample Introduction
6. Detectors
6.1. Optical Spectroscopy
6.2. Mass Spectrometry
6.3. Need to Incorporate Appropriate Supporting Technologies
7. Competing Technologies
8. Putting the Pieces Together on a Plasma Instrument Feasibility
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Rector, J.; Lin, J.Q.; Young, J.H.; Sans, M.; Katta, N.; Giese, N.; Yu, W.; Nagi, C.; Suliburk, J. Nondestructive tissue analysis for ex vivo and in vivo cancer diagnosis using a handheld mass spectrometry system. Sci. Transl. Med. 2017, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ewing, R.G.; Miller, C.J. Detection of volatile vapors emitted from explosives with a handheld ion mobility spectrometer. Field Anal. Chem. Technol. 2001, 5, 215–221. [Google Scholar] [CrossRef]
- Wolf, J.-C.; Etter, R.; Schaer, M.; Siegenthaler, P.; Zenobi, R. Direct and sensitive detection of CWA simulants by active capillary plasma ionization coupled to a handheld ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 2016, 27, 1197–1202. [Google Scholar] [CrossRef]
- Ettinger, A.S.; Leonard, M.L.; Mason, J. CDC’s Lead Poisoning Prevention Program: A long-standing responsibility and commitment to protect children from lead exposure. J. Public Health Manag. Pract. Jphmp 2019, 25, S5. [Google Scholar] [CrossRef]
- Kostrubiak, M. World Trade Center Dust: Composition and Spatial-Temporal Considerations for Health. In World Trade Center Pulmonary Diseases and Multi-Organ System Manifestations; Springer: Berlin/Heidelberg, Germany, 2018; pp. 107–120. [Google Scholar]
- Durmus, N.; Pehlivan, S.; Zhang, Y.; Shao, Y.; Arslan, A.A.; Corona, R.; Henderson, I.; Sterman, D.H.; Reibman, J. Lung Cancer Characteristics in the World Trade Center Environmental Health Center. Int. J. Environ. Res. Public Health 2021, 18, 2689. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, N.; Boumans, P.W.J.M. Theory of Spectrochemical Excitation; Hilger & Watts: London, UK; Plenum Press: New York, NY, USA, 1966. [Google Scholar]
- Gray, A.L. It all depends on the source. Proc. Soc. Anal. Chem. 1974, 11, 182–183. [Google Scholar] [CrossRef]
- Date, A.R.; Gray, A.L. Plasma source mass spectrometry using an inductively coupled plasma and a high resolution quadrupole mass filter. Analyst 1981, 106, 1255–1267. [Google Scholar] [CrossRef]
- Houk, R.S.; Fassel, V.A.; Flesch, G.D.; Svec, H.J.; Gray, A.L.; Taylor, C.E. Inductively coupled argon plasma as an ion source for mass spectrometric determination of trace elements. Anal. Chem. 1980, 52, 2283–2289. [Google Scholar] [CrossRef] [Green Version]
- Reed, T.B. Growth of refractory crystals using the induction plasma torch. J. Appl. Phys. 1961, 32, 2534–2535. [Google Scholar] [CrossRef]
- Webb, B.D.; Denton, M.B. Comparison of a very high frequency 148 MHz inductively coupled plasma to a 27 MHz ICP. Spectrochim. Acta Part B At. Spectrosc. 1986, 41, 361–376. [Google Scholar] [CrossRef]
- Fassel, V.A.; Kniseley, R.N. Inductively coupled plasmas. Anal. Chem. 1974, 46, 1155A–1164a. [Google Scholar] [CrossRef]
- Margoshes, M.; Scribner, B.F. Emission spectrometry. Anal. Chem. 1968, 40, 223–246. [Google Scholar] [CrossRef]
- Hammer, M.R. A magnetically excited microwave plasma source for atomic emission spectroscopy with performance approaching that of the inductively coupled plasma. Spectrochim. Acta Part B At. Spectrosc. 2008, 63, 456–464. [Google Scholar] [CrossRef]
- Polyakova, E.; Pelipasov, O. Plasma molecular species and matrix effects in the Hummer cavity microwave induced plasma optical emission spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2020, 173, 105988. [Google Scholar] [CrossRef]
- Xing, Z.; Wang, J.; Zhang, S.; Zhang, X. Determination of bismuth in solid samples by hydride generation atomic fluorescence spectrometry with a dielectric barrier discharge atomizer. Talanta 2009, 80, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.-J.; Wu, D.-F.; Yu, Y.-L.; Wang, J.-H. Nonthermal optical emission spectrometry: Direct atomization and excitation of cadmium for highly sensitive determination. Anal. Chem. 2016, 88, 4192–4195. [Google Scholar] [CrossRef] [Green Version]
- Anghel, S.D.; Simon, A.; Frentiu, T. Characterization of a very low power argon CCP. J. Anal. At. Spectrom. 2005, 20, 966–973. [Google Scholar] [CrossRef]
- Sharma, T.; Litoria, P.; Bajwa, B.; Kaur, I. Appraisal of groundwater quality and associated risks in Mansa district (Punjab, India). Environ. Monit. Assess. 2021, 193, 1–21. [Google Scholar] [CrossRef]
- Rice, G.; D’silva, A.; Fassel, V. A new He discharge-afterglow and its application as a gas chromatographic detector. Spectrochim. Acta Part B At. Spectrosc. 1985, 40, 1573–1584. [Google Scholar] [CrossRef]
- Deng, X.; Nikiforov, A.Y.; Vanraes, P.; Leys, C. Direct current plasma jet at atmospheric pressure operating in nitrogen and air. J. Appl. Phys. 2013, 113, 023305. [Google Scholar] [CrossRef] [Green Version]
