
Biocompatibility Study of Electrospun Nanocomposite Membranes Based on Chitosan/Polyvinyl Alcohol/Oxidized Carbon Nano-Onions

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Nanocomposite Characterization
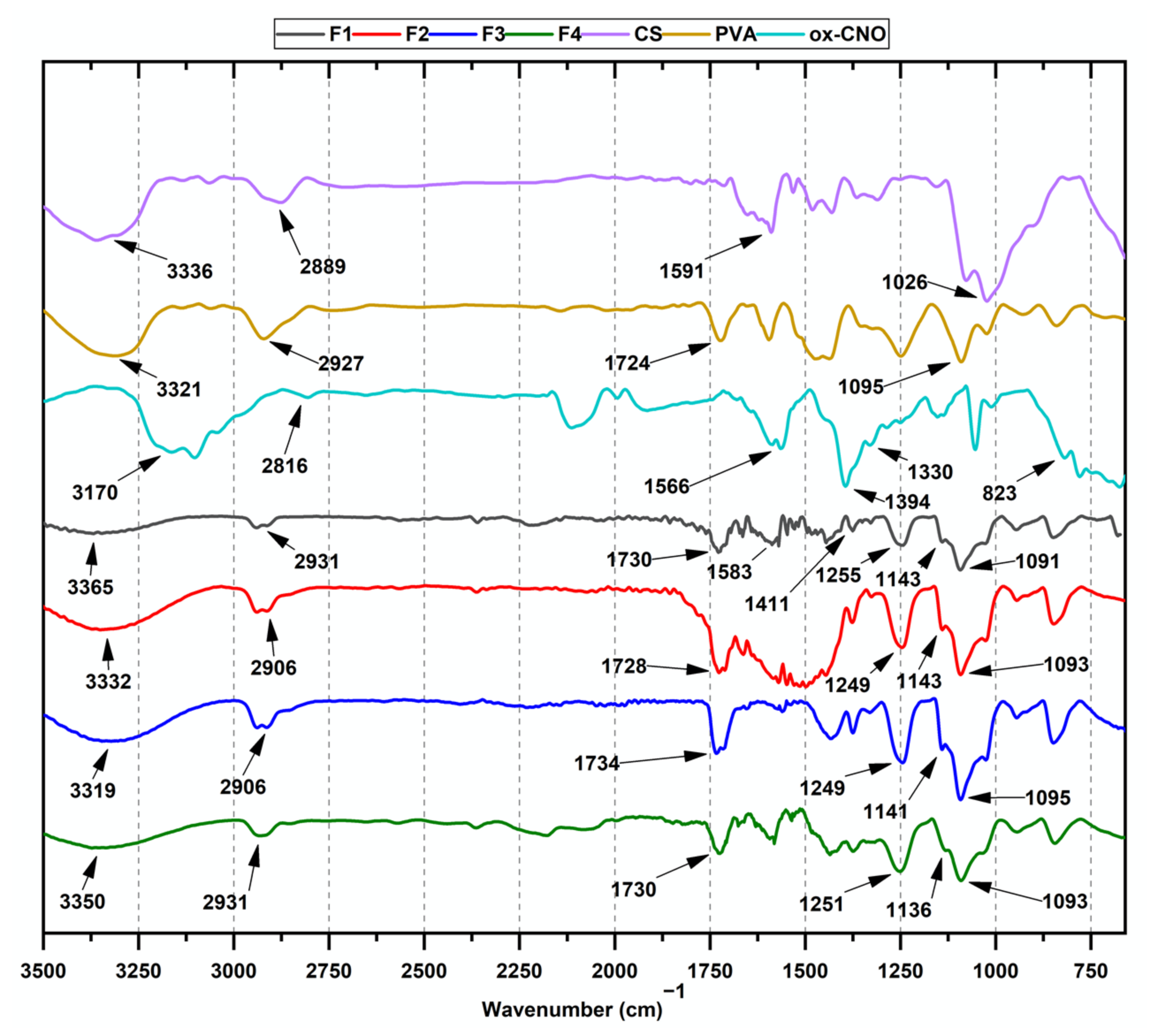
2.1.1. Fourier-Transform Infrared Spectroscopy (FT-IR)
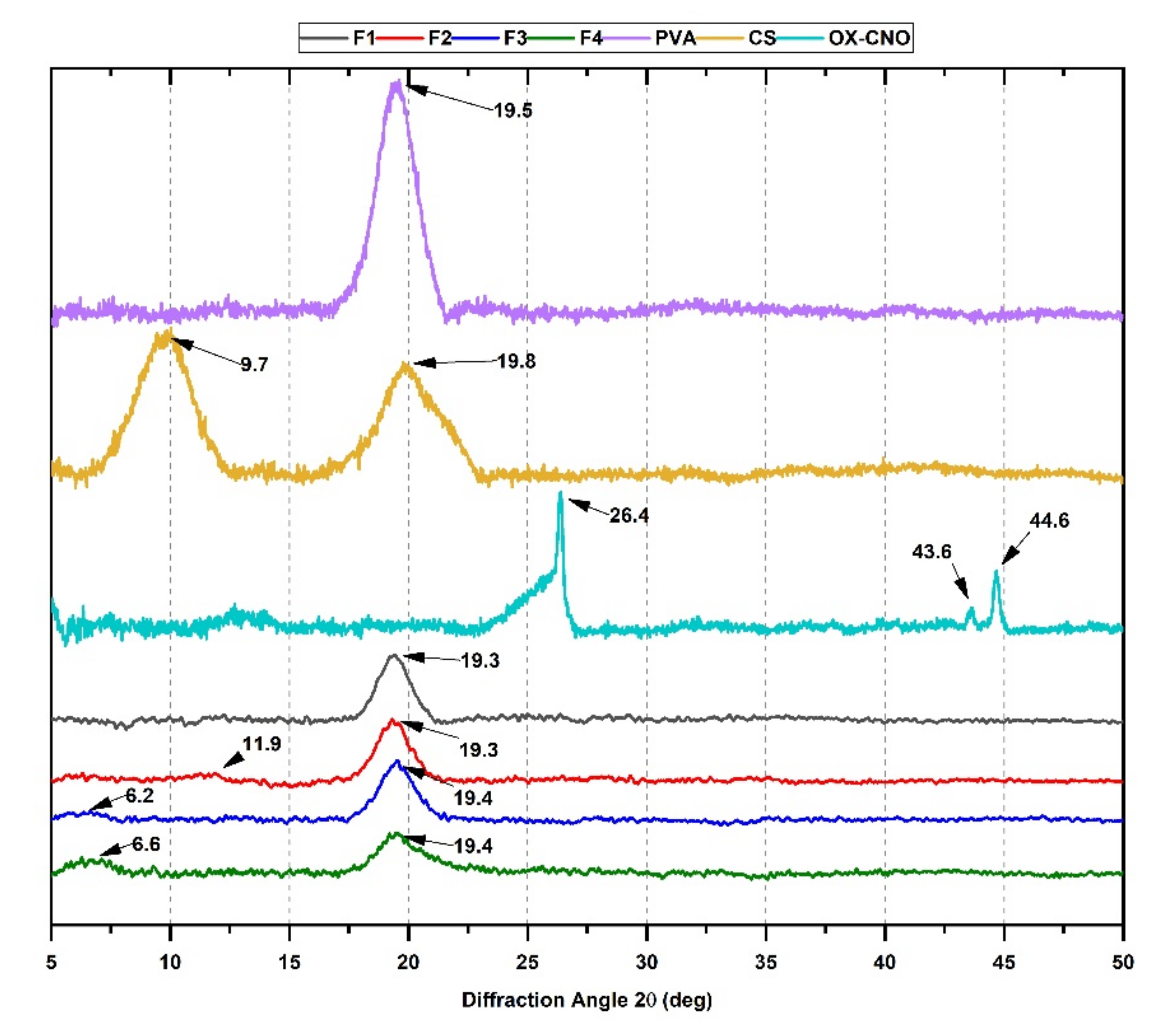
2.1.2. X-ray Diffraction (XRD)
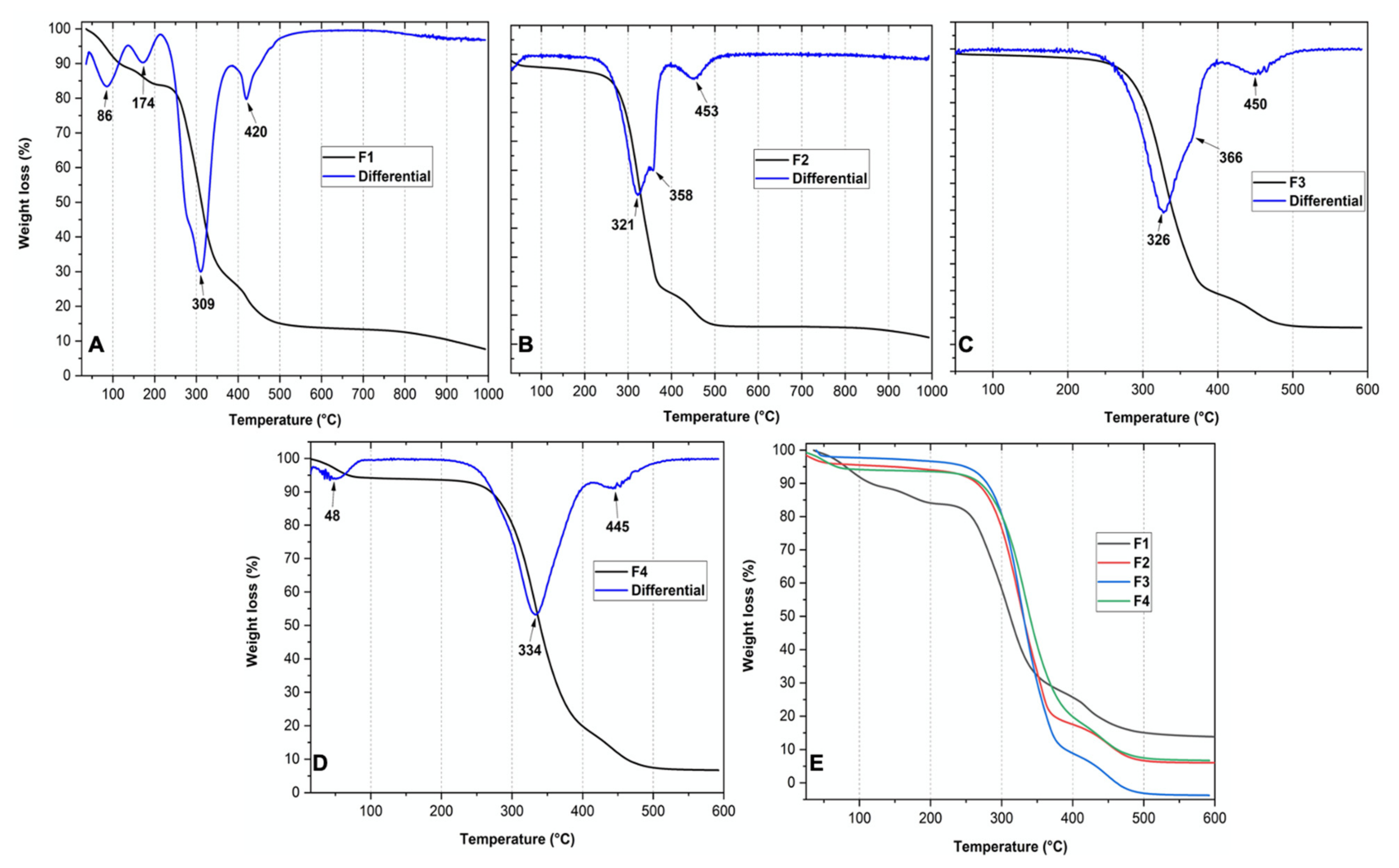
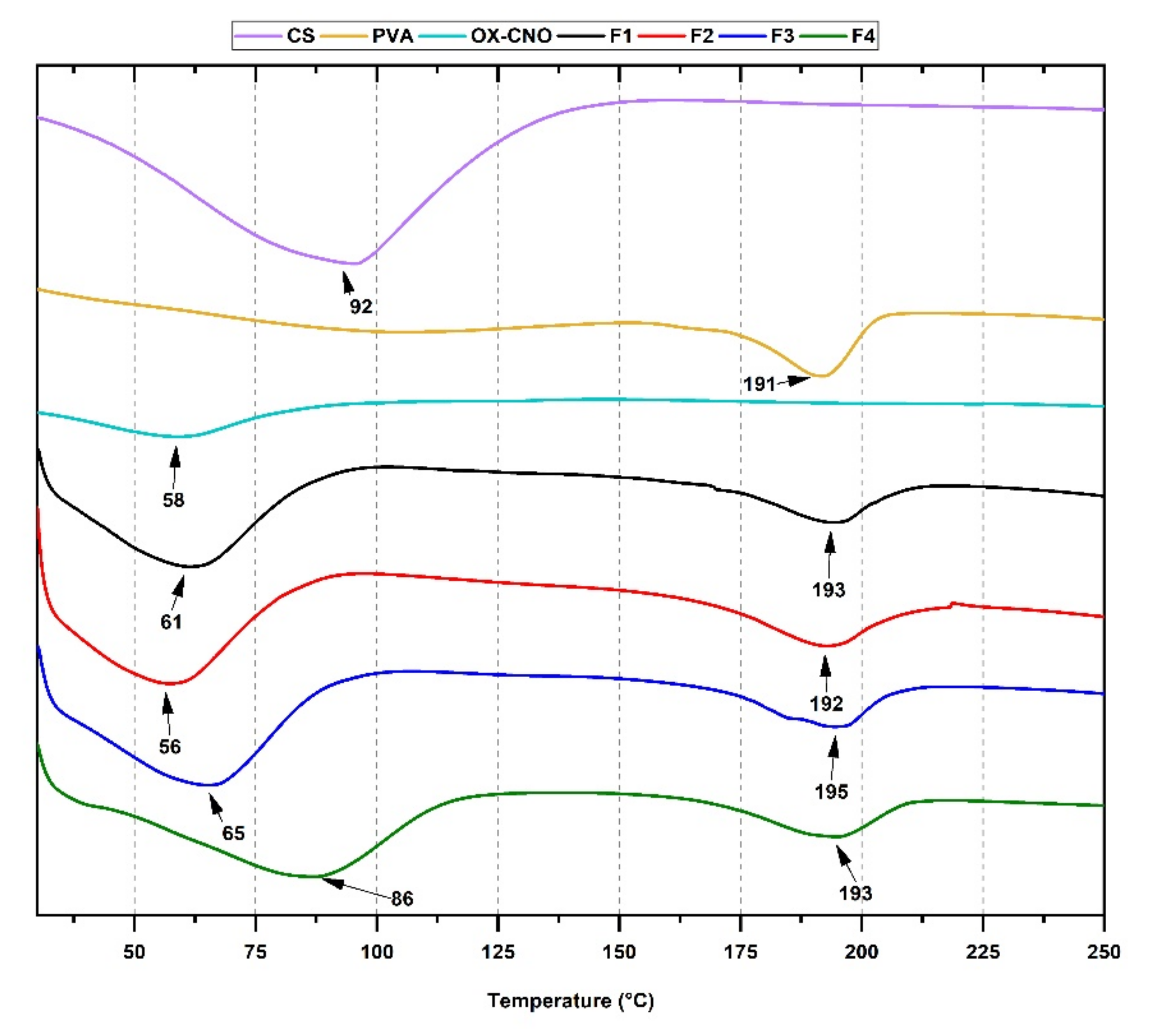
2.1.3. Thermal Analysis
2.1.4. Scanning Electron Microscopy (SEM)
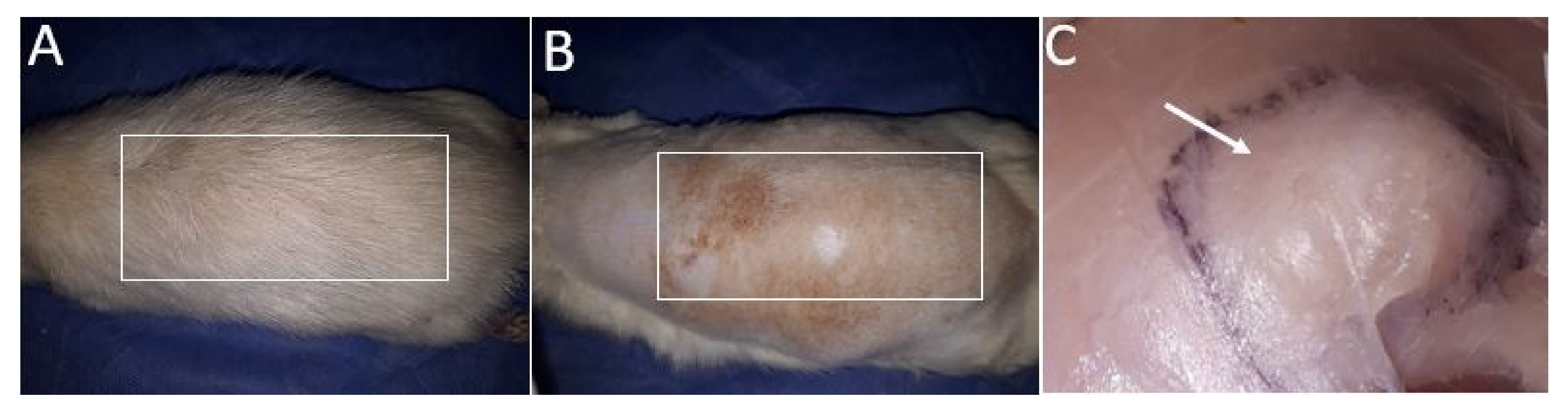
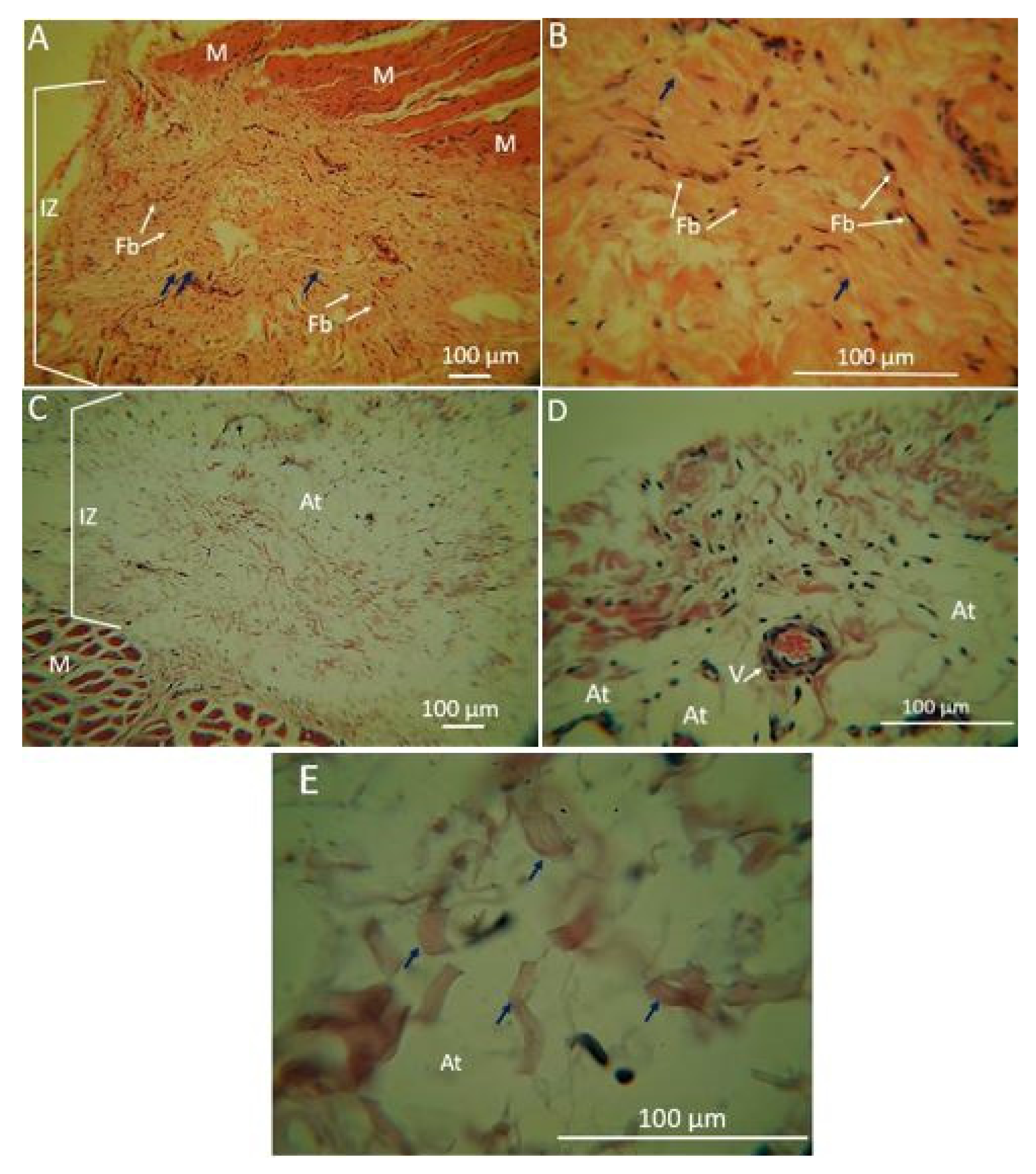
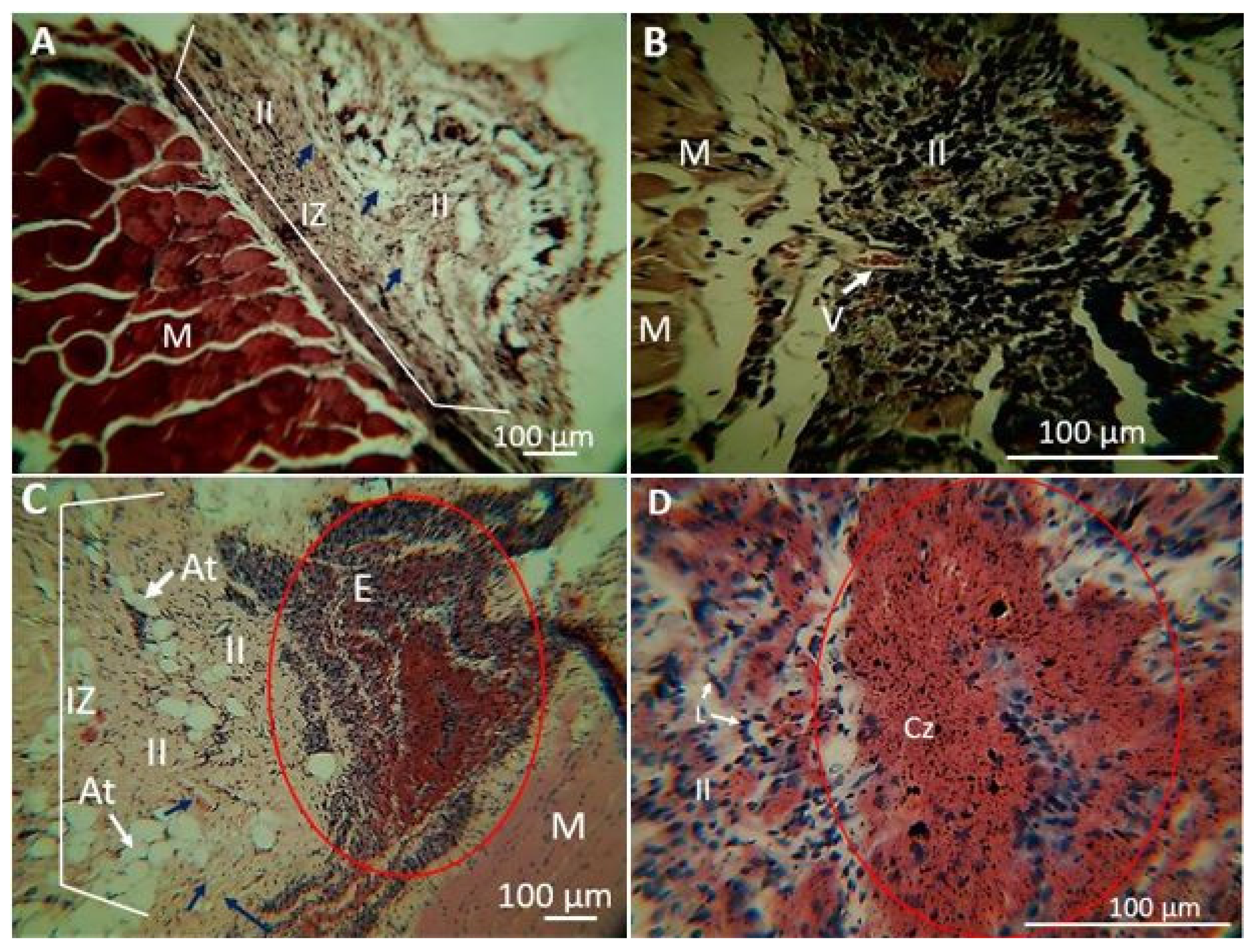
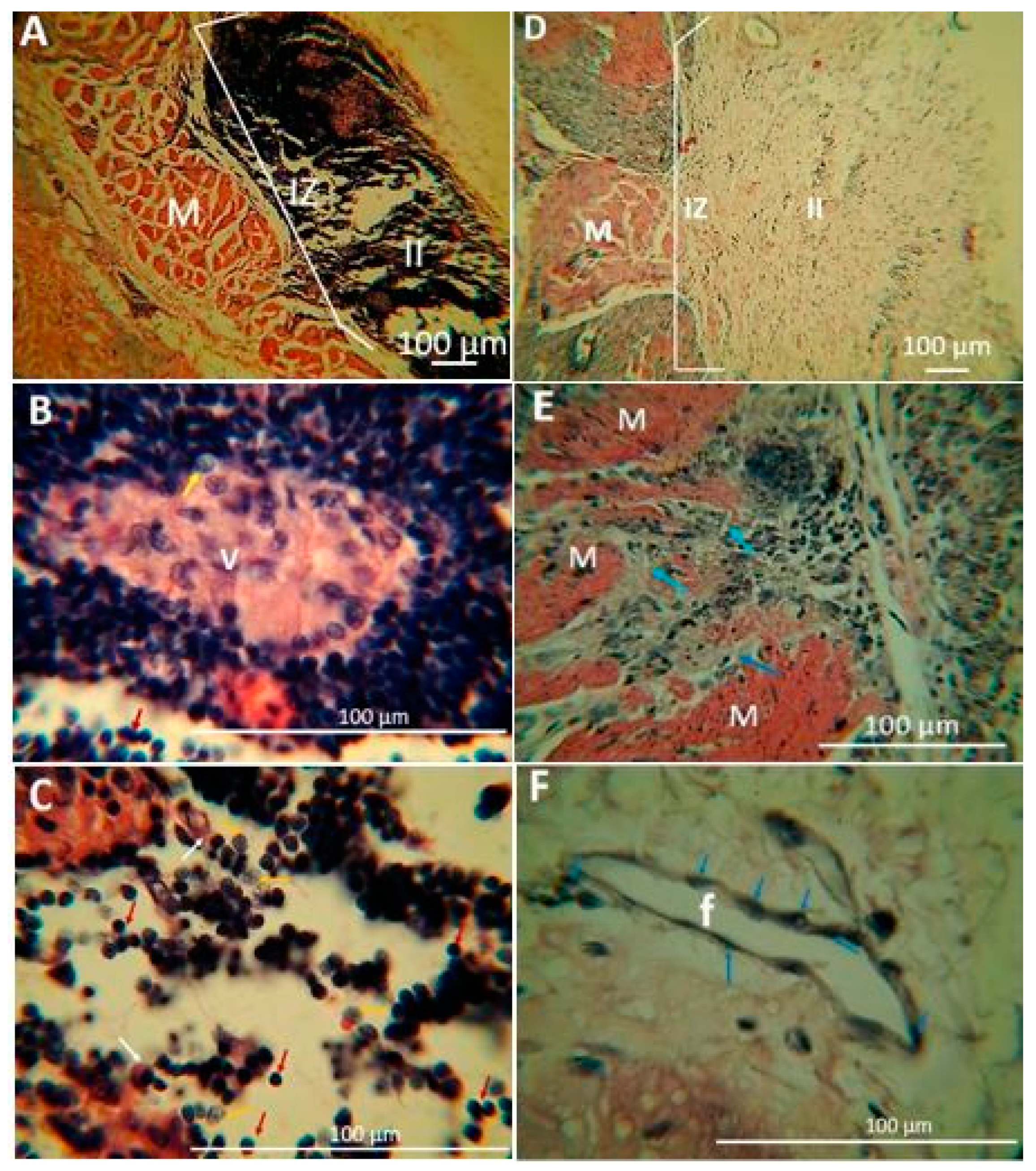
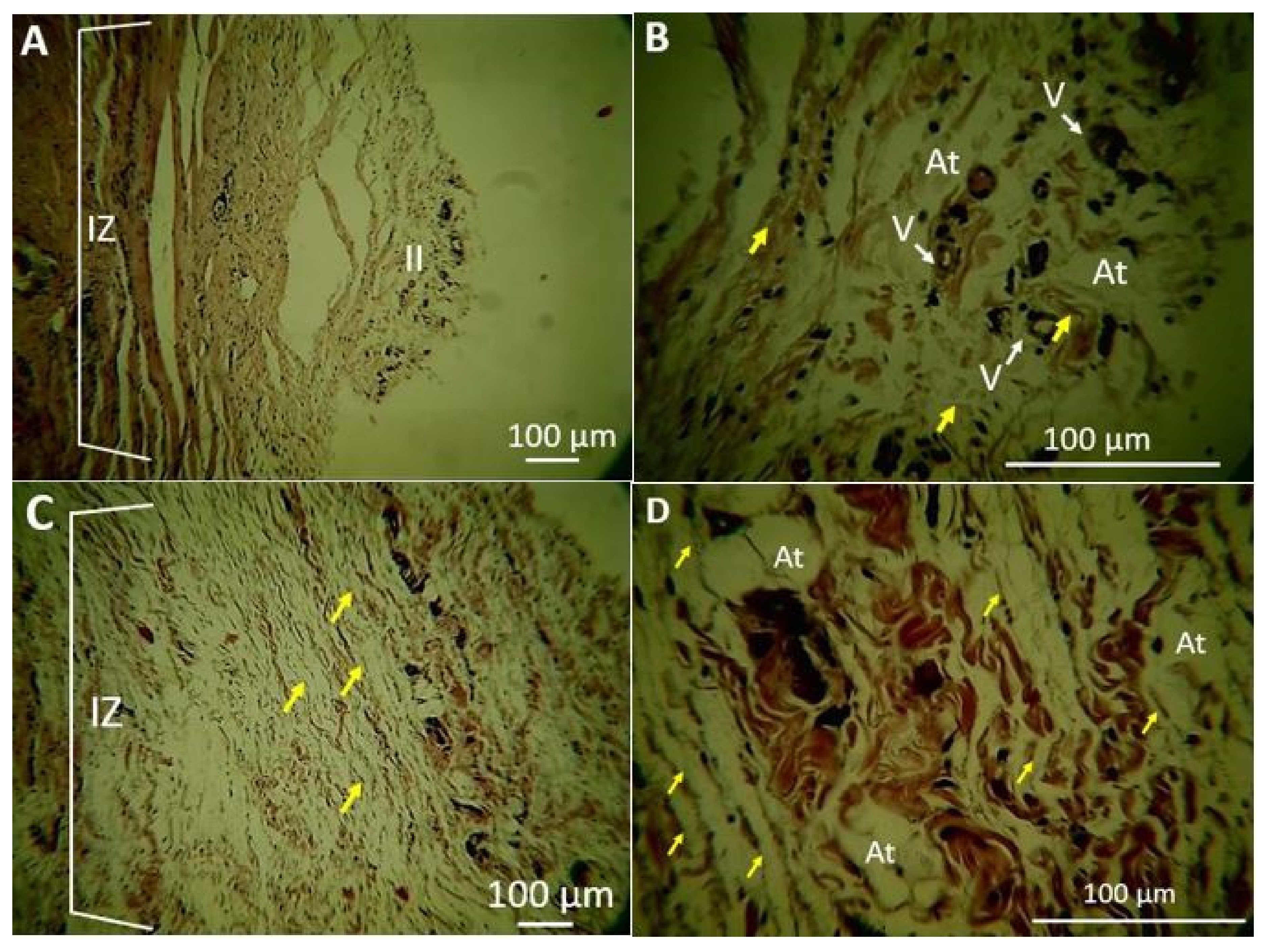
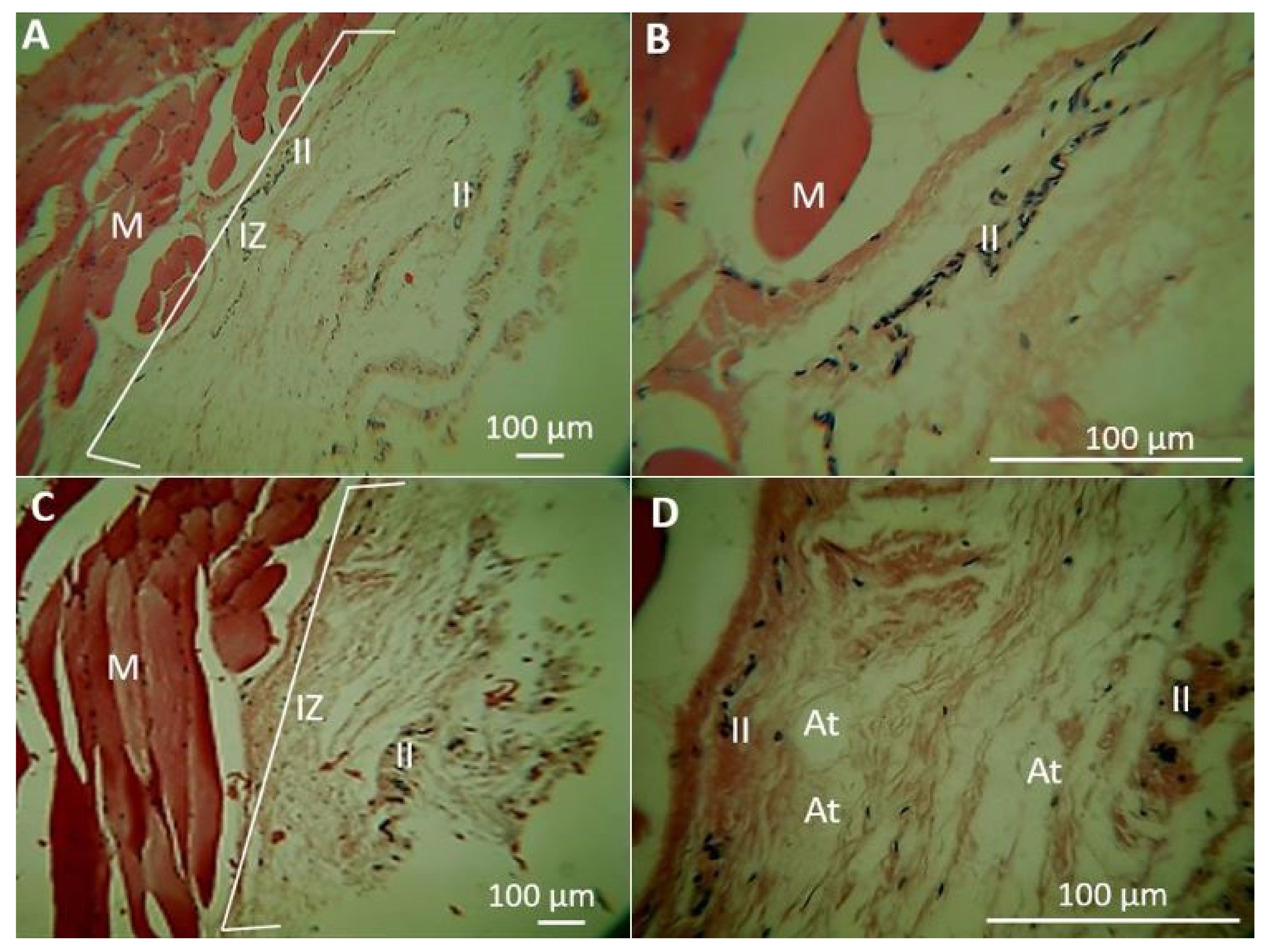
2.1.5. Biomodel Tests In Vivo
3. Materials and Methods
3.1. Materials
3.2. Methods
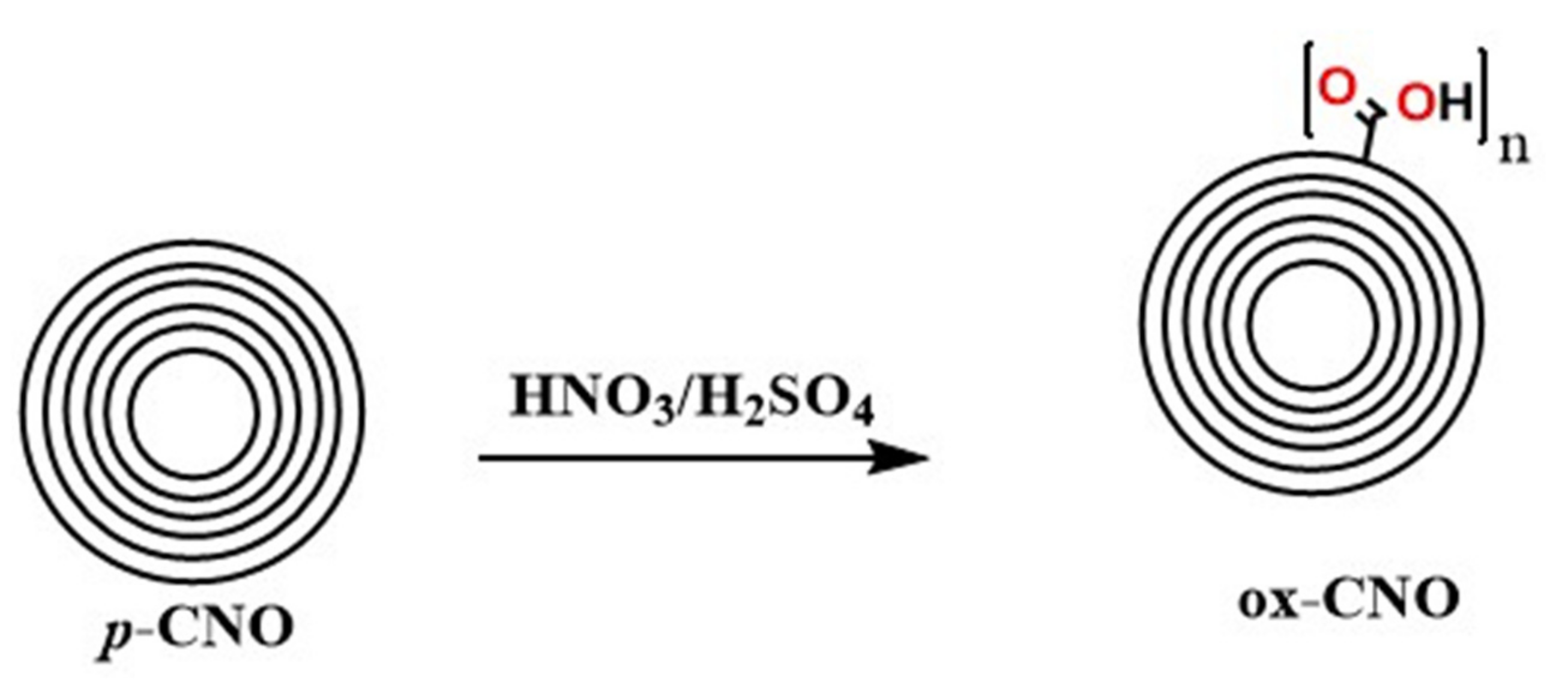
3.2.1. Synthesis of Carbon Nano-Onions and Oxidized Carbon Nano-Onions
3.2.2. Preparation of Electrospun CS/PVA/ox-CNO Nanocomposites
3.2.3. Electrospun CS/PVA/ox-CNO Nanofibers Characterization
X-ray Diffraction (DRX)
Scanning Electron Microscopy (SEM)
Thermal Studies
Biomodels Tests In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D bioactive composite scaffolds for bone tissue engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef]
- Ashman, O.; Phillips, A.M. Treatment of non-unions with bone defects: Which option and why? Injury 2013, 44, S43–S45. [Google Scholar] [CrossRef]
- Brydone, A.S.; Meek, D.; Maclaine, S. Bone grafting, orthopaedic biomaterials, and the clinical need for bone engineering. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 2010, 224, 1329–1343. [Google Scholar] [CrossRef]
- Kim, B.S.; Mooney, D.J. Development of biocompatible synthetic extracellular matrices for tissue engineering. Trends Biotechnol. 1998, 16, 224–230. [Google Scholar] [CrossRef]
- Stratton, S.; Shelke, N.B.; Hoshino, K.; Rudraiah, S.; Kumbar, S.G. Bioactive polymeric scaffolds for tissue engineering. Bioact. Mater. 2016, 1, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Jamróz, E.; Kulawik, P.; Kopel, P. The effect of nanofillers on the functional properties of biopolymer-based films: A review. Polymers 2019, 11, 675. [Google Scholar] [CrossRef]
- George, J.; Ishida, H. A review on the very high nanofiller-content nanocomposites: Their preparation methods and properties with high aspect ratio fillers. Prog. Polym. Sci. 2018, 86, 1–39. [Google Scholar] [CrossRef]
- Venkatesan, J.; Bhatnagar, I.; Manivasagan, P.; Kang, K.-H.; Kim, S.-K. Alginate composites for bone tissue engineering: A review. Int. J. Biol. Macromol. 2015, 72, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Leena, R.S.; Selvamurugan, N. Chitosan based biocomposite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1354–1365. [Google Scholar] [CrossRef]
