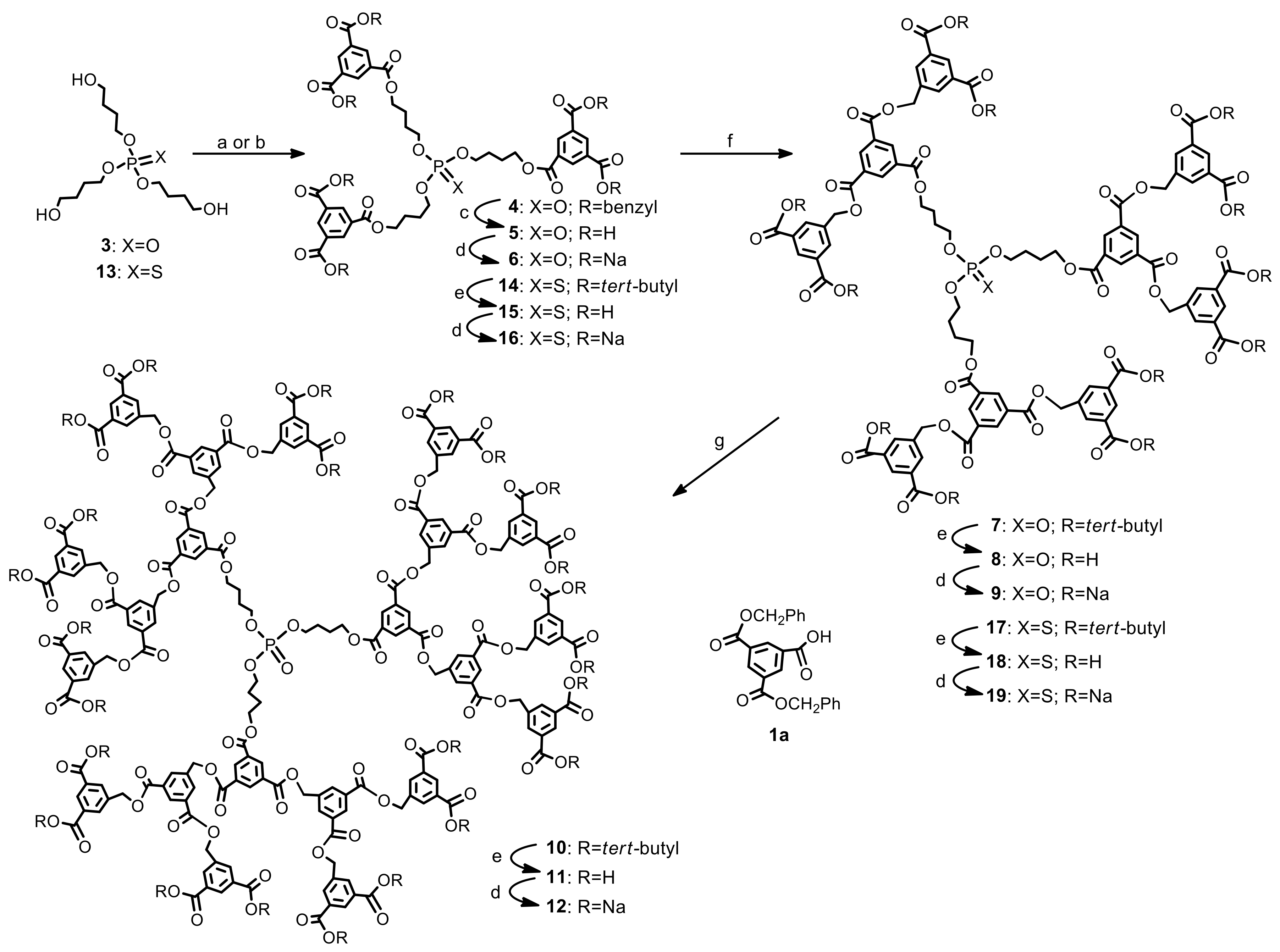
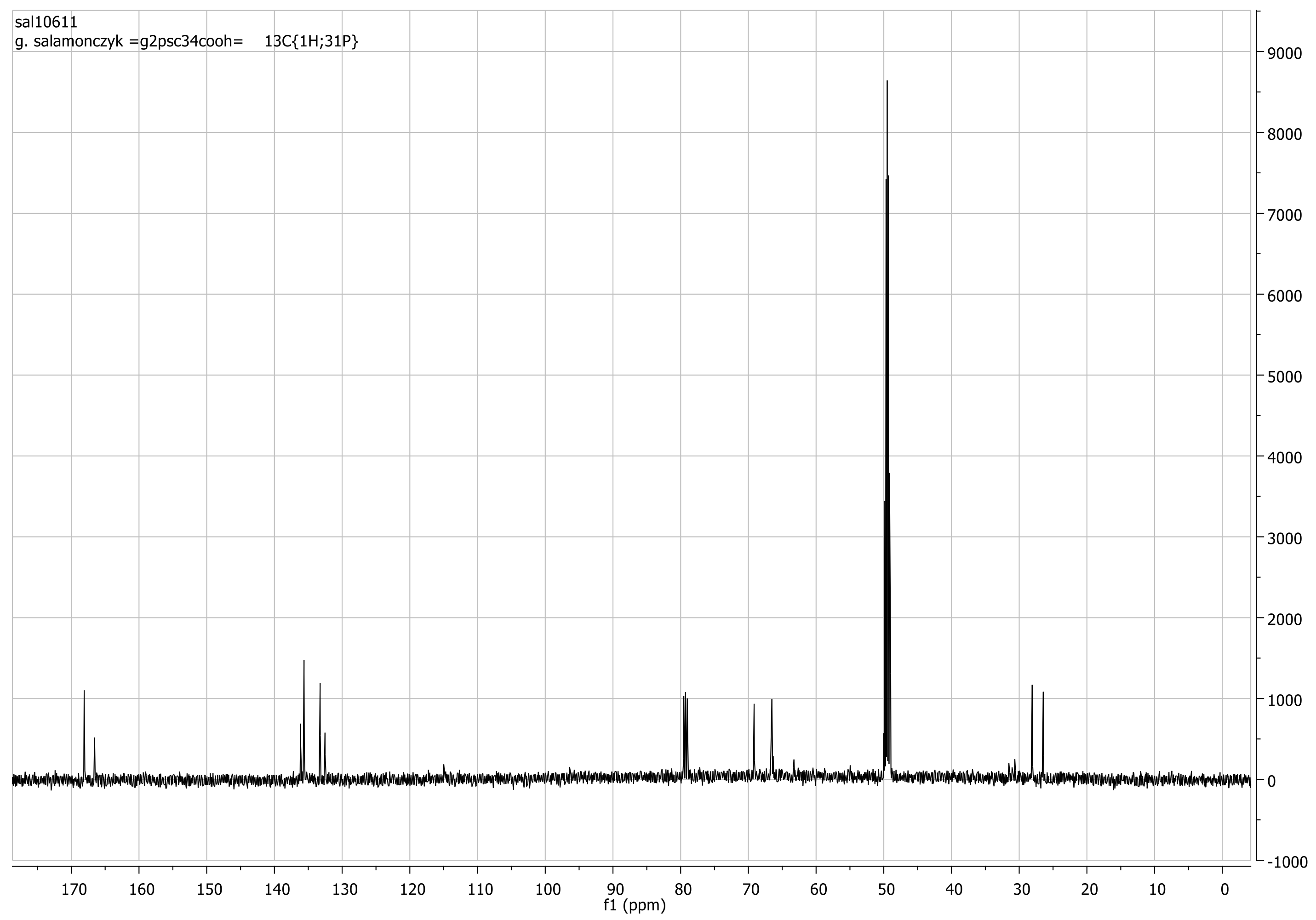
3.3. Synthetic Procedures and Analyses for Dendrimers: 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19
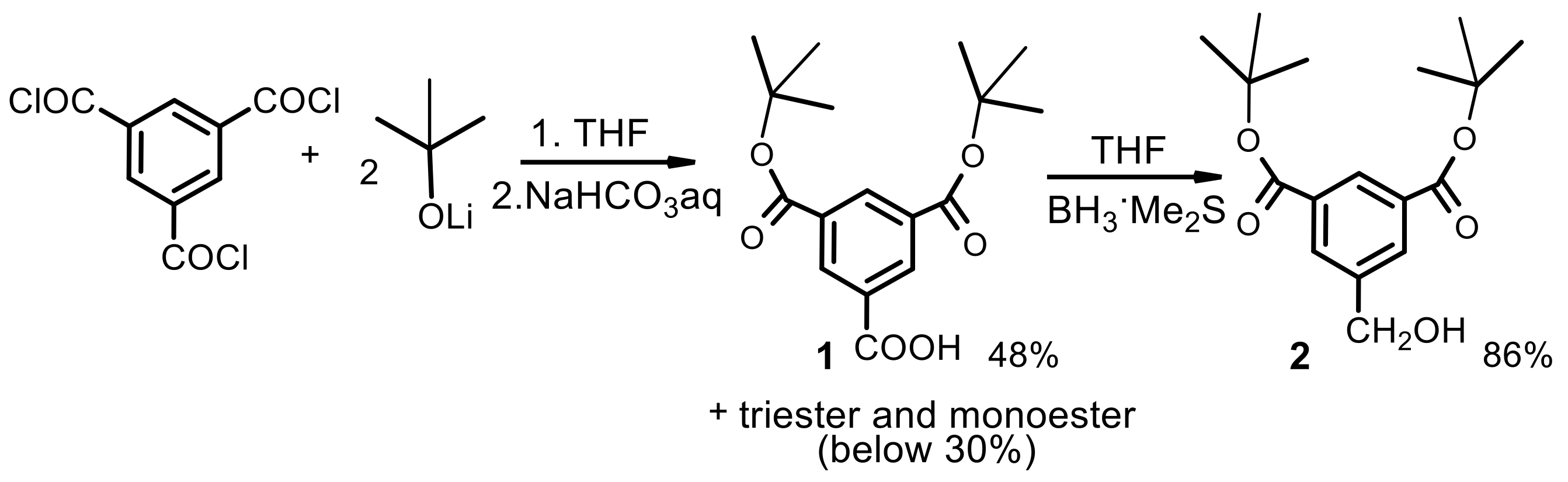
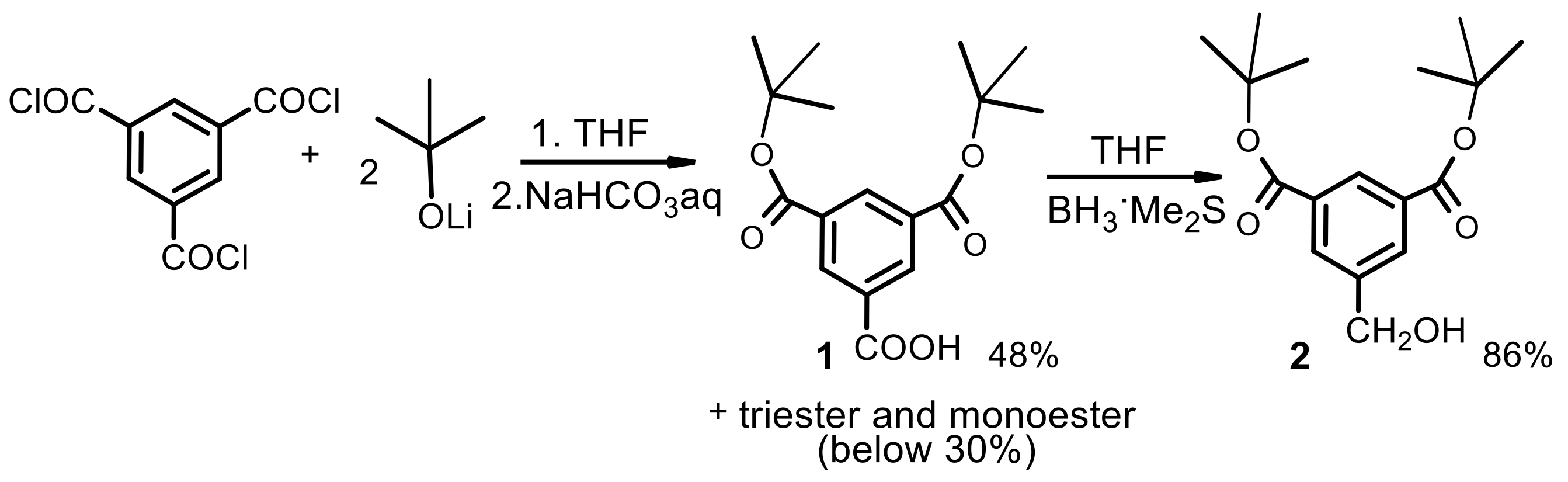
First generation polyester dendrimer4. Tris(4-hydroxybutyl) phosphate 3 (FW 314.3, 190 mg, 0.6 mmol), was dispersed in dry dichloromethane (6 mL). Dibenzyl trimesoate (1a) (800 mg, 2.05 mmol, 3.4 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (FW 191.7, 403 mg, 2.1 mmol, 3.5 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (FW 122.2, 36 mg, 0.3 mmol, 0.4 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (60 mL), washed with 0.1 M citric acid (15 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the CH2Cl2–MeOH 100:1 mixture and gradually increasing the polarity to CH2Cl2–MeOH 20:1, gave the title ester 4 (790 mg). Yield 92%. Rf 0.3 (CH2Cl2-MeOH 20:1); NMR: δH (500 MHz, CDCl3) 1.86-1.87 [m, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 1.91–1.92 [m, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.14 [dt, 3J(H,H) = 5.8 Hz, 3J(P,H) = 11.2 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.42 [t, 3J(H,H) = 5.8 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.39 [s, 12H, (PhCH2)], 7.32–7.47 (m, 30H, Ph), 8.85 [d, 4J(H,H) = 1.5 Hz, 6H, Ar], 8.88 [t, 4J(H,H) = 1.5 Hz, 3H, Ar] ppm; δC (125 MHz, CDCl3) 25.05 [3C, O=COCH2CH2CH2CH2O)], 26.94 [d, 3J(C,P) = 6.3 Hz, 3C, (OCH2CH2CH2CH2OP)], 65.05 [3C, (OCH2CH2CH2CH2OP)], 67.20 [d, 2J(C,P) = 5.0 Hz, 3C, (OCH2CH2CH2CH2OP)], 67.43 [6C, (PhCH2)], 128.4 [12C, (PhCα)], 128.5 [6C, (PhCγ)], 128.7 [12C, (PhCβ)], 131.3 [6C, (ipso Ph)], 134.4 [3C, (Ar)], 134.7 [6C, (Ar)], 135.5 [9C, (ipso Ar)], 164.8 [6C, (C=O)], 164.9 [3C, (C=O)] ppm; δP {1H} (202 MHz, CDCl3) −0.51 ppm; MALDI TOF MS calcd. for C81H75O22P, M = 1430.4. Found m/z = 1453.9 M + Na, 1469.9 M + K.