- Parzen, P.; Goldstein, L. Current fluctuations in the direct-current gas discharge plasma. Phys. Rev. 1951, 82, 724–726. [Google Scholar] [CrossRef]
- Olsen, H. The electric arc as a light source for quantitative spectroscopy. J. Quant. Spectrosc. Radiat. Transf. 1963, 3, 305–333. [Google Scholar] [CrossRef]
- Olsen, H. Thermal and electrical properties of an argon plasma. Phys. Fluids 1959, 2, 614–623. [Google Scholar] [CrossRef]
- Domitz, S. Experimental Evaluation of a Direct-Current Low-Pressure Plasma Source; National Aeronautics and Space Administration, Lewis Research Center: Cleveland, OH, USA, 1963; pp. 1–34, NASA-TN-D-1659 United States DTIE English. [Google Scholar]
- Coleman, G.; Braun, W.; Allen, A. Characterization of an improved dc plasma excitation source. Appl. Spectrosc. 1980, 34, 24–30. [Google Scholar] [CrossRef]
- Tendero, C.; Tixier, C.; Tristant, P.; Desmaison, J.; Leprince, P. Atmospheric pressure plasmas: A review. Spectrochim. Acta Part B At. Spectrosc. 2006, 61, 2–30. [Google Scholar] [CrossRef]
- Pankaj, S.K.; Wan, Z.; Keener, K.M. Effects of cold plasma on food quality: A review. Foods 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, S.; Von Woedtke, T.; Weltmann, K.-D. The kINPen—A review on physics and chemistry of the atmospheric pressure plasma jet and its applications. J. Phys. D: Appl. Phys. 2018, 51, 233001. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shashurin, A. Study of atmospheric pressure plasma jet parameters generated by DC voltage driven cold plasma source. J. Appl. Phys. 2017, 122, 063301. [Google Scholar] [CrossRef]
- Ji, H.; Dong, S.; Han, F.; Li, Y.; Chen, G.; Li, L.; Chen, Y. Effects of dielectric barrier discharge (DBD) cold plasma treatment on physicochemical and functional properties of peanut protein. Food Bioprocess. Technol. 2018, 11, 344–354. [Google Scholar] [CrossRef]
- Bourke, P.; Ziuzina, D.; Han, L.; Cullen, P.; Gilmore, B.F. Microbiological interactions with cold plasma. J. Appl. Microbiol. 2017, 123, 308–324. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.; Chevalier, C.; Blades, M. A capacitively coupled microplasma (CCµP) formed in a channel in a quartz wafer. J. Anal. At. Spectrom. 2001, 16, 919–921. [Google Scholar] [CrossRef]
- Liang, D.C.; Blades, M. Atmospheric pressure capacitively coupled plasma atomizer for atomic absorption spectrometry. Anal. Chem. 1988, 60, 27–31. [Google Scholar] [CrossRef]
- Zou, Z.; Deng, Y.; Hu, J.; Jiang, X.; Hou, X. Recent trends in atomic fluorescence spectrometry towards miniaturized instrumentation-A review. Anal. Chim. Acta 2018, 1019, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Tombrink, S.; Müller, S.; Heming, R.; Michels, A.; Lampen, P.; Franzke, J. Liquid analysis dielectric capillary barrier discharge. Anal. Bioanal. Chem. 2010, 397, 2917–2922. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, W.; Zheng, C.; Hou, X. Dielectric barrier discharge in analytical spectrometry. Appl. Spectrosc. Rev. 2011, 46, 368–387. [Google Scholar] [CrossRef]
- Krähling, T.; Müller, S.; Meyer, C.; Stark, A.-K.; Franzke, J. Liquid electrode dielectric barrier discharge for the analysis of solved metals. J. Anal. At. Spectrom. 2011, 26, 1974–1978. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, J.; Zhang, S.; Na, X.; Zhang, X. Determination of Se, Pb, and Sb by atomic fluorescence spectrometry using a new flameless, dielectric barrier discharge atomizer. Spectrochim. Acta Part. B At. Spectrosc. 2008, 63, 431–436. [Google Scholar] [CrossRef]
- Niu, G.; Knodel, A.; Burhenn, S.; Brandt, S.; Franzke, J. Miniature Dielectric Barrier Discharge (DBD) in Analytical Atomic Spectrometry. Anal. Chim. Acta 2020, 1147, 211–239. [Google Scholar] [CrossRef]
- Michels, A.; Tombrink, S.; Vautz, W.; Miclea, M.; Franzke, J. Spectroscopic characterization of a microplasma used as ionization source for ion mobility spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2007, 62, 1208–1215. [Google Scholar] [CrossRef]
- Vautz, W.; Michels, A.; Franzke, J. Micro-plasma: A novel ionisation source for ion mobility spectrometry. Anal. Bioanal. Chem. 2008, 391, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Klute, F.D.; Schütz, A.; Marggraf, U.; Drees, C.; Vogel, P.; Vautz, W.; Franzke, J. Flexible microtube plasma (FμTP) as an embedded ionization source for a microchip mass spectrometer interface. Anal. Chem. 2018, 90, 10111–10116. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.Y.; Kang, J.H.; Choi, Y.S.; Lee, J.Y.; Park, J.S. Analytical features of microwave plasma-atomic emission spectrometry (MP-AES) for the quantitation of manganese (Mn) in wild grape (Vitis coignetiae) red wines: Comparison with inductively coupled plasma-optical emission spectrometry (ICP-OES). Food Chem. 2019, 274, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Sing, R.; Salin, E. Introduction of liquid samples into the inductively coupled plasma by direct insertion on a wire loop. Anal. Chem. 1989, 61, 163–169. [Google Scholar] [CrossRef]