- Narayanan, G.; Gupta, B.S.; Tonelli, A.E. Poly(ε-caprolactone) Nanowebs Functionalized with α- and γ-Cyclodextrins. Biomacromolecules 2014, 15, 4122–4133. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.A.; Müller, L.; Hofmann, I.; Greil, P.; Wenzel, M.M.; Staudenmaier, R. Cellulose-based scaffold materials for cartilage tissue engineering. Biomaterials 2006, 27, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Du, Y.; Yu, J.; Huang, R.; Zhang, L. Preparation and characterization of alginate/poly(vinyl alcohol) blend fibers. J. Macromol. Sci. Part A 2005, 42, 41–50. [Google Scholar]
- Islam, M.M.; Shahruzzaman, M.; Biswas, S.; Nurus Sakib, M.; Rashid, T.U. Chitosan based bioactive materials in tissue engineering applications-A review. Bioact. Mater. 2020, 5, 164–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-Y.; Seo, S.-J.; Moon, H.-S.; Yoo, M.-K.; Park, I.-Y.; Kim, B.-C.; Cho, C.-S. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv. 2008, 26, 1–21. [Google Scholar] [CrossRef]
- Nasrin, R.; Biswas, S.; Rashid, T.U.; Afrin, S.; Jahan, R.A.; Haque, P.; Rahman, M.M. Preparation of Chitin-PLA laminated composite for implantable application. Bioact. Mater. 2017, 2, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Kwon, O.H.; Jang, J. Electrospinning of chitosan dissolved in concentrated acetic acid solution. Biomaterials 2005, 26, 5427–5432. [Google Scholar] [CrossRef]
- Mahoney, C.; McCullough, M.B.; Sankar, J.; Bhattarai, N. Nanofibrous structure of chitosan for biomedical applications. J. Nanomed. Biother. Discov. 2012, 2, 102. [Google Scholar]
- Pakravan, M.; Heuzey, M.C.; Ajji, A. A fundamental study of chitosan/PEO electrospinning. Polymer 2011, 52, 4813–4824. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Nie, J. Electrospinning of chitosan/poly(vinyl alcohol)/acrylic acid aqueous solutions. J. Appl. Polym. Sci. 2006, 102, 5692–5697. [Google Scholar] [CrossRef]
- Liu, Y.; Park, M.; Shin, H.K.; Pant, B.; Park, S.J.; Kim, H.Y. Preparation and characterization of chitosan-based nanofibers by ecofriendly electrospinning. Mater. Lett. 2014, 132, 23–26. [Google Scholar] [CrossRef]
- Rafique, A.; Mahmood Zia, K.; Zuber, M.; Tabasum, S.; Rehman, S. Chitosan functionalized poly(vinyl alcohol) for prospects biomedical and industrial applications: A review. Int. J. Biol. Macromol. 2016, 87, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimzadeh, S.; Bari, M.R.; Hamishehkar, H.; Kafil, H.S.; Lim, L.-T. Essential oils-loaded electrospun chitosan-poly(vinyl alcohol) nonwovens laminated on chitosan film as bilayer bioactive edible films. LWT 2021, 144, 111217. [Google Scholar] [CrossRef]
- Kozhenkov, V.I.; Fuks, N.A. Electrohydrodynamic Atomisation of Liquids. Russ. Chem. Rev. 1976, 45, 1179–1184. [Google Scholar] [CrossRef]
- Smith, D.P.H. The Electrohydrodynamic Atomization of Liquids. IEEE Trans. Ind. Appl. 1986, IA-22, 527–535. [Google Scholar] [CrossRef]
