
Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening

 ,
,  ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. VS Workflow Design and In Silico Validation
2.1.1. Random Forest Model
2.1.2. Protein-Ligand Docking
2.1.3. Pharmacophore
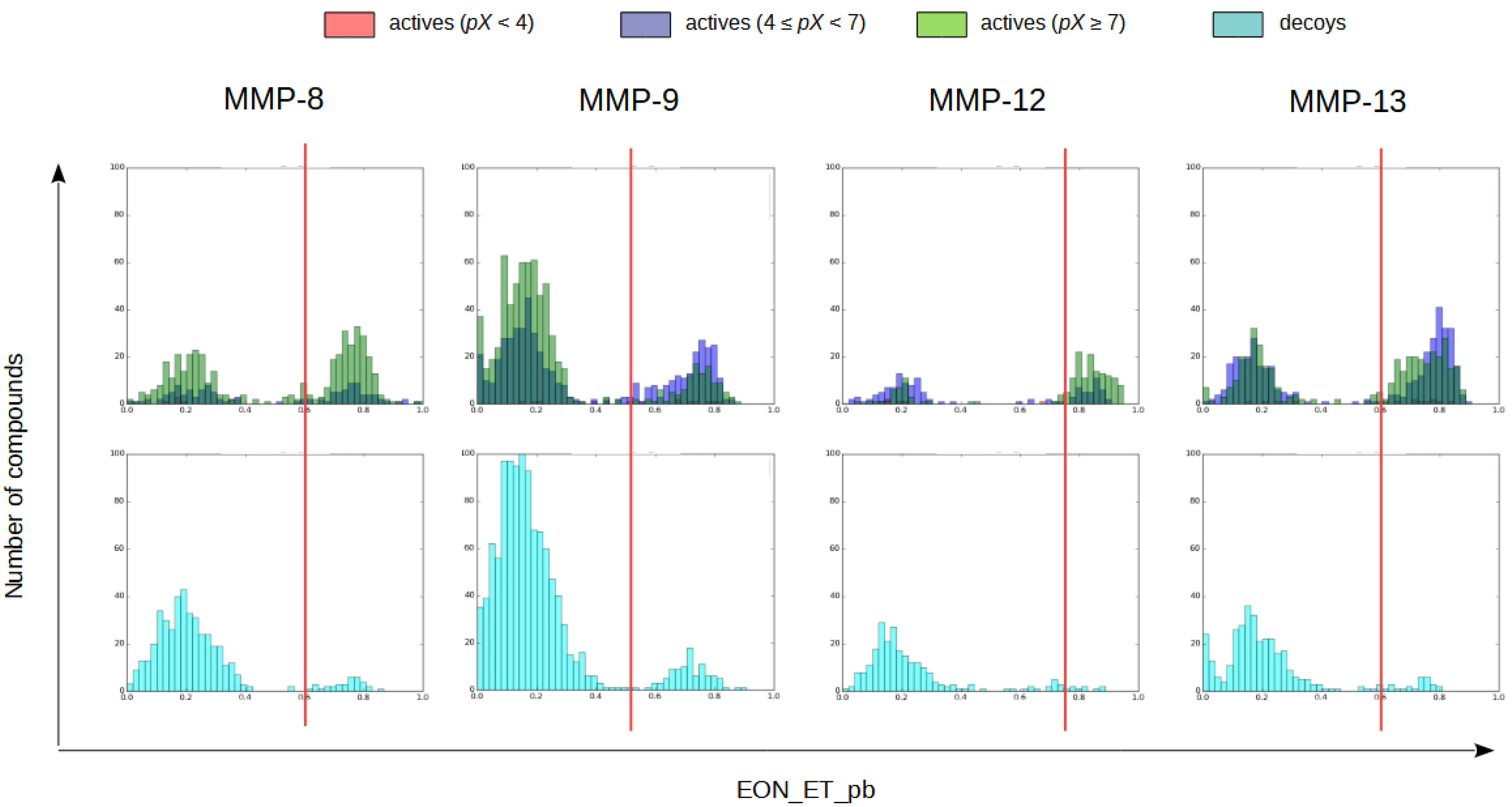
2.1.4. Electrostatic Similarity Analysis
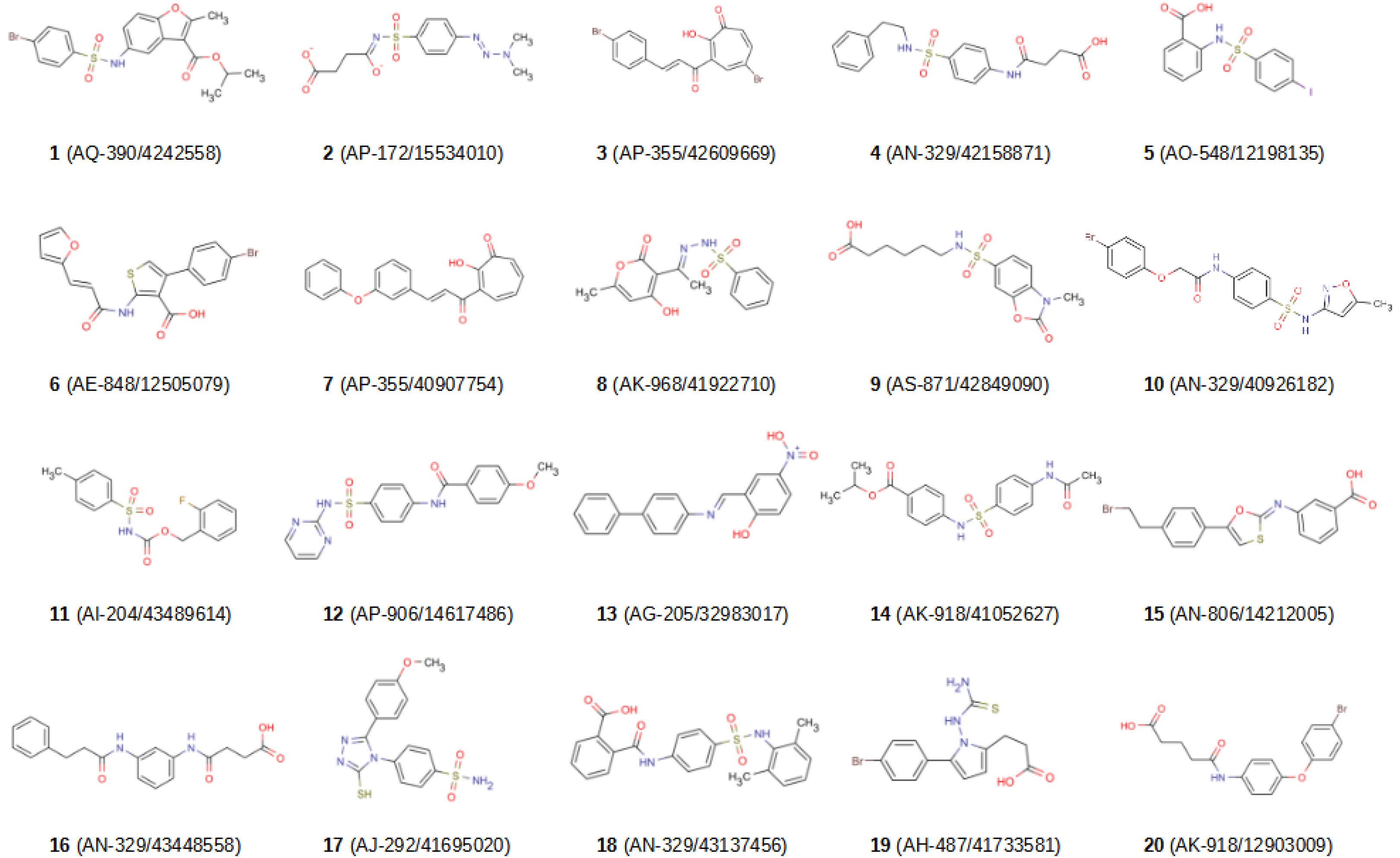
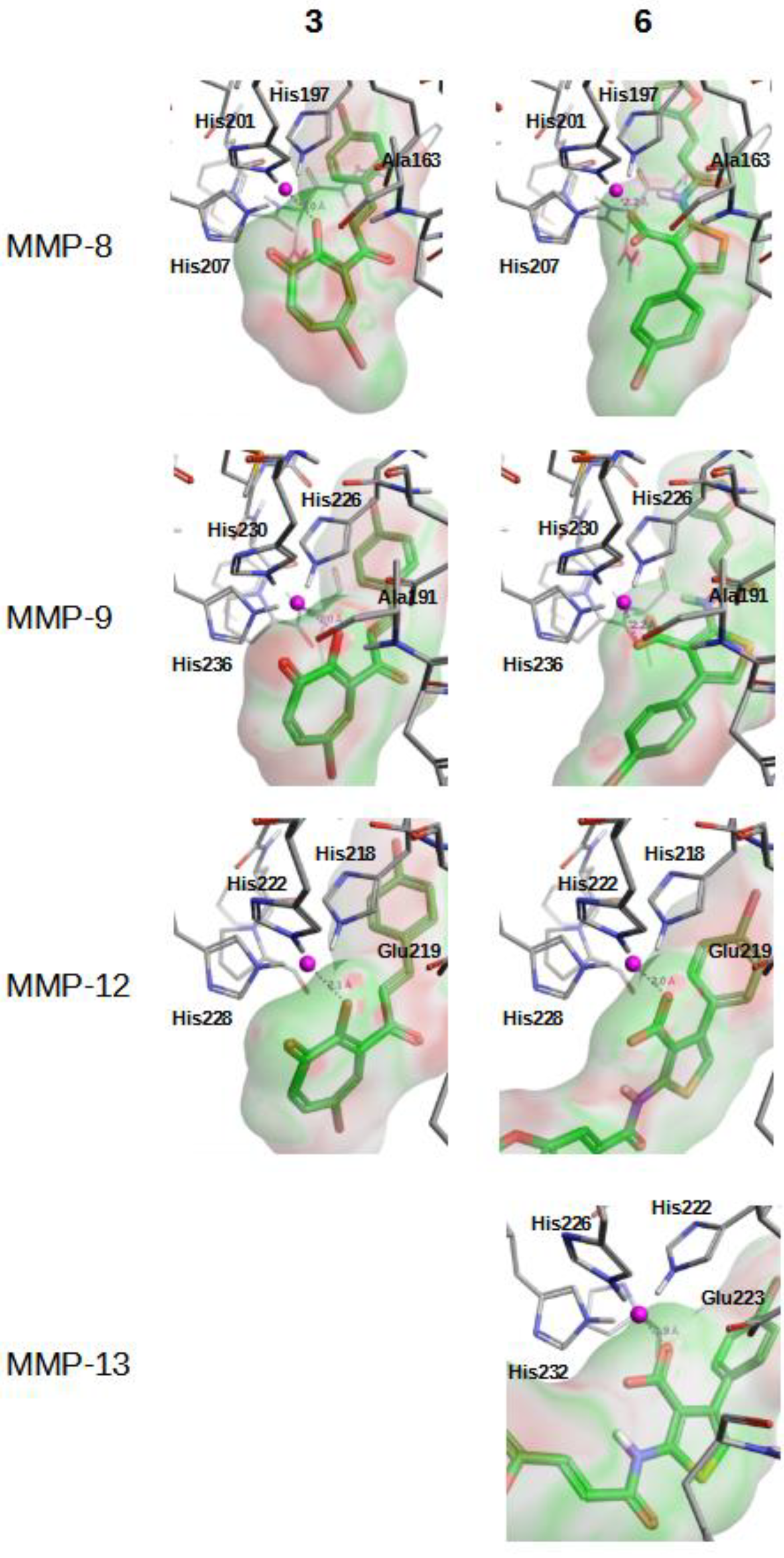
2.2. In Vitro Validation
2.3. VS of Natural Products
3. Materials and Methods
3.1. RF Model
3.2. Ligand Setup for Docking
3.3. Protein Preparation
3.4. Grid Generation
3.5. Protein-Ligand Docking
3.6. Pharmacophore Generation
3.7. Electrostatic Similarity Analysis
3.8. MMP Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Alberts, B.; Jonhson, A.D.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; W.W. Norton & Company: New York, NY, USA, 2014. [Google Scholar]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Rath, T.; Roderfeld, M.; Graf, J.; Wagner, S.; Vehr, A.-K.; Dietrich, C.; Geier, A.; Roeb, E. Enhanced expression of MMP-7 and MMP-13 in inflammatory bowel disease: A precancerous potential? Inflamm. Bowel Dis. 2006, 12, 1025–1035. [Google Scholar] [CrossRef]
- Siloşi, I.; Boldeanu, M.V.; Mogoantă, S.Ş.; Ghiluşi, M.; Cojocaru, M.; Biciuşcă, V.; Cojocaru, I.M.; Avrămescu, C.S.; Gheonea, D.I.; Siloşi, C.A.; et al. Matrix metalloproteinases (MMP-3 and MMP-9) implication in the pathogenesis of inflammatory bowel disease (IBD). Rom. J. Morphol. Embryol. 2014, 55, 1317–1324. [Google Scholar] [PubMed]