Polyanionic dendrimer6. Dendrimer 4 (FW 1431.4, 300 mg, 0.21 mmol) was dissolved in methanol (4 mL) and palladium on activated charcoal (10%, 100 mg) was then added. The reaction mixture was stirred under a hydrogen atmosphere for 10 h. The catalyst was filtered off, the filtrate concentrated to dryness in vacuo to furnish analytically pure acid 5 (181 mg, 97%) as a white solid. NMR: δH (500 MHz, CDCl3:CD3OD 1:1) 1.73-1.74 [m, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 1.83–1.84 [m, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.09 [dt, 3J(H,H) = 5.8 Hz, 3J(P,H) = 11.2 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.31 [t, 3J(H,H) = 5.8 Hz, 6H, (POCH2CH2CH2CH2OC)], 8.71 [d, 4J(H,H) = 1.6 Hz, 6H, Ar], 8.74 [t, 4J(H,H) = 1.6 Hz, 3H, Ar] ppm; δC {1H, 31P} (125 MHz, CD3OD-CDCl3 2:1) 24.35 [3C, O=COCH2CH2CH2CH2O)], 26.00 [3C, (OCH2CH2CH2CH2OP)], 64.45 [3C, (OCH2CH2CH2CH2OP)], 67.07 [3C, (OCH2CH2CH2CH2OP)], 130.4 [3C, (ipso Ar)], 131.2 [6C, (ipso Ar)], 133.5 [6C, (Ar)], 134.0 [3C, (Ar)], 164.5 [3C, (C=O)], 166.0 [6C, (C=O)] ppm; δP {1H} (202 MHz, CD3OD-CDCl3 2:1) −0.42 ppm; MALDI TOF MS calcd. for C39H39O22P, M = 890.2. Found m/z = 890.2. Next, acid 5 (60 mg, 0.066 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (34 mg, 0.4 mmol, 6 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mmHg) to give 6 as a hexasodium salt of 5 (68 mg), as a nonhygroscopic white powder.
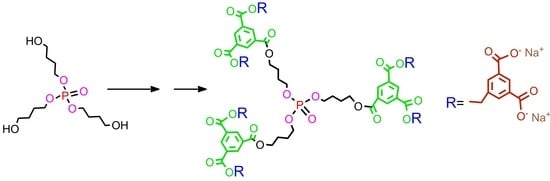
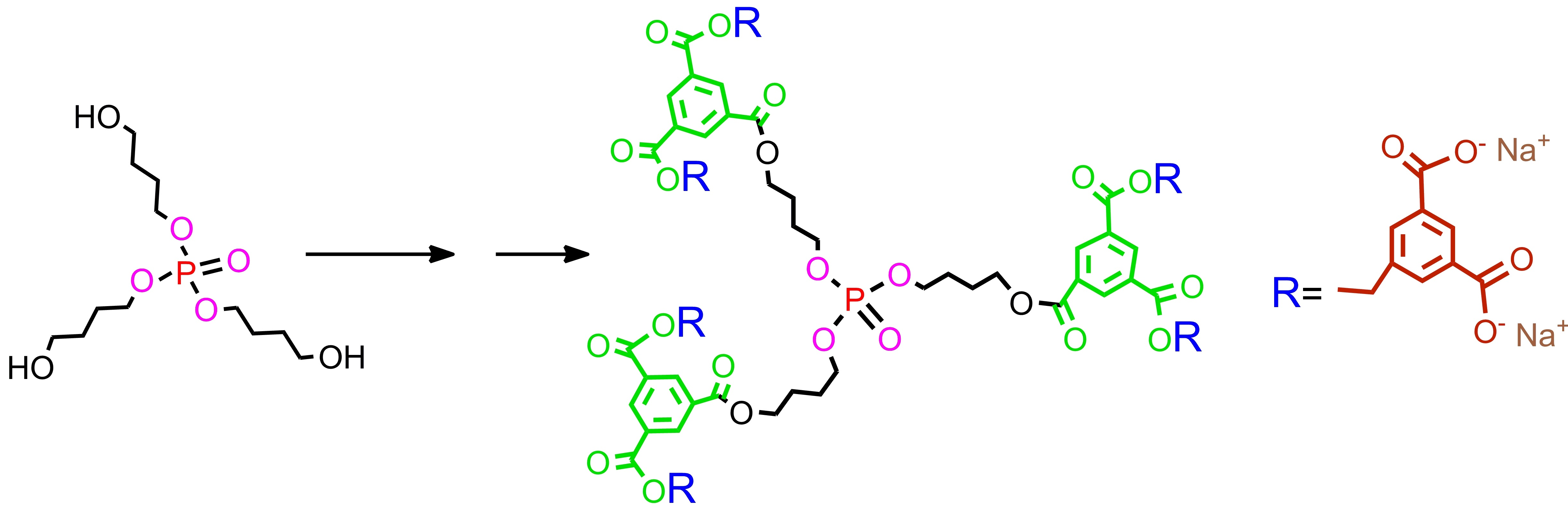
Second generation polyester dendrimer7. Carboxy-terminated, first generation phosphate dendrimer 5 (FW 890.7, 100 mg, 0.112 mmol), was dissolved in dry THF (2 mL). Then, the reaction mixture was diluted with dry dichloromethane (2 mL). Di-tert-butyl 5-hydroxymethylbenzene-1,3-dicarboxylate (2) (FW 308.4, 230 mg, 0.74 mmol, 6.6 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (172 mg, 0.896 mmol, 8.0 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (15 mg, 0.12 mmol, 1.0 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (50 mL), washed with 0.1 M citric acid (10 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the cyclohexane–acetone 20:1 mixture and gradually increasing the polarity to cyclohexane–acetone 8:1, gave the title ester 5 (260 mg). Yield 88%. NMR: δH (500 MHz, CDCl3-C6D6 2:1) 1.58 [s, 108H, C(CH3)3], 1.81–1.86 [m, 6H, (POCH2CH2CH2CH2OC)], 1.87–1.96 [m, 6H, (POCH2CH2CH2CH2OC)], 4.13 [dt, 3J(H,H) = 6.3 Hz, 3J(P,H) = 12.3 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.40 [t, 3J(H,H) = 6.5 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.29 [s, 12H, (ArCH2)], 8.20 [d, 4J(H,H) = 1.3 Hz, 12H, Ar2], 8.53 [t, 4J(H,H) = 1.3 Hz, 6H, Ar2], 8.85 [d, 3J(H,H) = 1.5 Hz, 6H, Ar1], 8.87 [t, 3J(H,H) = 1.5 Hz, 3H, Ar1] ppm; δC (125 MHz, CDCl3) 25.04 [3C, O=COCH2CH2CH2CH2O)], 26.96 [d, 3J(C,P) = 7.1 Hz, 3C, (OCH2CH2CH2CH2OP)], 28.21 [36C, C(CH3)3], 65.16 [3C, (OCH2CH2CH2CH2OP)], 67.43 [6C, (ArCH2)], 67.20 [d, 2J(C,P) = 5.7 Hz, 3C, (OCH2CH2CH2CH2OP)], 81.92 [12C, C(CH3)3], 130.6 [6C, (Ar2)], 131.1 [6C, (ipso Ar1)], 131.5 [3C, (ipso Ar1)], 132.9 [12C, (ipso Ar2)], 133.2 [12C, (Ar2)], 134.9 [3C, (Ar1)], 135.0 [6C, (Ar1)], 136.1 [6C, (ipso Ar2)], 164.7 [18C, (C=O)], 164.8 [3C, (C=O)] ppm; δP {1H} (202 MHz, CDCl3) 0.53 ppm; MALDI TOF MS calcd. for C141H171O46P, M = 2631.1. Found m/z = 2670.3 M + K.