- Michelson, D. Electrostatic Atomization; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Moore, A.D. Electrostatics and Its Applications; Wiley: Hoboken NJ, USA, 1973. [Google Scholar]
- Deitzel, J.M.; Kleinmeyer, J.; Harris, D.; Tan, B. The effect of processing variables on the morphology of electrospun nanofibers and textiles. Polymer 2001, 42, 261–272. [Google Scholar] [CrossRef]
- Shin, Y.M.; Hohman, M.M.; Brenner, M.P.; Rutledge, G.C. Experimental characterization of electrospinning: The electrically forced jet and instabilities. Polymer 2001, 42, 09955–09967. [Google Scholar] [CrossRef]
- Gibson, P.W.; Schreuder-Gibson, H.L.; Rivin, D. Electrospun fiber mats: Transport properties. AIChE J. 1999, 45, 190–195. [Google Scholar] [CrossRef]
- Deitzel, J.M.; Kosik, W.; McKnight, S.H.; Beck Tan, N.C.; DeSimone, J.M.; Crette, S. Electrospinning of polymer nanofibers with specific surface chemistry. Polymer 2001, 43, 1025–1029. [Google Scholar] [CrossRef]
- Deitzel, J.M.; Beck Tan, N.C.; Kleinmeyer, J.D.; Rehrmann, J.; Tevault, D.; Sendijarevic, I.; McHugh, A. Generation of Polymer Nanofibers Through Electrospinning; Army Research Lab Aberdeen Proving Ground Md: Aberdeen, MD, USA, 1999. [Google Scholar]
- Khan, W.S.; Asmatulu, R.; Ceylan, M.; Jabbarnia, A. Recent progress on conventional and non-conventional electrospinning processes. Fibers Polym. 2013, 14, 1235–1247. [Google Scholar] [CrossRef]
- Tamayo Marín, A.J.; Londoño, R.S.; Delgado, J.; Navia Porras, P.D.; Valencia Zapata, E.M.; Mina Hernandez, H.J.; Valencia, H.C.; Grande Tovar, D.C. Biocompatible and Antimicrobial Electrospun Membranes Based on Nanocomposites of Chitosan/Poly (Vinyl Alcohol)/Graphene Oxide. Int. J. Mol. Sci. 2019, 20, 2987. [Google Scholar] [CrossRef]
- Pakhira, B.; Ghosh, M.; Allam, A.; Sarkar, S. Carbon nano onions cross the blood brain barrier. RSC Adv. 2016, 6, 29779–29782. [Google Scholar] [CrossRef]
- Lettieri, S.; Camisasca, A.; D’Amora, M.; Diaspro, A.; Uchida, T.; Nakajima, Y.; Yanagisawa, K.; Maekawa, T.; Giordani, S. Far-red fluorescent carbon nano-onions as a biocompatible platform for cellular imaging. RSC Adv. 2017, 7, 45676–45681. [Google Scholar] [CrossRef]
- Bartolome, J.P.; Echegoyen, L.; Fragoso, A. Reactive Carbon Nano-Onion Modified Glassy Carbon Surfaces as DNA Sensors for Human Papillomavirus Oncogene Detection with Enhanced Sensitivity. Anal. Chem. 2015, 87, 6744–6751. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Redín, G.; Furuta, R.H.M.; Wilson, D.; Shimizu, F.M.; Materon, E.M.; Arantes, L.M.R.B.; Melendez, M.E.; Carvalho, A.L.; Reis, R.M.; Chaur, M.N. Screen-printed interdigitated electrodes modified with nanostructured carbon nano-onion films for detecting the cancer biomarker CA19-9. Mater. Sci. Eng. C 2019, 99, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Echegoyen, L.; Ortiz, A.; Chaur, M.N.; Palkar, A.J. Carbon Nano Onions. Chem. Nanocarbons 2010, 5, 463–483. [Google Scholar]