- Kurzepa, J.; Madro, A.; Czechowska, G.; Kurzepa, J.; Celiński, K.; Kazmierak, W.; Slstrokomka, M. Role of MMP-2 and MMP-9 and their natural inhibitors in liver fibrosis, chronic pancreatitis and non-specific inflammatory bowel diseases. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 570–579. [Google Scholar] [CrossRef]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint Diseases and Matrix Metalloproteinases: A Role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Solovуeva, N.I.; Timoshenko, O.S.; Gureeva, T.A.; Kugaevskaya, E.V. Matrix metalloproteinases and their endogenous regulators in squamous cervical carcinoma (review of the own data). Biomeditsinskaya Khimiya 2015, 61, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; López-Otín, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Cathcart, J.M.; Cao, J. MMP Inhibitors: Past, present and future. Front. Biosci. 2015, 20, 1164–1178. [Google Scholar]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Pulkoski-Gross, A.E. Historical Perspective of Matrix Metalloproteases. Front. Biosci. 2015, 7, 125–149. [Google Scholar] [CrossRef] [PubMed]
- Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.H.; Beckett, P.; Brown, P.D.; Bone, E.A.; Davidson, A.H.; Galloway, W.A.; Gearing, A.J.; Huxley, P.; Laber, D.; McCourt, M.; et al. Preclinical and clinical studies of MMP inhibitors in cancer. Ann. N. Y. Acad. Sci. 1999, 878, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.W.; Tierney, G.M.; Parsons, S.L.; Davis, T.R. Dupuytren’s disease and frozen shoulder induced by treatment with a matrix metalloproteinase inhibitor. J. Bone Jt. Surg. Br. 1998, 80, 907–908. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Fingleton, B. MMPs as therapeutic targets—Still a viable option? Semin. Cell Dev. Biol. 2008, 19, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Tampa, M.; Georgescu, S.R.; Mitran, M.I.; Mitran, C.I.; Matei, C.; Caruntu, A.; Scheau, C.; Nicolae, I.; Matei, A.; Caruntu, C.; et al. Current Perspectives on the Role of Matrix Metalloproteinases in the Pathogenesis of Basal Cell Carcinoma. Biomolecules 2021, 11, 903. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Kähäri, V.-M. Matrix metalloproteinases in keratinocyte carcinomas. Exp. Dermatol. 2021, 30, 50–61. [Google Scholar] [CrossRef]
- Quirt, I.; Bodurth, A.; Lohmann, R.; Rusthoven, J.; Belanger, K.; Young, V.; Wainman, N.; Stewar, W.; Eisenhauer, E. National Cancer Institute of Canada Clinical Trials Group Phase II study of marimastat (BB-2516) in malignant melanoma: A clinical and tumor biopsy study of the National Cancer Institute of Canada Clinical Trials Group. Investig. New Drugs 2002, 20, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. Understanding the variability of the S1′ pocket to improve matrix metalloproteinase inhibitor selectivity profiles. Drug Discov. Today 2020, 25, 38–57. [Google Scholar] [CrossRef]