Polyanionic dendrimer9. Dendrimer 7 (FW 2632.8, 134 mg, 0.05 mmol) was dissolved in dry dichloromethane (2 mL), and trifluoroacetic acid was added (2 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 5 h. All the volatiles were removed in vacuo, and the residue was dissolved in THF (1 mL). Next, acetone (10 mL) was added to that solution and the resulting mixture was kept in the refrigerator for about an hour. The precipitate was filtered off to provide acid 8 (88 mg, 90%) as a white solid. NMR: δH (500 MHz, CDCl3-CD3OD 2:1) 2.03–2.08 [m, 6H, (POCH2CH2CH2CH2OC)], 2.25–2.28 [m, 6H, (POCH2CH2CH2CH2OC)], 3.90-3.92 [m, 6H, (POCH2CH2CH2CH2OC)], 4.14–4.16 [m, 6H, (POCH2CH2CH2CH2OC)], 5.22 [s, 12H, (ArCH2)], 8.36 [s, 6H, Ar2], 8.48 [s, 12H, Ar2], 8.56 [s, 6H, Ar1], 8.61 [s, 3H, Ar1] ppm; δP {1H} (202 MHz, CD3OD-CDCl3 2:1) −2.80 ppm; MALDI TOF MS calcd. for C93H75O46P, M = 1959.3. Found m/z = 1982.6 M + Na. Later on, acid 8 (35 mg, 0.0179 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (18 mg, 0.2 mmol, 12 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mmHg) to give 9 as a dodecasodium salt of 8 (40 mg), as a nonhygroscopic white powder.
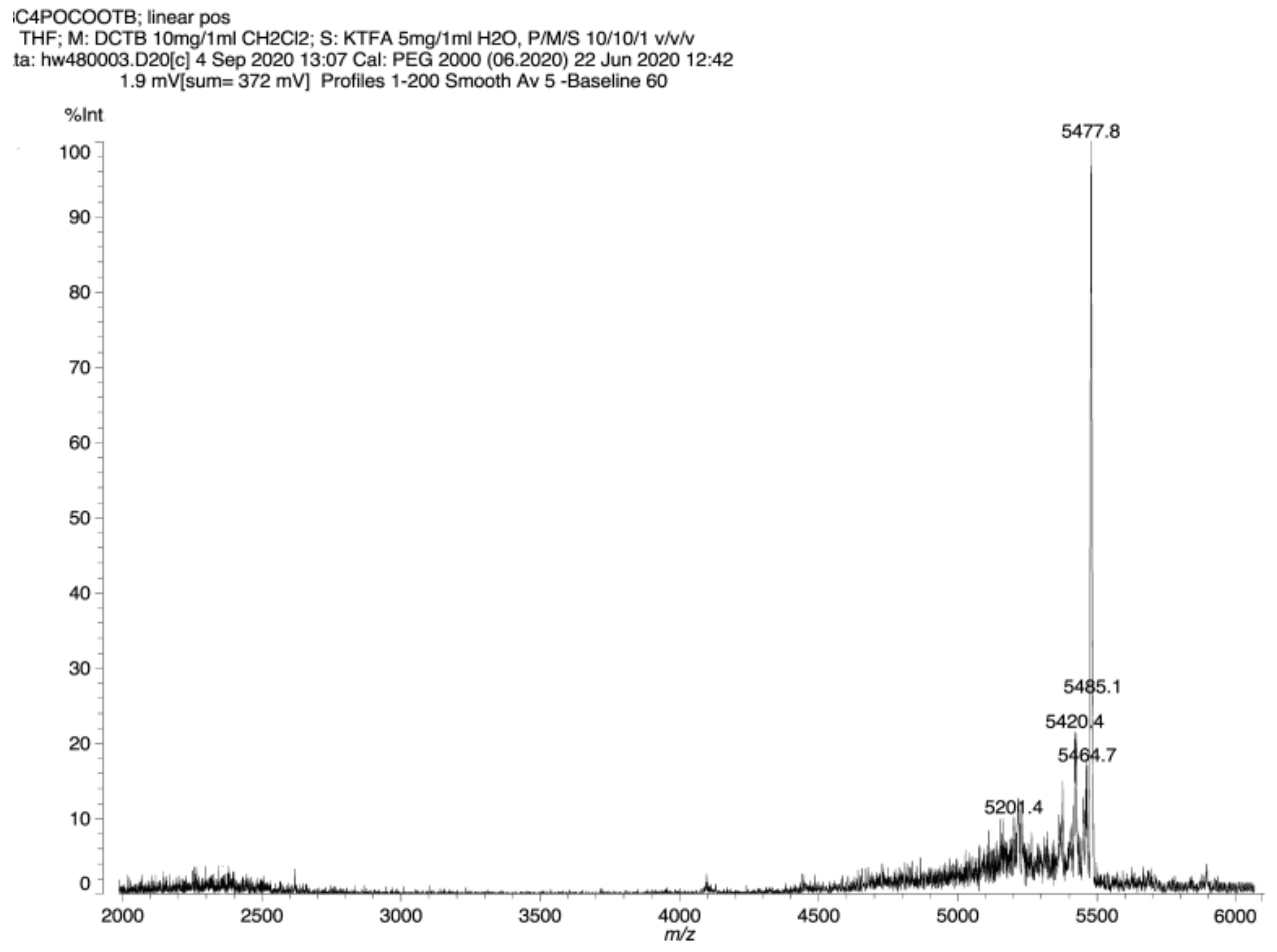
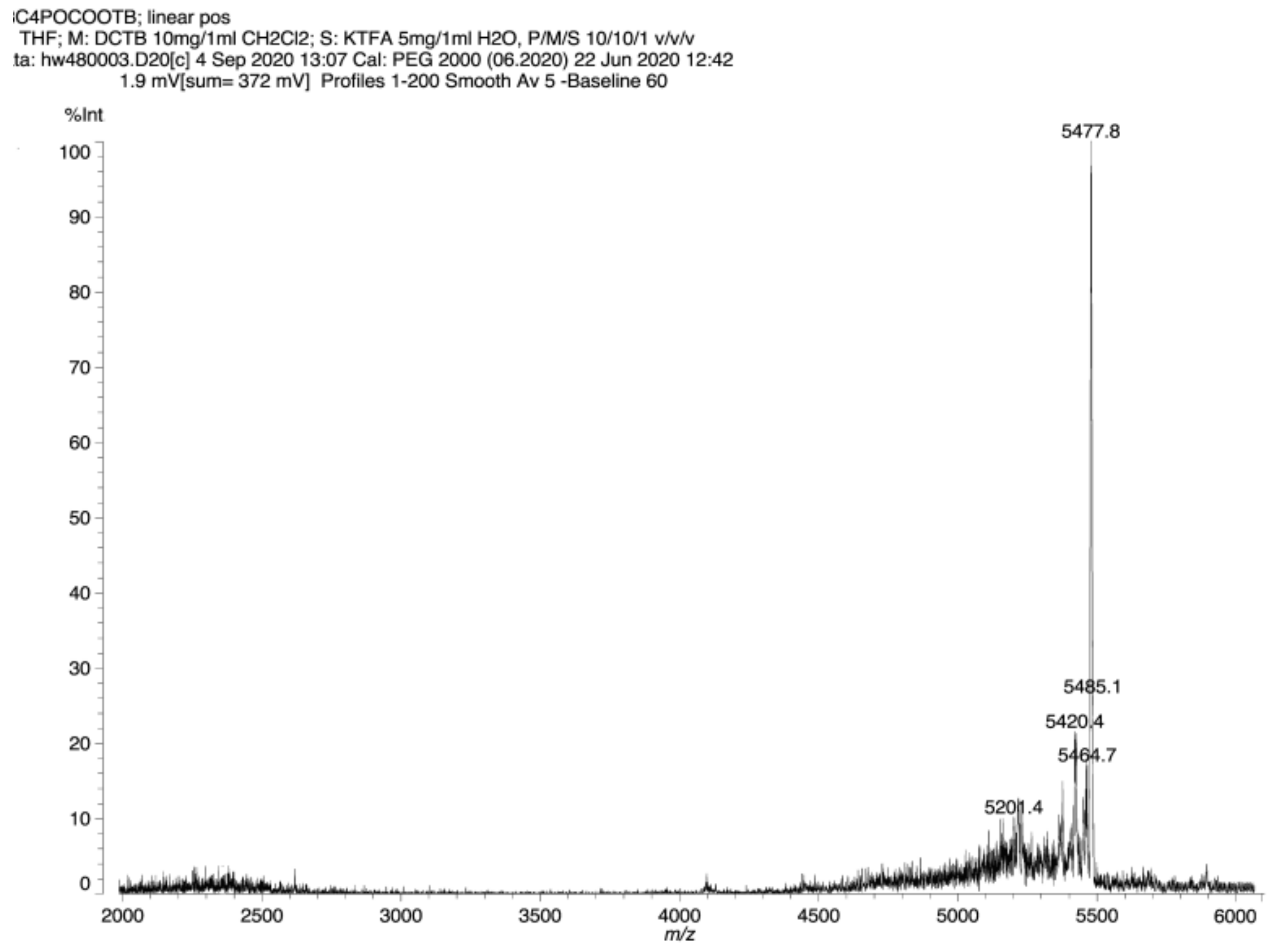
Third generation polyester dendrimer10. Carboxy-terminated, second generation phosphate dendrimer 8 (FW 1959.5, 40 mg, 0.02 mmol), was dissolved in dry DMF (1.5 mL). Then, the resulting solution was diluted with dry dichloromethane (2 mL). Di-tert-butyl 5-hydroxymethylbenzene-1,3-dicarboxylate (2) (FW 308.4, 82 mg, 0.264 mmol, 13.2 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (172 mg, 0.3 mmol, 15 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (5 mg, 0.04 mmol, 1.8 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (50 mL), washed with 0.1 M citric acid (10 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the cyclohexane–acetone 20:1 mixture and gradually increasing the polarity to cyclohexane–acetone 6:1, provided the title ester 10 (83 mg). Yield 76%. NMR: δH (500 MHz, CDCl3) 1.56 [s, 216H, C(CH3)3] 1.82–1.87 [m, 6H, (POCH2CH2CH2CH2OC)], 1.88–1.95 [m, 6H, (POCH2CH2CH2CH2OC)], 4.13 [dt, 3J(H,H) = 5.9 Hz, 3J(P,H) = 12.2 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.39 [t, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.44 [s, 24H, (Ar3CH2)], 5.46 [s, 12H, (Ar2CH2)], 8.19 [s, 24H, Ar3], 8.35 [s, 12H, Ar3], 8.51 [s, 12H, Ar2], 8.69 [s, 6H, Ar2], 8.83 [s, 6H, Ar1], 8.85 [s, 3H, Ar1] ppm; δC (125 MHz, CDCl3) 24.94 [3C, (O=COCH2CH2CH2CH2O)], 26.91 [d, 3J(C,P) = 7.4 Hz, 3C, (OCH2CH2CH2CH2OP)], 28.14 [72C, C(CH3)3], 65.12 [3C, (OCH2CH2CH2CH2OP)], 