- Buckley, B.T.; Boss, C.B. A tungsten filament vaporizer for sample introduction into a direct-current plasma. Appl. Spectrosc. 1990, 44, 505–512. [Google Scholar] [CrossRef]
- Kleinmann, I.; Svoboda, V. High-frequency excitation of independently vaporized samples in emission spectrometry. Anal. Chem. 1969, 41, 1029–1033. [Google Scholar] [CrossRef]
- Long, S.; Snook, R.; Browner, R. Some observations on electrothermal vaporisation for sample introduction into the inductively coupled plasma. Spectrochim. Acta Part B At. Spectrosc. 1985, 40, 553–568. [Google Scholar] [CrossRef]
- Gunn, A.; Millard, D.; Kirkbright, G. Optical emission spectrometry with an inductively coupled radiofrequency argon plasma source and sample introduction with a graphite rod electrothermal vaporisation device. Part I. Instrumental assembly and performance characteristics. Analyst 1978, 103, 1066–1073. [Google Scholar] [CrossRef]
- Qudus, H.I.; Purwadi, P.; Holilah, I.; Hadi, S. Analysis of Mercury in Skin Lightening Cream by Microwave Plasma Atomic Emission Spectroscopy (MP-AES). Molecules 2021, 26, 3130. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y. High-sensitivity microwave-induced plasma mass spectrometry for trace element analysis. J. Anal. At. Spectrom. 1994, 9, 745–749. [Google Scholar] [CrossRef]
- Rippetoe, W.; Johnson, E.; Vickers, T. Characterization of the plume of a direct current plasma arc for emission spectrometric analysis. Anal. Chem. 1975, 47, 436–440. [Google Scholar] [CrossRef]
- Braman, R.S.; Dynako, A. Direct current discharge spectral emission-type detector. Anal. Chem. 1968, 40, 95–106. [Google Scholar] [CrossRef]
- Gard, E.; Mayer, J.E.; Morrical, B.D.; Dienes, T.; Fergenson, D.P.; Prather, K.A. Real-time analysis of individual atmospheric aerosol particles: Design and performance of a portable ATOFMS. Anal. Chem. 1997, 69, 4083–4091. [Google Scholar] [CrossRef]
- Chen, H.; Gamez, G.; Zenobi, R. What can we learn from ambient ionization techniques? J. Am. Soc. Mass Spectrom. 2011, 20, 1947–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooks, R.G.; Ouyang, Z.; Takats, Z.; Wiseman, J.M. Ambient mass spectrometry. Science 2006, 311, 1566–1570. [Google Scholar] [CrossRef]
- Keil, A.; Talaty, N.; Janfelt, C.; Noll, R.J.; Gao, L.; Ouyang, Z.; Cooks, R.G. Ambient mass spectrometry with a handheld mass spectrometer at high pressure. Anal. Chem. 2007, 79, 7734–7739. [Google Scholar] [CrossRef]
- Yadav, B.; Spinelli, A.C.; Govindan, B.N.; Tsui, Y.Y.; McMullen, L.M.; Roopesh, M. Cold plasma treatment of ready-to-eat ham: Influence of process conditions and storage on inactivation of Listeria innocua. Food Res. Int. 2019, 123, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Rohner, U.; Whitby, J.A.; Wurz, P. A miniature laser ablation time-of-flight mass spectrometer for in situ planetary exploration. Meas. Sci. Technol. 2003, 14, 2159. [Google Scholar] [CrossRef]
- Afgan, M.S.; Hou, Z.; Wang, Z. Quantitative analysis of common elements in steel using a handheld μ-LIBS instrument. J. Anal. At. Spectrom. 2017, 32, 1905–1915. [Google Scholar] [CrossRef]
- Meuzelaar, H.L.; McClennen, W.; Dworzanski, J.; Sheya, S.; Snyder, A.; Harden, C.; Arnold, N. Hyphenated Techniques: The Next Generation of Field-Portable Analytical Instruments? Air and Waste Management Association: Pittsburgh, PA, USA, 1995. [Google Scholar]
- Fantz, U. Basics of plasma spectroscopy. Plasma Sources Sci. Technol. 2006, 15, S137. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.; Zhang, K.; Xue, Y.; Liu, W.; Chen, T.; Lu, Y.; Zhao, G. Review of MEMS Based Fourier Transform Spectrometers. Micromachines 2020, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.P. Product Review: Spectrometers Get Small. Anal. Chem. 2000, 72, 653A–658A. [Google Scholar] [CrossRef]
- Bacon, C.P.; Mattley, Y.; DeFrece, R. Miniature spectroscopic instrumentation: Applications to biology and chemistry. Rev. Sci. Instrum. 2004, 75, 1–16. [Google Scholar] [CrossRef]
- Cai, F.; Wang, Y.; Gao, M.; He, S. The design and implementation of a low-cost multispectral endoscopy through galvo scanning of a fiber bundle. Opt. Commun. 2018, 428, 1–6. [Google Scholar] [CrossRef]
- Sigernes, F.; Syrjäsuo, M.; Storvold, R.; Fortuna, J.; Grøtte, M.E.; Johansen, T.A. Do it yourself hyperspectral imager for handheld to airborne operations. Opt. Express 2018, 26, 6021–6035. [Google Scholar] [CrossRef] [PubMed]
- Crocombe, R.A. Handheld spectrometers: The state of the art. In Proceedings of the Next-Generation Spectroscopic Technologies VI, 8726, 87260R, International Society for Optics and Photonics, Baltimore, MD, USA, 29 May 2013. [Google Scholar]
- Frentiu, T.; Petreus, D.; Senila, M.; Mihaltan, A.I.; Darvasi, E.; Ponta, M.; Plaian, E.; Cordos, E.A. Low power capacitively coupled plasma microtorch for simultaneous multielemental determination by atomic emission using microspectrometers. Microchem. J. 2011, 97, 188–195. [Google Scholar] [CrossRef]