- Plonska-Brzezinska, M.E. Carbon Nano-Onions: A Review of Recent Progress in Synthesis and Applications. ChemNanoMat 2019, 5, 568–580. [Google Scholar] [CrossRef]
- Grande Tovar, C.D.; Castro, J.I.; Valencia, C.H.; Navia Porras, D.P.; Hernandez, M.; Herminsul, J.; Valencia, M.E.; Velásquez, J.D.; Chaur, M.N. Preparation of Chitosan/Poly (Vinyl Alcohol) Nanocomposite Films Incorporated with Oxidized Carbon Nano-Onions (Multi-Layer Fullerenes) for Tissue-Engineering Applications. Biomolecules 2019, 9, 684. [Google Scholar] [CrossRef]
- Grande Tovar, D.C.; Castro, I.J.; Valencia, H.C.; Navia Porras, P.D.; Herminsul Mina Hernandez, J.; Valencia Zapata, E.M.; Chaur, N.M. Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications. Molecules 2020, 25, 1203. [Google Scholar] [CrossRef] [PubMed]
- Pandele, A.M.; Ionita, M.; Crica, L.; Dinescu, S.; Costache, M.; Iovu, H. Synthesis, characterization, and in vitro studies of graphene oxide/chitosan-polyvinyl alcohol films. Carbohydr. Polym. 2014, 102, 813–820. [Google Scholar] [CrossRef]
- Nilsen-Nygaard, J.; Strand, S.P.; Vårum, K.M.; Draget, K.I.; Nordgård, C.T. Chitosan: Gels and interfacial properties. Polymers 2015, 7, 552–579. [Google Scholar] [CrossRef]
- Liao, K.-H.; Lin, Y.-S.; Macosko, C.W.; Haynes, C.L. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl. Mater. Interfaces 2011, 3, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Shao, K.; Li, Z.; Guo, N.; Zuo, Y.; Li, Q.; Lu, Z.; Chen, L.; He, Q.; Han, H. Antiviral activity of graphene oxide: How sharp edged structure and charge matter. ACS Appl. Mater. Interfaces 2015, 7, 21571–21579. [Google Scholar] [CrossRef]
- Bartkowski, M.; Giordani, S. Supramolecular chemistry of carbon nano-onions. Nanoscale 2020, 12, 9352–9358. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Tariq, M.; Ali, I.; Asghar, R.; Noorunnisa Khanam, P.; Augustine, R.; Hasan, A. Novel electrospun chitosan/polyvinyl alcohol/zinc oxide nanofibrous mats with antibacterial and antioxidant properties for diabetic wound healing. Int. J. Biol. Macromol. 2018, 120, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Koosha, M.; Mirzadeh, H. Electrospinning, mechanical properties, and cell behavior study of chitosan/PVA nanofibers. J. Biomed. Mater. Res. Part A 2015, 103, 3081–3093. [Google Scholar] [CrossRef]
- Luszczyn, J.; Plonska-Brzezinska, M.E.; Palkar, A.; Dubis, A.T.; Simionescu, A.; Simionescu, D.T.; Kalska-Szostko, B.; Winkler, K.; Echegoyen, L. Small noncytotoxic carbon nano-onions: First covalent functionalization with biomolecules. Chem. Eur. J. 2010, 16, 4870–4880. [Google Scholar] [CrossRef]
- Nakane, K.; Yamashita, T.; Iwakura, K.; Suzuki, F. Properties and structure of poly (vinyl alcohol)/silica composites. J. Appl. Polym. Sci. 1999, 133–138. [Google Scholar] [CrossRef]