66.27 [6C, (Ar2CH2)], 66.33 [12C, (Ar3CH2)], 67.09 [d, 2J(C,P) = 6.7 Hz, 3C, (OCH2CH2CH2CH2OP)], 81.81 [24C, C(CH3)3], 130.5 [12C, (Ar3)], 131.0 [6C, (Ar2)], 133.0 [24C, (Ar3)], 130.5 [12C, (Ar3)], 130.8 [6C, (ipso Ar1)], 131.0 [12C, (ipso Ar2)], 131.5 [3C, (ipso Ar1)], 132.8 [24C, (ipso Ar3)], 134.2 [12C, (Ar2)], 135.0 [9C, (Ar1)], 136.2 [12C, (ipso Ar3)], 136.7 [6C, (ipso Ar2)], 164.5 [9C, (C1=O)], 164.6 [24C, (C3=O)], 165.0 [12C, (C2=O)] ppm; δP {1H} (202 MHz, CDCl3) -0.46 ppm. MALDI TOF MS calcd. for C297H339O94P, M = 5440.1. Found m/z = 5477.8 M + K.
Polyanionic dendrimer12. Dendrimer 10 (80 mg, 0.015 mmol) was dissolved in dry dichloromethane (1.5 mL), and trifluoroacetic acid was added (2 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 5 h. All the volatiles were removed in vacuo, and the residue was dissolved in THF (1 mL). Next, acetone (10 mL) was added to that solution and the resulting mixture was kept in the refrigerator for about an hour. The precipitate was filtered off to provide acid 11 (54 mg, 88%) as a white solid. NMR: δH (500 MHz, THF-d8) 1.75–1.80 [m, 6H, (POCH2CH2CH2CH2OC)], 1.82–1.88 [m, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.07 [dt, 3J(P,H) = 12.3 Hz, 3J(H,H) = 6.2 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.33 [t, 3J(H,H) = 6.2 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.48 [s, 24H, (Ar3CH2)], 5.49 [s, 12H, (Ar2CH2)], 8.30 [d, 4J(H,H) = 1.3 Hz, 33H, 9HAr1, 24HAr3], 8.58 [s, 12H, Ar3], 8.76 [d, 4J(H,H) = 1.6 Hz, 12H, Ar2], 8.81 [t, 4J(H,H) = 1.5 Hz, 6H, Ar2] ppm; δC (125 MHz, THF-d8) 26.60 [d, 3J(C,P) = 7.2 Hz, 3C, (OCH2CH2CH2CH2OP)], 29.24 [3C, (O=COCH2CH2CH2CH2O)], 64.84 [6C, (Ar2CH2)], 66.08 [12C, (Ar3CH2)], 67.09 [3C, (OCH2CH2CH2CH2OP)], 67.34 [d, 2J(C,P) = 6.7 Hz, 3C, (OCH2CH2CH2CH2OP)], 130.6 [12C, (Ar3)], 131.1 [12C, (Ar2)], 131.4 [6C, (Ar2)], 131.7 [24C, (ipso Ar3)], 133.3 [12C, (ipso Ar2)], 133.3 [6C, (Ar1)], 133.4 [24C, (Ar3)], 133.6 [3C, (Ar1)], 134.1 [6C, (ipso Ar2)], 134.2 [12C, (ipso Ar3)], 136.8 [9C, (Ar1)], 164.0 [12C, (C2=O)], 164.1 [9C, (C1=O)], 165.9 [24C, (C3=O)] ppm; δP {1H} (202 MHz, THF-d8) −0.95 ppm; MALDI TOF MS calcd. for C201H147O94P, M = 4096.7.3. Found m/z, fragmentation: 4129.6, 3965.6, 3949.9, 3844.2, 3731.4, 3700.0, 3601.9 (major peaks). Elemental analysis: Found (acid): C, 58.87; H, 3.66; C201H147O94P requires C, 58.92; H, 3.62; P, 0.76%. Afterwards, acid 11 (FW 4097.2, 40 mg, 0.0098 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (20 mg, 0.235 mmol, 24 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mmHg) to give 12 as a tetracosasodium salt of 11 (45 mg), as a nonhygroscopic white powder.