- Ye, E.; Atabaki, A.H.; Han, N.; Ram, R.J. Miniature, sub-nanometer resolution Talbot spectrometer. Opt. Lett. 2016, 41, 2434–2437. [Google Scholar] [CrossRef] [PubMed]
- McGonigle, A.J.; Wilkes, T.C.; Pering, T.D.; Willmott, J.R.; Cook, J.M.; Mims, F.M.; Parisi, A.V. Smartphone spectrometers. Sensors 2018, 18, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, D.; Wang, B.; Huang, H.; Meng, X.; Liu, C.; Xue, L.; Liu, F.; Wang, S. Sunlight based handheld smartphone spectrometer. Biosens. Bioelectron. 2019, 143, 111632. [Google Scholar] [CrossRef]
- You, D.J.; San Park, T.; Yoon, J.-Y. Cell-phone-based measurement of TSH using Mie scatter optimized lateral flow assays. Biosens. Bioelectron. 2013, 40, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jeon, M.; Kim, Y.-J.; Choi, S. Open-source, handheld, wireless spectrometer for rapid biochemical assays. Sens. Actuators B Chem. 2020, 306, 127537. [Google Scholar] [CrossRef]
- Xu, W.; Manicke, N.E.; Cooks, G.R.; Ouyang, Z. Miniaturization of mass spectrometry analysis systems. JALA J. Assoc. Lab. Autom. 2010, 15, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Z.; Cooks, R.G. Miniature mass spectrometers. Annu. Rev. Anal. Chem. 2009, 2, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Noll, R.J.; Cooks, R.G. Handheld Miniature Ion Trap Mass Spectrometers. Anal. Chem. 2009, 81, 2421–2425. [Google Scholar] [CrossRef] [Green Version]
- Snyder, D.T.; Pulliam, C.J.; Ouyang, Z.; Cooks, R.G. Miniature and fieldable mass spectrometers: Recent advances. Anal. Chem. 2016, 88, 2–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Guo, M.; Cheng, Y.; Wang, Y.; Sun, W.; Zhang, H.; Dong, M.; Li, G. A review of the development and application of space miniature mass spectrometers. Vacuum 2018, 155, 108–117. [Google Scholar] [CrossRef]
- Guo, Q.; Gao, L.; Zhai, Y.; Xu, W. Recent developments of miniature ion trap mass spectrometers. Chin. Chem. Lett. 2018, 29, 1578–1584. [Google Scholar] [CrossRef]
- Miller, P.E.; Denton, M.B. The quadrupole mass filter: Basic operating concepts. J. Chem. Educ. 1986, 63, 617. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Yates III, J.R. The quadrupole ion trap mass spectrometer—A small solution to a big challenge. Anal. Biochem. 1997, 244, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blakeman, K. Development of High Pressure Mass Spectrometry for Handheld Instruments. Ph.D. Dissertation, University of North Carolina at Chapel Hill Graduate School, Chapel Hill, NC, USA, 2015. [Google Scholar]
- Gao, L.; Song, Q.; Patterson, G.E.; Cooks, R.G.; Ouyang, Z. Handheld rectilinear ion trap mass spectrometer. Anal. Chem. 2006, 78, 5994–6002. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.B.; Brokenshire, J.L. Hand-held ion mobility spectrometers. TrAC Trends Anal. Chem. 1994, 13, 275–280. [Google Scholar] [CrossRef]
- Hoaglund, C.S.; Valentine, S.J.; Sporleder, C.R.; Reilly, J.P.; Clemmer, D.E. Three-dimensional ion mobility/TOFMS analysis of electrosprayed biomolecules. Anal. Chem. 1998, 70, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Hollerbach, A.; Fedick, P.W.; Cooks, R.G. Ion mobility–mass spectrometry using a dual-gated 3D printed ion mobility spectrometer. Anal. Chem. 2018, 90, 13265–13272. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, T.; Clowers, B.H. Implementation of a flexible, open-source platform for ion mobility spectrometry. HardwareX 2018, 4, e00030. [Google Scholar] [CrossRef]
- Kabir, K.M.; Donald, W.A. Microscale differential ion mobility spectrometry for field deployable chemical analysis. TrAC Trends Anal. Chem. 2017, 97, 399–427. [Google Scholar] [CrossRef]
- Merenbloom, S.I.; Bohrer, B.C.; Koeniger, S.L.; Clemmer, D.E. Assessing the peak capacity of IMS—IMS separations of tryptic peptide ions in He at 300 K. Anal. Chem. 2007, 79, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Mielczarek, P.; Silberring, J.; Smoluch, M. Miniaturization in mass spectrometry. Mass Spectrom. Rev. 2020, 39, 453–470. [Google Scholar] [CrossRef]
- Yang, M.; Kim, T.-Y.; Hwang, H.-C.; Yi, S.-K.; Kim, D.-H. Development of a palm portable mass spectrometer. J. Am. Soc. Mass Spectrom. 2008, 19, 1442–1448. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, P.I.; Dalgleish, J.K.; Shelley, J.T.; Kirleis, M.A.; McNicholas, M.T.; Li, L.; Chen, T.-C.; Chen, C.-H.; Duncan, J.S.; Boudreau, F. Autonomous in situ analysis and real-time chemical detection using a backpack miniature mass spectrometer: Concept, instrumentation development, and performance. Anal. Chem. 2014, 86, 2900–2908. [Google Scholar] [CrossRef]
- Li, L.; Chen, T.-C.; Ren, Y.; Hendricks, P.I.; Cooks, R.G.; Ouyang, Z. Mini 12, Miniature Mass Spectrometer for Clinical and Other Applications—Introduction and Characterization. Anal. Chem. 2014, 86, 2909–2916. [Google Scholar] [CrossRef]