- Mallakpour, S.; Zadehnazari, A. A facile, efficient, and rapid covalent functionalization of multi-walled carbon nanotubes with natural amino acids under microwave irradiation. Prog. Org. Coatings 2014, 77, 679–684. [Google Scholar] [CrossRef]
- Samuels, R.J. Solid state characterization of the structure of chitosan films. J. Polym. Sci. Polym. Phys. Ed. 1981, 19, 1081–1105. [Google Scholar] [CrossRef]
- Jia, Y.T.; Gong, J.; Gu, X.H.; Kim, H.Y.; Dong, J.; Shen, X.Y. Fabrication and characterization of poly (vinyl alcohol)/chitosan blend nanofibers produced by electrospinning method. Carbohydr. Polym. 2007, 67, 403–409. [Google Scholar] [CrossRef]
- Sangeetha, K.; Sudha, P.N.; Sukumaran, A. Novel chitosan based thin sheet nanofiltration membrane for rejection of heavy metal chromium. Int. J. Biol. Macromol. 2019, 132, 939–953. [Google Scholar]
- Vrentas, J.S.; Jarzebski, C.M.; Duda, J.L. A Deborah number for diffusion in polymer-solvent systems. AIChE J. 1975, 21, 894–901. [Google Scholar] [CrossRef]
- Yang, X.; Tu, Y.; Li, L.; Shang, S.; Tao, X. Well-Dispersed Chitosan/Graphene Oxide Nanocomposites. ACS Appl. Mater. Interfaces 2010, 2, 1707–1713. [Google Scholar] [CrossRef]
- Tang, C.; Chen, N.; Zhang, Q.; Wang, K.; Fu, Q.; Zhang, X. Preparation and properties of chitosan nanocomposites with nanofillers of different dimensions. Polym. Degrad. Stab. 2009, 94, 124–131. [Google Scholar] [CrossRef]
- Navarro-Pardo, F.; Martínez-Barrera, G.; Martínez-Hernández, A.L.; Castaño, V.M.; Rivera-Armenta, J.L.; Medellín-Rodríguez, F.; Velasco-Santos, C. Effects on the thermo-mechanical and crystallinity properties of nylon 6, 6 electrospun fibres reinforced with one dimensional (1D) and two dimensional (2D) carbon. Materials 2013, 6, 3494–3513. [Google Scholar] [CrossRef] [PubMed]
- Dassios, K.G. Poly(Vinyl Alcohol)-Infiltrated Carbon Nanotube Carpets. Mater. Sci. Appl. 2012, 3, 658–663. [Google Scholar] [CrossRef][Green Version]
- Mucha, M.; Pawlak, A. Thermal analysis of chitosan and its blends. Thermochim. Acta 2005, 427, 69–76. [Google Scholar] [CrossRef]
- Müller, K.; Zollfrank, C.; Schmid, M. Natural Polymers from Biomass Resources as Feedstocks for Thermoplastic Materials. Macromol. Mater. Eng. 2019, 304, 1–17. [Google Scholar] [CrossRef]
- Mohammed, G.; El Sayed, A.M.; Morsi, W.M. Spectroscopic, thermal, and electrical properties of MgO/polyvinyl pyrrolidone/polyvinyl alcohol nanocomposites. J. Phys. Chem. Solids 2018, 115, 238–247. [Google Scholar] [CrossRef]
- Finch, C.A. Polyvinyl Alcohol; Properties and Applications; John Wiley & Sons: Hoboken, NJ, USA, 1973; ISBN 047125892X. [Google Scholar]
- Fong, H.; Chun, I.; Reneker, D.H. Beaded nanofibers formed during electrospinning. Polymer 1999, 40, 4585–4592. [Google Scholar] [CrossRef]
- Zong, X.; Kim, K.; Fang, D.; Ran, S.; Hsiao, B.S.; Chu, B. Structure and process relationship of electrospun bioabsorbable nanofiber membranes. Polymer 2002, 43, 4403–4412. [Google Scholar] [CrossRef]