First generation polyester thiophosphate dendrimer14. Tris(4-hydroxybutyl) thiophosphate 13 (FW 330.4, 166 mg, 0.5 mmol), was dissolved in dry dichloromethane (5 mL). Di-tert-butyl trimesoate (1) (322.4, 560 mg, 1.7 mmol, 3.4 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (FW 191.7, 336 mg, 1.75 mmol, 3.5 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (FW 122.2, 25 mg, 0.2 mmol, 0.4 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (60 mL), washed with 0.1 M citric acid (15 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the cyclohexane–acetone 100:1 mixture and gradually increasing the polarity to cyclohexane–acetone 20:1, gave the title ester 14 (560 mg). Yield 90%. Rf 0.25 (cyclohexane–acetone 25:1). NMR: δH (500 MHz, CDCl3) 1.59 [s, 54H, C(CH3)3], 1.79-1.84 [m, 3J(H,H) = 6.4 Hz, 12H, (POCH2CH2CH2CH2OC) and (POCH2CH2CH2CH2OC)], 4.12 [dt, 3J(H,H) = 5.8 Hz, 3J(P,H) = 12.6 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.42 [t, 3J(H,H) = 5.7 Hz, 6H, (POCH2CH2CH2CH2OC)], 8.70 [s, 3H, Ar], 8.71 [s, 6H, Ar] ppm; δC (125 MHz, CDCl3) 25.13 [3C, O=COCH2CH2CH2CH2O)], 26.77 [d, 3J(C,P) = 7.8 Hz, 3C, (OCH2CH2CH2CH2OP)], 28.19 [18C, C(CH3)3], 64.93 [3C, (OCH2CH2CH2CH2OP)], 67.70 [d, 2J(C,P) = 5.4 Hz, 3C, (OCH2CH2CH2CH2OP)], 82.24 [6C, C(CH3)3], 130.9 [3C, (ipso Ar)], 132.8 [6C, (ipso Ar)], 134.0 [6C, (Ar)], 134.4 [3C, (Ar)], 164.3 [6C, (C=O)], 165.3 [3C, (C=O)] ppm; δP {1H} (202 MHz, CDCl3) 68.52 ppm; MALDI TOF MS calcd. for C63H87O21PS, M = 1242.5. Found m/z = 1282.5 M + K.
Polyanionic dendrimer16. Dendrimer 14 (FW 1243.4, 125 mg, 0.1 mmol) was dissolved in dry dichloromethane (1.5 mL), and trifluoroacetic acid was added (2 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 5 h. All the volatiles were removed in vacuo, and the residue was dissolved in THF (1 mL). Next, acetone (10 mL) was added to that solution and the resulting mixture was kept in the refrigerator for about an hour. The precipitate was filtered off to provide acid 15 (84 mg, 93%) as a white solid. NMR: δH (500 MHz, CDCl3-THF-d8 1:5) 1.60–1.69 [m, 12H, (POCH2CH2CH2CH2OC) and (POCH2CH2CH2CH2OC)], 3.86 [dt, 3J(H,H) = 6.0 Hz, 3J(P,H) = 12.6 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.11 [t, 3J(H,H) = 6.0 Hz, 6H, (POCH2CH2CH2CH2OC)], 8.52 [s, 6H, Ar], 8.55 [s, 3H, Ar] ppm; δC (125 MHz, CDCl3-THF-d8 1:5) 25.06 [3C, O=COCH2CH2CH2CH2O)], 26.22 [d, 3J(C,P) = 7.5 Hz, 3C, (OCH2CH2CH2CH2OP)], 67.04 [d, 2J(C,P) = 5.2 Hz, 3C, (OCH2CH2CH2CH2OP)], 67.25 [3C, (OCH2CH2CH2CH2OP)], 130.6 [9C, (ipso Ar)], 131.3 [9C, (Ar)], 164.3 [3C, (C=O)], 165.6 [6C, (C=O)] ppm; δP {1H} (202 MHz, CDCl3-THF-d8 1:5) 68.52 ppm; HRMS (TOF MS ES) M − H+: found 905.1371. C39H39O21PS requires 905.1364. Soon after, acid 15 (35 mg, 0.0179 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (10 mg, 0.11 mmol, 6 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mmHg) to give 16 as a hexasodium salt of 15 (40 mg), as a nonhygroscopic white powder.
Second generation polyester thiophosphate dendrimer17. Carboxy-terminated, first generation thiophosphate dendrimer 15 (FW 906.8, 40 mg, 0.044 mmol) was dissolved in dry THF (1.0 mL). Then, the reaction mixture was diluted with dry dichloromethane (2 mL). Di-tert-butyl 5-hydroxymethylbenzene-1,3-dicarboxylate (2) (FW 308.4, 67 mg, 0.3 mmol, 6.6 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (70 mg, 0.36 mmol, 8.0 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (FW 122.2, 5.0 mg, 0.04 mmol, 0.9 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (20 mL), washed with 0.1 M citric acid (10 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the cyclohexane–acetone 100:1 mixture and gradually increasing the polarity to cyclohexane–acetone 15:1, afforded the title ester 17 (95 mg). Yield 84%. δH (500 MHz, CDCl3) 1.58 [s, 108H, C(CH3)3], 1.80–1.87 [m, 6H, (POCH2CH2CH2CH2OC)], 1.88–1.96 [m, 6H, (POCH2CH2CH2CH2OC)], 4.12 [dt, 3J(H,H) = 6.3 Hz, 3J(P,H) =14.3 Hz, 6H, (POCH2CH2CH2CH2OC)], 4.40 [t, 3J(H,H) = 6.4 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.46 [s, 12H, (ArCH2)], 8.20 (s, 12H, Ar2), 8.53 (s, 6H, Ar2), 8.85 (s, 3H, Ar1), 8.87 (s, 6H, Ar1) ppm; δC (125 MHz, CDCl3) 25.01 [3C, O=COCH2CH2CH2CH2O)], 26.66 [d, 3J(C,P) = 7.6 Hz, 3C, (OCH2CH2CH2CH2OP)], 28.16 [36C, C(CH3)3], 65.13 [3C, (OCH2CH2CH2CH2OP)], 66.51 [6C, (ArCH2)], 67.61 [d, 2J(C,P) = 5.7 Hz, 3C, (OCH2CH2CH2CH2OP)], 81.87 [12C, C(CH3)3], 130.6 [6C, (Ar2)], 130.9 [6C, (ipso Ar1)], 131.6 [3C, (ipso Ar1)], 132.8 [12C, (ipso Ar2)], 133.1 [12C, (Ar2)], 134.9 [3C, (Ar1)], 135.0 [6C, (Ar1)], 136.0 [6C, (ipso Ar2)], 164.6 [21C, (C=O)] ppm; δP {1H} (202 MHz, CD3OD-CDCl3 2:1) 68.60 ppm; MALDI TOF MS calcd. for C141H171O45PS, M = 2647.0. Found m/z = 2670.4 M + Na, 2686.7 M + K.