- Maas, J.D.; Hendricks, P.I.; Ouyang, Z.; Cooks, R.G.; Chappell, W.J. Miniature monolithic rectilinear ion trap arrays by stereolithography on printed circuit board. J. Microelectromech. Syst. 2010, 19, 951–960. [Google Scholar] [CrossRef]
- Hendricks, P.; Duncan, J.; Noll, R.J.; Ouyang, Z.; Cooks, R.G. Performance of a low voltage ion trap. Int. J. Mass Spectrom. 2011, 305, 69–73. [Google Scholar] [CrossRef]
- Stravs, M.A.; Stamm, C.; Ort, C.; Singer, H. Transportable Automated HRMS Platform “MS2field” Enables Insights into Water-Quality Dynamics in Real Time. Environ. Sci. Technol. Lett. 2021, 8. [Google Scholar] [CrossRef]
- Wang, J. Portable electrochemical systems. Trac Trends Anal. Chem. 2002, 21, 226–232. [Google Scholar] [CrossRef]
- Motalebizadeh, A.; Bagheri, H.; Asiaei, S.; Fekrat, N.; Afkhami, A. New portable smartphone-based PDMS microfluidic kit for the simultaneous colorimetric detection of arsenic and mercury. RSC Adv. 2018, 8, 27091–27100. [Google Scholar] [CrossRef] [Green Version]
- Cremers, D.A.; Ferris, M.J.; Davies, M. Transportable laser-induced breakdown spectroscopy (LIBS) instrument for field-based soil analysis. In Advanced Technologies for Environmental Monitoring and Remediation; Society of Photo-Optical Instrumentation Engineers: Bellingham, WA, USA, 1996; pp. 190–200. [Google Scholar]
- Myers, M.J.; Myers, J.D.; Sarracino, J.T.; Hardy, C.R.; Guo, B.; Christian, S.M.; Myers, J.A.; Roth, F.; Myers, A.G. LIBS system with compact fiber spectrometer, head mounted spectra display and hand held eye-safe erbium glass laser gun. In Solid State Lasers XIX: Technology and Devices; International Society for Optics and Photonics: San Francisco, CA, USA, 2010. [Google Scholar]
- Ripoll, L.; Hidalgo, M. Electrospray deposition followed by laser-induced breakdown spectroscopy (ESD-LIBS): A new method for trace elemental analysis of aqueous samples. J. Anal. At. Spectrom. 2019, 34, 2016–2026. [Google Scholar] [CrossRef]
- Senesi, G.S.; Harmon, R.S.; Hark, R.R. Field-portable and handheld LIBS: Historical review, current status and future prospects. Spectrochim. Acta Part B At. Spectrosc. 2020, 175, 106013. [Google Scholar] [CrossRef]
- Borrill, A.J.; Reily, N.E.; Macpherson, J.V. Addressing the practicalities of anodic stripping voltammetry for heavy metal detection: A tutorial review. Analyst 2019, 144, 6834–6849. [Google Scholar] [CrossRef] [PubMed]
- Kruusma, J.; Banks, C.E.; Compton, R.G. Mercury-free sono-electroanalytical detection of lead in human blood by use of bismuth-film-modified boron-doped diamond electrodes. Anal. Bioanal. Chem. 2004, 379, 700–706. [Google Scholar] [CrossRef]
- Kalnicky, D.J.; Singhvi, R. Field portable XRF analysis of environmental samples. J. Hazard. Mater. 2001, 83, 93–122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Yuan, Z.; Cheng, Q.; Zhang, Z.; Yang, J. Rapid in situ determination of heavy metal concentrations in polluted water via portable XRF: Using Cu and Pb as example. Environ. Pollut. 2018, 243, 1325–1333. [Google Scholar] [CrossRef]
- You, R.; Li, P.; Jing, G.; Cui, T. Ultrasensitive micro ion selective sensor arrays for multiplex heavy metal ions detection. Microsyst. Technol. 2019, 25, 845–849. [Google Scholar] [CrossRef]
- Szigeti, Z.; Vigassy, T.; Bakker, E.; Pretsch, E. Approaches to improving the lower detection limit of polymeric membrane ion-selective electrodes. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2006, 18, 1254–1265. [Google Scholar] [CrossRef]
- Sobin, C.; Parisi, N.; Schaub, T.; de la Riva, E. A Bland–Altman comparison of the Lead Care® System and inductively coupled plasma mass spectrometry for detecting low-level lead in child whole blood samples. J. Med Toxicol. 2011, 7, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.; Ortiz, D.; Pappas, S.; Quigley, S.; Yendell, S.; Ettinger, A.S. Response to the US FDA LeadCare Testing Systems Recall and CDC Health Alert. J. Public Health Manag. Pract. JPHMP 2019, 25, S91. [Google Scholar] [CrossRef] [PubMed]
- Montaño, M.D.; Olesik, J.W.; Barber, A.G.; Challis, K.; Ranville, J.F. Single Particle ICP-MS: Advances toward routine analysis of nanomaterials. Anal. Bioanal. Chem. 2016, 408, 5053–5074. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Traub, H.; Jakubowski, N.; Drescher, D.; Baranov, V.I.; Kneipp, J. Trends in single-cell analysis by use of ICP-MS. Anal. Bioanal. Chem. 2014, 406, 6963–6977. [Google Scholar] [CrossRef]
- Barreiros, M.; Carvalho, M.; Costa, M.; Marques, M.; Ramos, M. Application of total reflection XRF to elemental studies of drinking water. X-ray Spectrom. Int. J. 1997, 26, 165–168. [Google Scholar] [CrossRef]
- Kang, J.; Li, R.; Wang, Y.; Chen, Y.; Yang, Y. Ultrasensitive detection of trace amounts of lead in water by LIBS-LIF using a wood-slice substrate as a water absorber. J. Anal. At. Spectrom. 2017, 32, 2292–2299. [Google Scholar] [CrossRef]
- Wang, J. Stripping analysis at bismuth electrodes: A review. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2005, 17, 1341–1346. [Google Scholar] [CrossRef]