- Liu, S.; Wei, L.; Hao, L.; Fang, N.; Chang, M.W.; Xu, R.; Yang, Y.; Chen, Y. Sharper and faster “nano darts” kill more bacteria: A study of antibacterial activity of individually dispersed pristine single-walled carbon nanotube. ACS Nano 2009, 3, 3891–3902. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Miranda, O.R.; Thompson, M.A.; Pabelick, C.M.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Prakash, Y.S.; Mukherjee, P. Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 2010, 10, 2543–2548. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, F.; Wang, Q.; Wu, X. Fabrication of chitosan/graphene oxide polymer nanofiber and its biocompatibility for cartilage tissue engineering. Mater. Sci. Eng. C 2017, 79, 697–701. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.; Duan, B.; Wu, L.; Li, S.; Yuan, X. Preparation of electrospun chitosan/poly(vinyl alcohol) membranes. Colloid Polym. Sci. 2007, 285, 855–863. [Google Scholar] [CrossRef]
- Malafaya, P.B.; Santos, T.C.; van Griensven, M.; Reis, R.L. Morphology, mechanical characterization and in vivo neo-vascularization of chitosan particle aggregated scaffolds architectures. Biomaterials 2008, 29, 3914–3926. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W.; Goh, J.C.H.; Teoh, S.H. An introduction to biodegradable materials for tissue engineering applications. Ann. Acad. Med. Singap. 2001, 30, 183–191. [Google Scholar] [PubMed]
- Han, J.; Zhou, Z.; Yin, R.; Yang, D.; Nie, J. Alginate–chitosan/hydroxyapatite polyelectrolyte complex porous scaffolds: Preparation and characterization. Int. J. Biol. Macromol. 2010, 46, 199–205. [Google Scholar] [CrossRef]
- Nishikawa, H.; Ueno, A.; Nishikawa, S.; Kido, J.; Ohishi, M.; Inoue, H.; Nagata, T. Sulfated glycosaminoglycan synthesis and its regulation by transforming growth factor-β in rat clonal dental pulp cells. J. Endod. 2000, 26, 169–171. [Google Scholar] [CrossRef]
- Correia, C.R.; Moreira-Teixeira, L.S.; Moroni, L.; Reis, R.L.; van Blitterswijk, C.A.; Karperien, M.; Mano, J.F. Chitosan scaffolds containing hyaluronic acid for cartilage tissue engineering. Tissue Eng. Part C Methods 2011, 17, 717–730. [Google Scholar] [CrossRef]
- Mezzana, P. Clinical efficacy of a new chitin nanofibrils-based gel in wound healing. Acta Chir. Plast. 2008, 50, 81–84. [Google Scholar]
- Kumar, P.T.S.; Praveen, G.; Raj, M.; Chennazhi, K.P.; Jayakumar, R. Flexible, micro-porous chitosan–gelatin hydrogel/nanofibrin composite bandages for treating burn wounds. RSC Adv. 2014, 4, 65081–65087. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Chitosan and alginate wound dressings: A short review. Trends Biomater. Artif. Organs 2004, 18, 18–23. [Google Scholar]