Polyanionic dendrimer19. Dendrimer 17 (FW 2648.9, 70 mg, 0.0264 mmol) was dissolved in dry dichloromethane (1.5 mL), and trifluoroacetic acid was added (1.5 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 5 h. All the volatiles were removed in vacuo, and the residue was dissolved in THF (1 mL). Next, acetone (10 mL) was added to that solution and the resulting mixture was kept in the refrigerator for about an hour. The precipitate was filtered off to provide acid 18 (48 mg, 92%) as a white solid. NMR: δH (500 MHz, THF-d8) 1.82–1.88 [m, 6H, (POCH2CH2CH2CH2OC)], 1.90-1.98 [m, 6H, (POCH2CH2CH2CH2OC)], 3.59-3.64 [m, 6H, (POCH2CH2CH2CH2OC)], 3.80 [t, 3J(H,H) = 6.8 Hz, 6H, (POCH2CH2CH2CH2OC)], 5.47 [s, 12H, (ArCH2)], 8.30 (s, 12H, Ar2), 8.36 (s, 6H, Ar2), 8.54 (s, 6H, Ar1), 8.61 (s, 3H, Ar1), 10.87 (brs, 12H, COOH) ppm; δC (125 MHz, THF-d8) 25.40 [3C, O=COCH2CH2CH2CH2O)], 28.96 [3C, (OCH2CH2CH2CH2OP)], 65.92 [3C, (OCH2CH2CH2CH2OP)], 66.96, [3C, (OCH2CH2CH2CH2OP)], 67,24 [6C, (ArCH2)], 128.6 [9C, (Ar1)], 130.5 [12C, (ipso Ar2)], 130.9 [6C, (ipso Ar2)], 131.6 [6C, (Ar2)], 133.5 [12C, (Ar2)], 137.1 [9C, (ipso Ar1)], 163.9 [3C, (C=O)], 164.5 [6C, (C=O)], 165.8 [12C, (HOC=O)] ppm δP {1H} (202 MHz, THF-d8) 68.71 ppm; MALDI TOF MS calcd. for C93H75O45PS, M = 1974.3. Found m/z, fragmentation: 1149.8, 1121.7, 1093.5, 1065.5 (major peaks). Elemental analysis: Found (acid): C, 56.51; H, 3.85; S, 1.62%. C93H75O45PS requires C, 56.54; H, 3.83; S, 1.62%. Afterwards, acid 18 (35 mg, 0.0179 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (18 mg, 0.2 mmol, 12 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mmHg) to give 19 as a dodecasodium salt of 18 (40 mg), as a nonhygroscopic white powder.
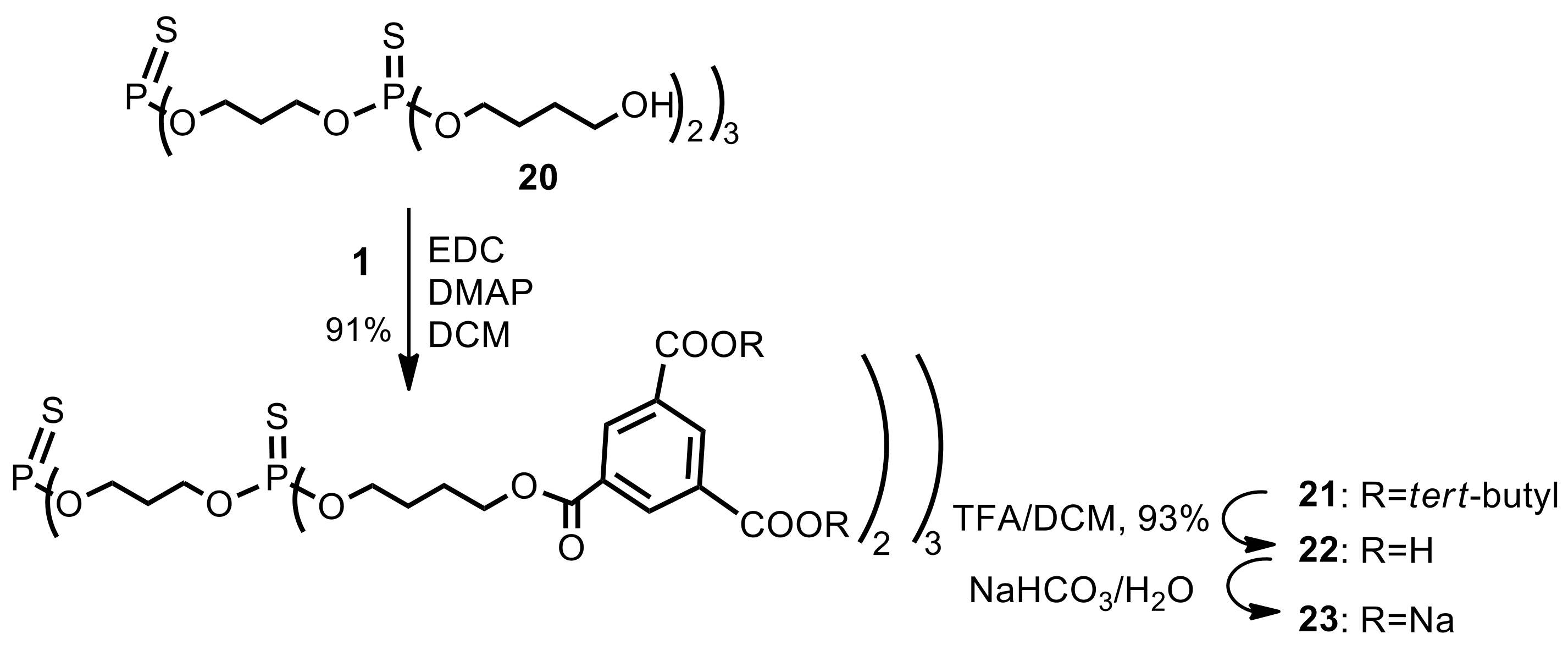
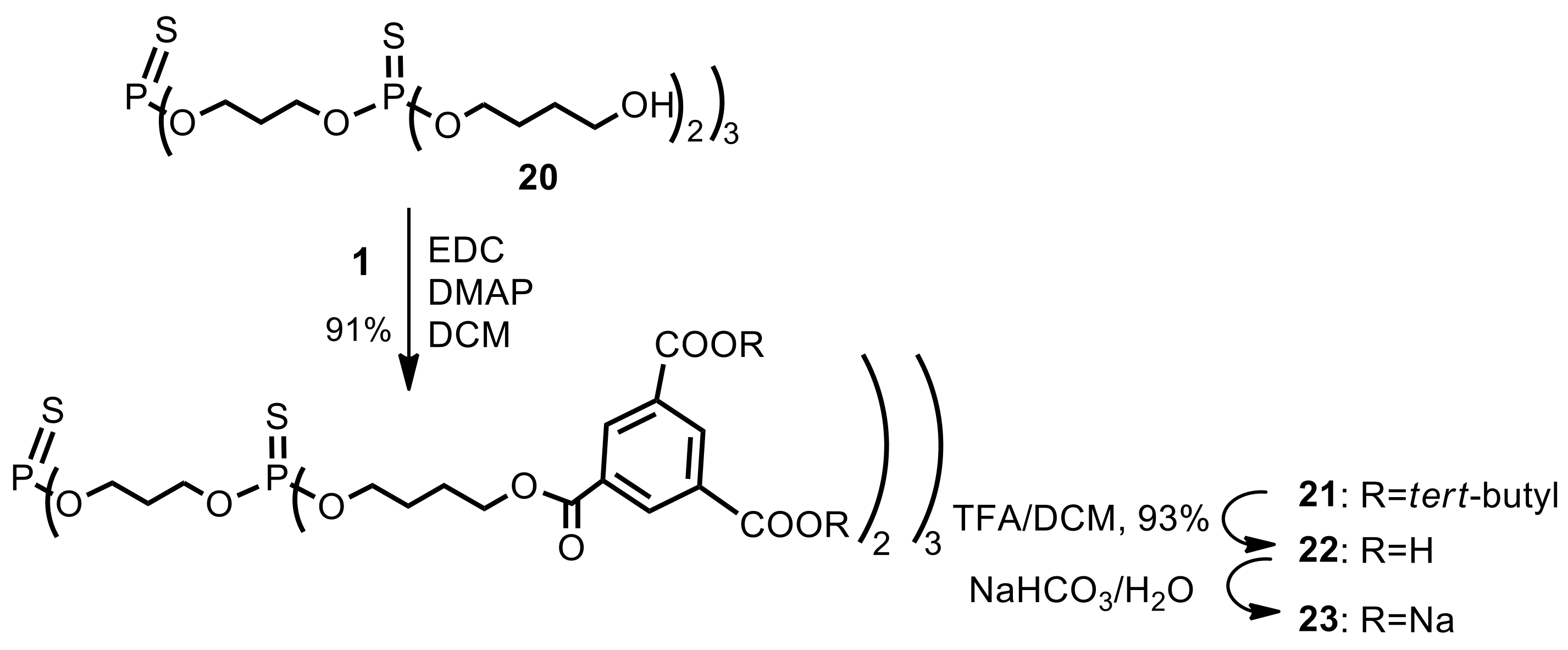
Second generation fully protected dendrimer21. Hydroxy-terminated, first generation thiophosphate dendrimer12 20 (FW 1009.1, 51 mg, 0.050 mmol) was suspended in dry dichloromethane (3.0 mL). Di-tert-butyl 1.3.5-benzenetricarboxylate (1) (FW 322.4, 105 mg, 0.325 mmol, 6.5 equiv.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (77 mg, 0.4 mmol, 8.0 equiv.) were added under argon atmosphere. The suspension was stirred vigorously and quickly dissolved. After 5 min, 4-dimethylaminopyridine (DMAP) (FW 122.2, 5.0 mg, 0.04 mmol, 0.9 equiv.) was added and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (20 mL), washed with 0.1 M citric acid (10 mL), dried (MgSO4), and evaporated under reduced pressure. The residue was placed on a short plug of silica gel. Elution with the neat dichloromethane provided the title dendrimer 21 (127 mg). Yield 90%. δH (500 MHz, CDCl3) 1.58 [s, 108H, C(CH3)3], 1.78–1.87 [m, 12H, (POCH2CH2CH2CH2OC)], 1.88–1.95 [m, 