- Xing, G.; Sardar, M.R.; Lin, B.; Lin, J.-M. Analysis of trace metals in water samples using NOBIAS chelate resins by HPLC and ICP-MS. Talanta 2019, 204, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Milne, A.; Landing, W.; Bizimis, M.; Morton, P. Determination of Mn, Fe, Co, Ni, Cu, Zn, Cd and Pb in seawater using high resolution magnetic sector inductively coupled mass spectrometry (HR-ICP-MS). Anal. Chim. Acta 2010, 665, 200–207. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Z. Wavelet smoothing applied to the determination of trace arsenic, lead, antimony and selenium in environmental water by ICP-OES. J. Anal. At. Spectrom. 2004, 19, 738–742. [Google Scholar] [CrossRef]
- Bakker, E.; Pretsch, E. Modern potentiometry. Angew. Chem. Int. Ed. 2007, 46, 5660–5668. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Xie, Q.; Ming, H. Simultaneous sensitive analysis of Cd (ii), Pb (ii) and As (iii) using a dual-channel anodic stripping voltammetry approach. New J. Chem. 2020, 44, 5739–5745. [Google Scholar] [CrossRef]
- Tanvir, E.; Whitfield, K.M.; Ng, J.C.; Shaw, P.N. Development and validation of an ICP-MS method and its application to determine multiple trace elements in small volumes of whole blood and plasma. J. Anal. Toxicol. 2020, 44, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Massadeh, A.; Gharibeh, A.; Omari, K.; Al-Momani, I.; Alomari, A.; Tumah, H.; Hayajneh, W. Simultaneous determination of Cd, Pb, Cu, Zn, and Se in human blood of Jordanian smokers by ICP-OES. Biol. Trace Elem. Res. 2010, 133, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soleymanpour, A.; Shafaatian, B.; Kor, K.; Hasaninejad, A.R. Coated wire lead (II)-selective electrode based on a Schiff base ionophore for low concentration measurements. Mon. Für Chem. Chem. Mon. 2012, 143, 181–188. [Google Scholar] [CrossRef]
- Landes, F.C.; Paltseva, A.; Sobolewski, J.M.; Cheng, Z.; Ellis, T.K.; Mailloux, B.J.; van Geen, A. A field procedure to screen soil for hazardous lead. Anal. Chem. 2019, 91, 8192–8198. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Coles, B.J.; Ramsey, M.H.; Thornton, I. Sequential extraction of soils for multielement analysis by ICP-AES. Chem. Geol. 1995, 124, 109–123. [Google Scholar] [CrossRef]
- Rehan, I.; Gondal, M.; Rehan, K. Determination of lead content in drilling fueled soil using laser induced spectral analysis and its cross validation using ICP/OES method. Talanta 2018, 182, 443–449. [Google Scholar] [CrossRef]
- Kadachi, A.N.; Al-Eshaikh, M.A. Limits of detection in XRF spectroscopy. X-ray Spectrom. 2012, 41, 350–354. [Google Scholar] [CrossRef]
- Tian, K.; Huang, B.; Xing, Z.; Hu, W. In situ investigation of heavy metals at trace concentrations in greenhouse soils via portable X-ray fluorescence spectroscopy. Environ. Sci. Pollut. Res. 2018, 25, 11011–11022. [Google Scholar] [CrossRef]
- Kadara, R.O.; Tothill, I.E. Stripping chronopotentiometric measurements of lead (II) and cadmium (II) in soils extracts and wastewaters using a bismuth film screen-printed electrode assembly. Anal. Bioanal. Chem. 2004, 378, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Riondato, J.; Vanhaecke, F.; Moens, L.; Dams, R. Fast and reliable determination of (ultra-) trace and/or spectrally interfered elements in water by sector field ICP-MS. J. Anal. At. Spectrom. 2000, 15, 341–345. [Google Scholar] [CrossRef]
- Fichet, P.; Tabarant, M.; Salle, B.; Gautier, C. Comparisons between libs and ICP/OES. Anal. Bioanal. Chem. 2006, 385, 338–344. [Google Scholar] [CrossRef]
- Mages, M.; Woelfl, S.; Óvári, M. The use of a portable total reflection X-ray fluorescence spectrometer for field investigation. Spectrochim. Acta Part B At. Spectrosc. 2003, 58, 2129–2138. [Google Scholar] [CrossRef]
- Kallithrakas-Kontos, N.; Koulouridakis, P.; Hatzistavros, V.; Aretaki, I. Chromium speciation by TXRF analysis. X-ray Spectrom. Int. J. 2009, 38, 152–156. [Google Scholar] [CrossRef]
- Leśniewska, B.; Godlewska-Żyłkiewicz, B. Speciation of chromium in alkaline soil extracts by an ion-pair reversed phase HPLC-ICP MS method. Molecules 2019, 24, 1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamatha, P.; Venkateswarlu, G.; Swamy, A.; Sahayam, A. Microwave assisted extraction of Cr (III) and Cr (VI) from soil/sediments combined with ion exchange separation and inductively coupled plasma optical emission spectrometry detection. Anal. Methods 2014, 6, 9653–9657. [Google Scholar] [CrossRef]
- Dell’Aglio, M.; Gaudiuso, R.; Senesi, G.S.; De Giacomo, A.; Zaccone, C.; Miano, T.M.; De Pascale, O. Monitoring of Cr, Cu, Pb, V and Zn in polluted soils by laser induced breakdown spectroscopy (LIBS). J. Environ. Monit. 2011, 13, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Hartyani, Z.; Dávid, E.; Szabó, S.; Szilágyi, V.; Horváth, T.; Tóth, Á.H. Determination of the trace elements distribution of polluted soils in Hungary by X-ray methods. Microchem. J. 2000, 67, 195–200. [Google Scholar] [CrossRef]