- Chandorkar, Y.; Basu, B. The foreign body response demystified. ACS Biomater. Sci. Eng. 2018, 5, 19–44. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Manchola, J.F.; Medina-Cárdenas, S.; Moreno, S. Reacción granulomatosa de tipo cuerpo extraño secundaria a biomateriales y su respuesta inmunológica: Revisión sistemática de la literatura. Salut. Sci. Spiritus 2016, 2, 21–28. [Google Scholar]
- Tomihata, K.; Ikada, Y. In Vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 1997, 18, 567–575. [Google Scholar] [CrossRef]
- Pella, M.C.G.; Lima-Tenório, M.K.; Tenorio-Neto, E.T.; Guilherme, M.R.; Muniz, E.C.; Rubira, A.F. Chitosan-based hydrogels: From preparation to biomedical applications. Carbohydr. Polym. 2018, 196, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Ishihara, M.; Simizu, M.; Obara, K.; Ishizuka, T.; Saito, Y.; Yura, H.; Morimoto, Y.; Takase, B.; Matsui, T. Vascularization in vivo caused by the controlled release of fibroblast growth factor-2 from an injectable chitosan/non-anticoagulant heparin hydrogel. Biomaterials 2004, 25, 699–706. [Google Scholar] [CrossRef]
- Pawar, V.; Bulbake, U.; Khan, W.; Srivastava, R. Chitosan sponges as a sustained release carrier system for the prophylaxis of orthopedic implant-associated infections. Int. J. Biol. Macromol. 2019, 134, 100–112. [Google Scholar] [CrossRef]
- Crawford, L.; Wyatt, M.; Bryers, J.; Ratner, B. Biocompatibility Evolves: Phenomenology to Toxicology to Regeneration. Adv. Healthc. Mater. 2021, 2002153. [Google Scholar] [CrossRef]
- Hall, M.; Bansal, P.; Lee, J.H.; Realff, M.J.; Bommarius, A.S. Cellulose crystallinity—A key predictor of the enzymatic hydrolysis rate. FEBS J. 2010, 277, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Kastellorizios, M.; Tipnis, N.; Burgess, D.J. Foreign body reaction to subcutaneous implants. In Immune Responses to Biosurfaces; Lambris, J.D., Ekdahl, K.N., Ricklin, D., Nilsson, B., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 93–108. [Google Scholar]
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 2015, 8, 5671–5701. [Google Scholar] [CrossRef]
- Chhabra, P.; Tyagi, P.; Bhatnagar, A.; Mittal, G.; Kumar, A. Optimization, characterization, and efficacy evaluation of 2% chitosan scaffold for tissue engineering and wound healing. J. Pharm. Bioallied Sci. 2016, 8, 300. [Google Scholar]
- Scarritt, M.E.; Londono, R.; Badylak, S.F. Host response to implanted materials and devices: An overview. In The Immune Response to Implanted Materials and Devices; Springer: Berlin, Germany, 2017; pp. 1–14. [Google Scholar]
- Lynd, L.R.; Weimer, P.J.; Van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef]
- Duarte, A.R.C.; Mano, J.F.; Reis, R.L. Preparation of chitosan scaffolds loaded with dexamethasone for tissue engineering applications using supercritical fluid technology. Eur. Polym. J. 2009, 45, 141–148. [Google Scholar] [CrossRef]