12H, (POCH2CH2CH2CH2OC)], 2.12–2.21 [m, 6H, (POCH2CH2CH2OP)], 4.12 [dt, 3J(H,H) = 6.3 Hz, 3J(P,H) = 13.6 Hz, 12H, (POCH2CH2CH2CH2OC)], 4.24 [dt, 3J(H,H) = 6.3 Hz, 3J(P,H) = 14.3 Hz, 12H, (POCH2CH2CH2OP)], 4.45 [t, 3J(H,H) = 6.1 Hz, 12H, (POCH2CH2CH2CH2OC)], 8.70 (s, 6H, Ar), 8.71 (s, 12H, Ar) ppm; δC (125 MHz, CDCl3), 25.07 [6C, O=COCH2CH2CH2CH2O)], 26.71 [d, 3J(C,P) = 7.8 Hz, 6C, (OCH2CH2CH2CH2OP)], 28.13 [36C, C(CH3)3], 29.41–29.49 [m, 3J(C,P) = 7.8 Hz, 3C, (POCH2CH2CH2OP)], 61.62–61.73 [m, 6C, (POCH2CH2CH2OP)], 64.87 [6C, (OCH2CH2CH2CH2OP)], 67.64 [d, 2J(C,P) = 5.4 Hz, 6C, (OCH2CH2CH2CH2OP)], 82.17 [12C, C(CH3)3], 130.9 [6C, (ipso Ar)], 132.8 [12C, (ipso Ar)], 134.0 [12C, (Ar)], 134.4 [6C, (Ar)], 164.2 [12C, (C=O)], 165.2 [6C, (C=O)] ppm; δP {1H} (202 MHz, CDCl3), 68.29 (P0), 68.58 (3P1) ppm; MALDI TOF MS calcd. for C135H192O48P4S4, M = 2833.0. Found m/z = 2845.8 M + Na.
Polyanionic dendrimer23. Dendrimer 21 (80 mg, 0.028 mmol) was dissolved in dry dichloromethane (1.5 mL), and trifluoroacetic acid was added (1.5 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 5 h. All the volatiles were removed in vacuo, and the residue was dissolved in THF (1 mL). Next, acetone (11 mL) was added to that solution and the resulting mixture was kept in the refrigerator for about an hour. The precipitate was filtered off to provide acid 22 (56 mg, 93%) as a white solid. NMR: δH (200 MHz, CD3OD) 1.75–1.96 [m, 24H, (POCH2CH2CH2CH2OC)], 2.12–2.21 [m, 6H, (POCH2CH2CH2OP)], 4.09–4.21 [m, 3J(H,H) = 6.3 Hz, 12H, (POCH2CH2CH2CH2OC)], 4.24–4.30 [m, 12H, (POCH2CH2CH2OP)], 4.37 [t, 3J(H,H) = 6.0 Hz, 12H, (POCH2CH2CH2CH2OC)], 8.70 (s, 12H, Ar), 8.74 (s, 6H, Ar) ppm; δC (125 MHz, CD3OD-CDCl3 3:1), 26.44 [6C, O=COCH2CH2CH2CH2O)], 28.09 [d, 3J(C,P) = 7.4 Hz, 6C, (OCH2CH2CH2CH2OP)], 30.60–30.70 [m, 3C, (POCH2CH2CH2OP)], 66.27–66.40 [m, 6C, (POCH2CH2CH2OP)], 66.53 [6C, (OCH2CH2CH2CH2OP)], 69.15 [d, 2J(C,P) = 5.6 Hz, 6C, (OCH2CH2CH2CH2OP)], 132.5 [6C, (ipso Ar)], 133.3 [6C, (Ar)], 135.6 [12C, (Ar)], 136.1 [12C, (ipso Ar)], 166.6 [6C, (C=O)], 167.9 [12C, (C=O)] ppm; δP {1H} (202 MHz, CD3OD-CDCl3 3:1), 68.61 (P0), 68.80 (3P1) ppm; MALDI TOF MS calcd. for C87H96O48P4S4, M = 2160.3. Found m/z, fragmentation: 1470.0, 1450.8, 1005.7, 947.5, 930.7 (major peaks). Elemental analysis: Found (acid): C, 48.39; H, 4.41; S, 6.00%. C87H96O48P4S4 requires C, 48.34; H, 4.48; S, 5.93%. Finally, acid 22 (40 mg, 0.0185 mmol) was dispersed in water (2 mL) and solid sodium bicarbonate (19 mg, 0.22 mmol, 12 equiv.) was added. After 20 min, the resulting solution was frozen and lyophilized under high vacuum (0.1 mm Hg) to give 23 as a dodecasodium salt of 22 (47 mg), as a nonhygroscopic white powder.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}