- Terán-Baamonde, J.; Soto-Ferreiro, R.-M.; Carlosena, A.; Andrade, J.-M.; Prada, D. Determination of cadmium in sediments by diluted HCI extraction and isotope dilution ICP-MS. Talanta 2018, 186, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.; Fassel, V.; D’silva, A. Computer-controlled scanning monochromator for the determination of 50 elements in geochemical and environmental samples by inductively coupled plasma-atomic emission spectrometry. Anal. Chem. 1980, 52, 2168–2172. [Google Scholar] [CrossRef]
- Santos, D., Jr.; Nunes, L.C.; Trevizan, L.C.; Godoi, Q.; Leme, F.O.; Braga, J.W.B.; Krug, F.J. Evaluation of laser induced breakdown spectroscopy for cadmium determination in soils. Spectrochim. Acta Part B At. Spectrosc. 2009, 64, 1073–1078. [Google Scholar] [CrossRef]
- Knopp, R.; Scherbaum, F.; Kim, J. Laser induced breakdown spectroscopy (LIBS) as an analytical tool for the detection of metal ions in aqueous solutions. Fresenius’ J. Anal. Chem. 1996, 355, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Marguí, E.; Queralt, I.; Hidalgo, M. Determination of cadmium at ultratrace levels in environmental water samples by means of total reflection X-ray spectrometry after dispersive liquid–liquid microextraction. J. Anal. At. Spectrom. 2013, 28, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Essi, M.; Kouame, N.; Cisse, G. Cd-ISe and membrane technique device for in situ monitoring. Chalcogenide Lett. 2018, 15, 371–378. [Google Scholar]
- Venus, M.; Puntarić, D.; Gvozdić, V.; Vidosavljević, D.; Bijelić, L.; Puntarić, A.; Puntarić, E.; Vidosavljević, M.; Matijana, J.; Jasenka, Š. Determinations of uranium concentrations in soil, water, vegetables and biological samples from inhabitants of war affected areas in eastern Croatia (ICP-MS method). J. Environ. Radioact. 2019, 203, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, G.; Jabbari, A.; Yamini, Y.; Paki, D. Determination of uranium and thorium in natural waters by ICP-OES after on-line solid phase extraction and preconcentration in the presence of 2, 3-dihydro-9, 10-dihydroxy-1, 4-antracenedion. J. Anal. Chem. 2009, 64, 602–608. [Google Scholar] [CrossRef]
- Sarkar, A.; Alamelu, D.; Aggarwal, S.K. Determination of thorium and uranium in solution by laser-induced breakdown spectrometry. Appl. Opt. 2008, 47, G58–G64. [Google Scholar] [CrossRef]
- Alsecz, A.; Osan, J.; Kurunczi, S.; Alföldy, B.; Varhegyi, A.; Török, S. Analytical performance of different X-ray spectroscopic techniques for the environmental monitoring of the recultivated uranium mine site. Spectrochim. Acta Part B At. Spectrosc. 2007, 62, 769–776. [Google Scholar] [CrossRef]
- Agrahari, S.; Kumar, S.; Srivastava, A. Ion selective electrode for uranium based on composite multiwalled carbon nanotube-benzo-15-crown-5 in PVC matrix coated on graphite rod. J. Anal. Chem. 2014, 69, 36–44. [Google Scholar] [CrossRef]
- Trivelpiece, A.; Gould, R. Space charge waves in cylindrical plasma columns. J. Appl. Phys. 1959, 30, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Moisan, M.; Beaudry, C.; Lepprince, P. A new HF device for the production of long plasma columns at a high electron density. Phys. Lett. A 1974, 50, 125–126. [Google Scholar] [CrossRef]
- Moisan, M.; Leprince, P.; Beaudry, C.; Bloyet, E. Devices and Methods of Using HF Waves to Energize a Column of Gas Enclosed in an Insulating Casing. U.S. Patent No. 4,049,940, 20 September 1977. [Google Scholar]
- Moisan, M.; Beaudry, C.; Leprince, P. A small microwave plasma source for long column production without magnetic field. IEEE Trans. Plasma Sci. 1975, 3, 55–59. [Google Scholar] [CrossRef]
- Moisan, M.; Zakrzewski, Z.; Pantel, R. The theory and characteristics of an efficient surface wave launcher (surfatron) producing long plasma columns. J. Phys. D Appl. Phys. 1979, 12, 219. [Google Scholar] [CrossRef]
- Selby, M.; Hieftje, G.M. Taming the surfatron. Spectrochim. Acta Part B At. Spectrosc. 1987, 42, 285–298. [Google Scholar] [CrossRef]
- Long, G.L.; Perkins, L.D. Direct introduction of aqueous samples into a low-powered microwave-induced plasma for atomic emission spectrometry. Appl. Spectrosc. 1987, 41, 980–985. [Google Scholar] [CrossRef]
- Deng, Y.; Zeng, W.; Jiang, X.; Hou, X. Portable photochemical vapor generation-microwave plasma optical emission spectrometer. J. Anal. At. Spectrom. 2020, 35, 1316–1319. [Google Scholar] [CrossRef]
- Haertel, B.; Von Woedtke, T.; Weltmann, K.-D.; Lindequist, U. Non-thermal atmospheric-pressure plasma possible application in wound healing. Biomol. Ther. 2014, 22, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keidar, M. Plasma for cancer treatment. Plasma Sources Sci. Technol. 2015, 24, 033001. [Google Scholar] [CrossRef]
- Misra, N.; Moiseev, T.; Patil, S.; Pankaj, S.; Bourke, P.; Mosnier, J.; Keener, K.; Cullen, P. Cold plasma in modified atmospheres for post-harvest treatment of strawberries. Food Bioprocess Technol. 2014, 7, 3045–3054. [Google Scholar] [CrossRef]