- Raftery, R.; O’brien, F.J.; Cryan, S.-A. Chitosan for gene delivery and orthopedic tissue engineering applications. Molecules 2013, 18, 5611–5647. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Koo, D.W.; Lee, J.S. Late Onset Foreign Body Reaction due to Poly-l-Lactic Acid Facial Injections for Cosmetic Purpose. Ann. Dermatol. 2020, 32, 519. [Google Scholar] [CrossRef]
- Lehman, J.S.; Sokumbi, O.; Peters, M.S.; Bridges, A.G.; Comfere, N.I.; Gibson, L.E.; Wieland, C.N. Histopathologic features of noninfectious granulomatous disorders involving the skin. Hum. Pathol. 2020, 103, 127–145. [Google Scholar] [CrossRef]
- Rinaudo, M.; Milas, M.; Le Dung, P. Characterization of chitosan. Influence of ionic strength and degree of acetylation on chain expansion. Int. J. Biol. Macromol. 1993, 15, 281–285. [Google Scholar] [CrossRef]
- Nara, S.; Komiya, T. Studies on the Relationship Between Water-satured State and Crystallinity by the Diffraction Method for Moistened Potato Starch. Starch Stärke 1983, 35, 407–410. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Xc (%) | Tg (°C) | Tm (°C) |
|---|---|---|---|
| F1 | 0.86 | 93 | 192 |
| F2 | 1.68 | 94 | 193 |
| F3 | 8.17 | 92 | 195 |
| F4 | 12.5 | 89 | 193 |
| Formulation * | Average Diameter (nm) | Standard Deviation (±nm) |
|---|---|---|
| F1 | 147.8 | 29.4 |
| F2 | 120.7 | 11.9 |
| F3 | 117.9 | 15.9 |
| F4 | 102.1 | 12.9 |
| Variable | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| TCD (cm) | 20 | 15 | 15 | 15 |
| Injection rate (mL/h) | 0.13 | 0.6 | 0.6 | 0.85 |
| Voltage (kV) | 16 | 18.5 | 17 | 15 |
| Component | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| CS (%) | 8 | 8 | 3.5 | 5.4 |
| PVA (%) | 92 | 91 | 95.5 | 92.6 |
| ox-CNO (%) | 0 | 1 | 1 | 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, J.I.; Chaur, M.N.; Llano, C.H.V.; Valencia Zapata, M.E.; Mina Hernandez, J.H.; Grande-Tovar, C.D. Biocompatibility Study of Electrospun Nanocomposite Membranes Based on Chitosan/Polyvinyl Alcohol/Oxidized Carbon Nano-Onions. Molecules 2021, 26, 4753. https://doi.org/10.3390/molecules26164753
Castro JI, Chaur MN, Llano CHV, Valencia Zapata ME, Mina Hernandez JH, Grande-Tovar CD. Biocompatibility Study of Electrospun Nanocomposite Membranes Based on Chitosan/Polyvinyl Alcohol/Oxidized Carbon Nano-Onions. Molecules. 2021; 26(16):4753. https://doi.org/10.3390/molecules26164753
Chicago/Turabian StyleCastro, Jorge Iván, Manuel N. Chaur, Carlos Humberto Valencia Llano, Mayra Eliana Valencia Zapata, José Herminsul Mina Hernandez, and Carlos David Grande-Tovar. 2021. "Biocompatibility Study of Electrospun Nanocomposite Membranes Based on Chitosan/Polyvinyl Alcohol/Oxidized Carbon Nano-Onions" Molecules 26, no. 16: 4753. https://doi.org/10.3390/molecules26164753
APA StyleCastro, J. I., Chaur, M. N., Llano, C. H. V., Valencia Zapata, M. E., Mina Hernandez, J. H., & Grande-Tovar, C. D. (2021). Biocompatibility Study of Electrospun Nanocomposite Membranes Based on Chitosan/Polyvinyl Alcohol/Oxidized Carbon Nano-Onions. Molecules, 26(16), 4753. https://doi.org/10.3390/molecules26164753