- Pavlovich, M.J.; Clark, D.S.; Graves, D.B. Quantification of air plasma chemistry for surface disinfection. Plasma Sources Sci. Technol. 2014, 23, 065036. [Google Scholar] [CrossRef]
- Vasilyak, L. Physical methods of disinfection (a review). Plasma Phys. Rep. 2021, 47, 318–327. [Google Scholar] [CrossRef]
- Moon, S.Y.; Choe, W. A comparative study of rotational temperatures using diatomic OH, O2 and N2+ molecular spectra emitted from atmospheric plasmas. Spectrochim. Acta Part B At. Spectrosc. 2003, 58, 249–257. [Google Scholar] [CrossRef]
- Workman, J.M.; Fleitz, P.; Fannin, H.B.; Caruso, J.A.; Seliskar, C. A comparative study of rotational temperatures in a microwave plasma: OH radical versus N2+. Appl. Spectrosc. 1988, 42, 96–100. [Google Scholar] [CrossRef]
- Sysolyatina, E.; Vasiliev, M.; Kurnaeva, M.; Kornienko, I.; Petrov, O.; Fortov, V.; Gintsburg, A.; Petersen, E.; Ermolaeva, S. Frequency of cell treatment with cold microwave argon plasma is important for the final outcome. J. Phys. D Appl. Phys. 2016, 49, 294002. [Google Scholar] [CrossRef]
- Duarte, S.; Kuo, S.; Murata, R.; Chen, C.; Saxena, D.; Huang, K.; Popovic, S. Air plasma effect on dental disinfection. Phys. Plasmas 2011, 18, 073503. [Google Scholar] [CrossRef]
- Iuchi, K.; Morisada, Y.; Yoshino, Y.; Himuro, T.; Saito, Y.; Murakami, T.; Hisatomi, H. Cold atmospheric-pressure nitrogen plasma induces the production of reactive nitrogen species and cell death by increasing intracellular calcium in HEK293T cells. Arch. Biochem. Biophys. 2018, 654, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Motley, C.B.; Long, G.L. Evaluation of sample introduction techniques of packed-column SFC into an MIP. Appl. Spectrosc. 1990, 44, 667–672. [Google Scholar] [CrossRef]
- Wind, M.; Eisenmenger, A.; Lehmann, W.D. Modified direct injection high efficiency nebulizer with minimized dead volume for the analysis of biological samples by micro-and nano-LC-ICP-MS. J. Anal. At. Spectrom. 2002, 17, 21–26. [Google Scholar] [CrossRef]
- Takats, Z.; Wiseman, J.M.; Cooks, R.G. Ambient mass spectrometry using desorption electrospray ionization (DESI): Instrumentation, mechanisms and applications in forensics, chemistry, and biology. J. Mass Spectrom. 2005, 40, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Houk, R. Arc nebulization for elemental analysis of conducting solids by inductively coupled plasma mass spectrometry. Anal. Chem. 1986, 58, 1739–1743. [Google Scholar] [CrossRef]
- Brenner, I.; Lorber, A.; Goldbart, Z. Trace element analysis of geological materials by direct solids insertion of a graphite cup into an inductively coupled plasma. Spectrochim. Acta Part B At. Spectrosc. 1987, 42, 219–225. [Google Scholar] [CrossRef]

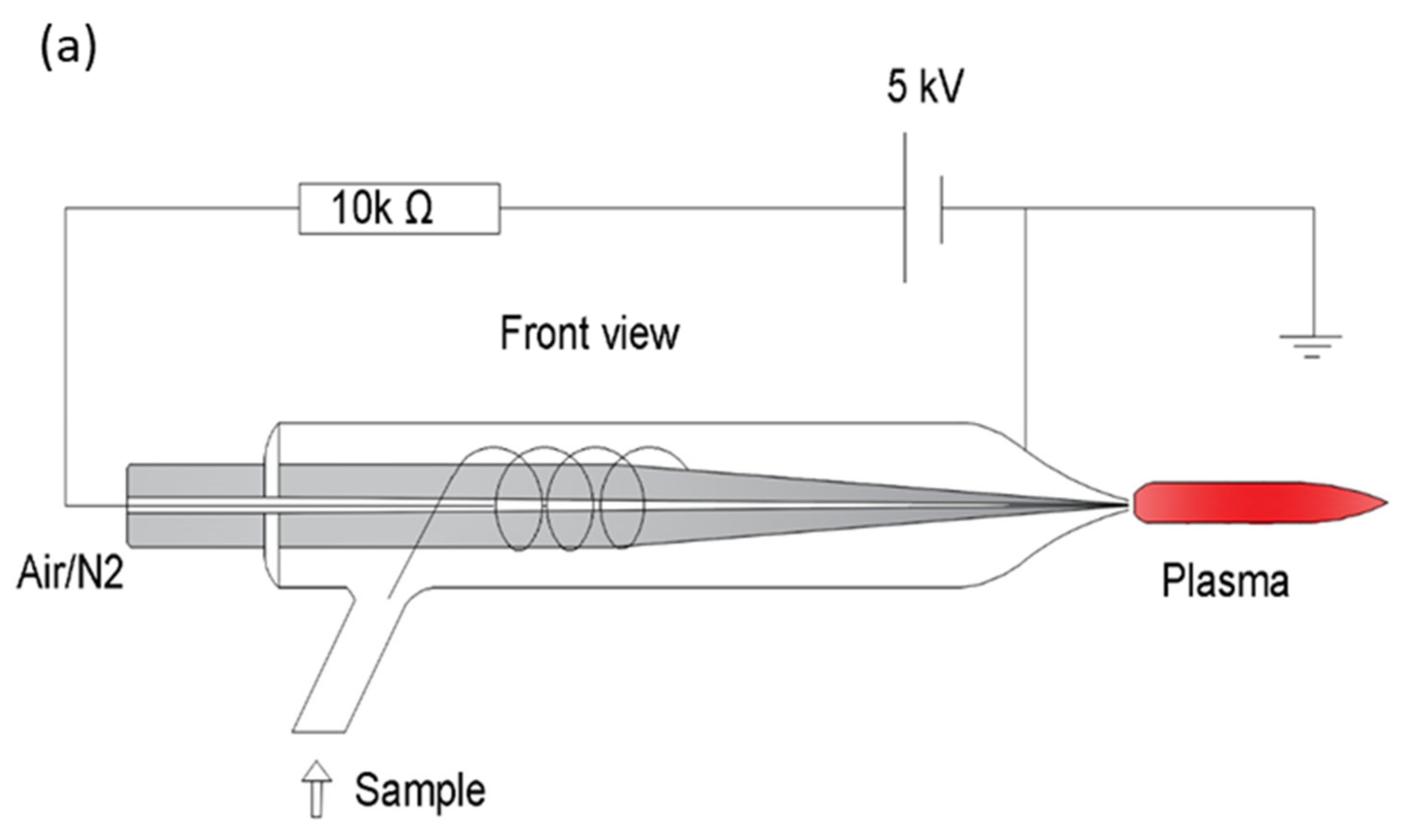
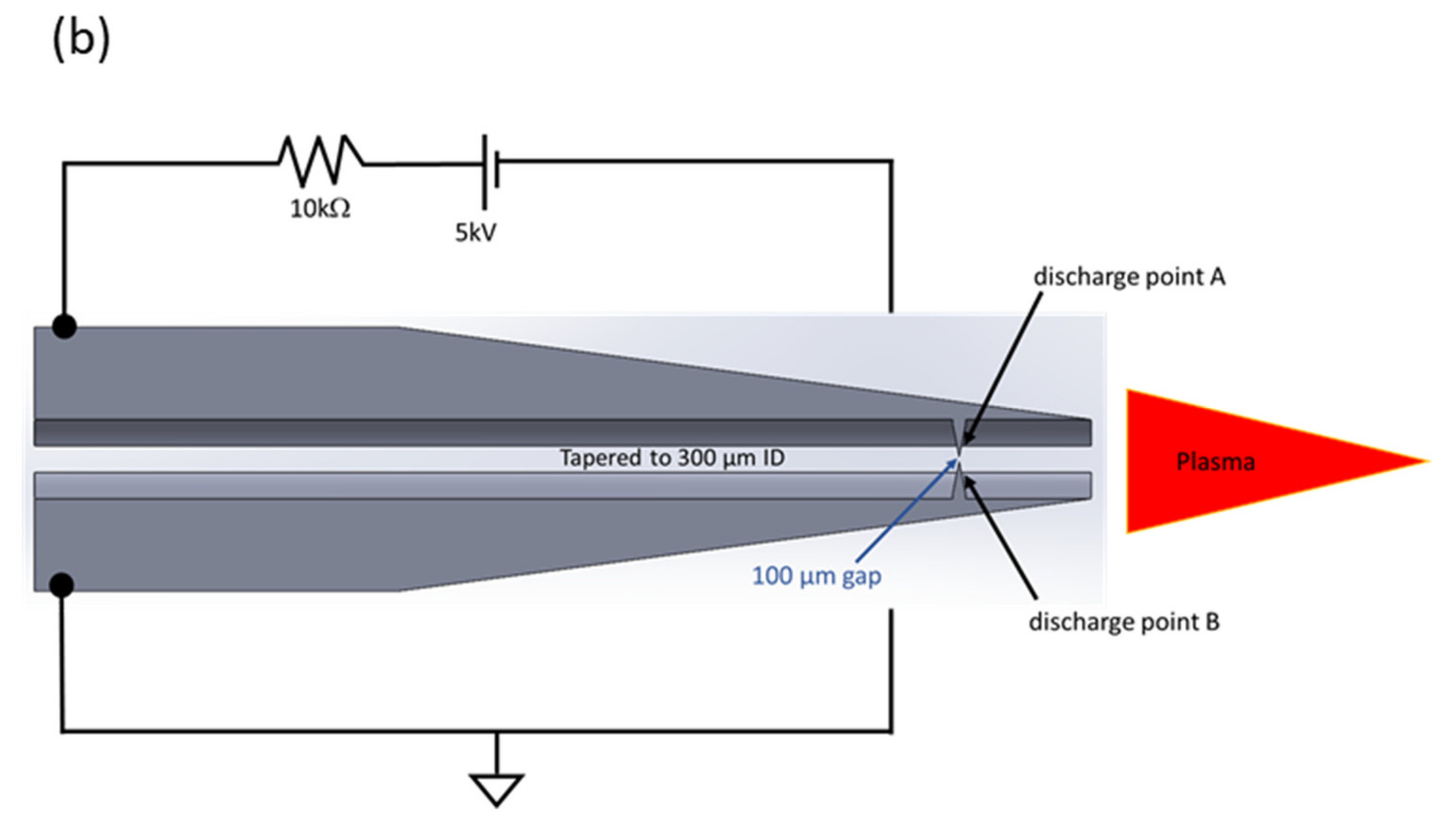

{kind=link}
{kind=link}
{kind=link}
| Analytical Method | |||||||
|---|---|---|---|---|---|---|---|
| Element | Matrix | ICPMS | ICPOES | LIBS | XRF | ISE | ASV/LeadCare |
| Pb | Water | 1.5 [118]; 0.0042 [119] | 2 [120] | 0.32 * [116] | 0.9 [115] | 0.012 [121] (buffer) | 0.05 [122] |
| Pb | Blood | 0.06 [123] | 1.5 [124] | N/A | N/A | 10,000 ** [125] | 0.2 [106]; 14 [4] |
| Pb | Solid | 100 [126] | 4400 [127] | 42,000 [104]; 125,000 [128] | 5 [129]; 5000 [130] | N/A | 10 [131] |
| Cr | Water | 3 [132] | 0.2 [133] | 700 [133] | 2 [134]; 0.6 [135] | 0.4 [109] | 0.1 [117] |
| Cr | Solid | 4 (Cr III); 4.5 (Cr VI) [136] | 5000 (Cr VI) [137] | 17,000 [138] | 2000 [139] | N/A | N/A |
| Cd | Solid | 1.8 [140] | 500 [141] | 4000 [142] | 46,000 [129] | N/A | N/A |
| Cd | Water | 1.2 [118] | 0.14 [115] | 500,000 [143] | 4.9 [115]; 0.04 [144] | 11 [145] | 0.03 [122] |
| U | Water | 0.001 [146] | 0.69 [147] | 19,000 [148] | 17 [149] | 13 [150] | 0.3 [117] |
| U | Solid | 480 [146] | 6000 [141] | 250,000 [104] | 10,000 [149] | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckley, B.T.; Buckley, R.; Doherty, C.L. Moving toward a Handheld “Plasma” Spectrometer for Elemental Analysis, Putting the Power of the Atom (Ion) in the Palm of Your Hand. Molecules 2021, 26, 4761. https://doi.org/10.3390/molecules26164761
Buckley BT, Buckley R, Doherty CL. Moving toward a Handheld “Plasma” Spectrometer for Elemental Analysis, Putting the Power of the Atom (Ion) in the Palm of Your Hand. Molecules. 2021; 26(16):4761. https://doi.org/10.3390/molecules26164761
Chicago/Turabian StyleBuckley, Brian T., Rachel Buckley, and Cathleen L. Doherty. 2021. "Moving toward a Handheld “Plasma” Spectrometer for Elemental Analysis, Putting the Power of the Atom (Ion) in the Palm of Your Hand" Molecules 26, no. 16: 4761. https://doi.org/10.3390/molecules26164761
APA StyleBuckley, B. T., Buckley, R., & Doherty, C. L. (2021). Moving toward a Handheld “Plasma” Spectrometer for Elemental Analysis, Putting the Power of the Atom (Ion) in the Palm of Your Hand. Molecules, 26(16), 4761. https://doi.org/10.3390/molecules26164761