- Rittié, L.; Fisher, G.J. UV-light-induced signal cascades and skin aging. Ageing Res. Rev. 2002, 1, 705–720. [Google Scholar] [CrossRef]
- Imokawa, G.; Nakajima, H.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging II: Over-expression of neprilysin plays an essential role. Int. J. Mol. Sci. 2015, 16, 7776–7795. [Google Scholar] [CrossRef] [PubMed]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Marrero, A.; Gomis-Rüth, F.X. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 20–28. [Google Scholar] [CrossRef]
- Fabre, B.; Ramos, A.; de Pascual-Teresa, B. Targeting matrix metalloproteinases: Exploring the dynamics of the S1′ pocket in the design of selective, small molecule inhibitors. J. Med. Chem. 2014, 57, 10205–10219. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pochetti, G.; Gavuzzo, E.; Campestre, C.; Agamennone, M.; Tortorella, P.; Consalvi, V.; Gallina, C.; Hiller, O.; Tschesche, H.; Tucker, P.A.; et al. Structural insight into the stereoselective inhibition of MMP-8 by enantiomeric sulfonamide phosphonates. J. Med. Chem. 2006, 49, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Vera, L.; Devel, L.; Catalani, M.P.; Czarny, B.; Cassar-Lajeunesse, E.; Nuti, E.; Rossello, A.; Dive, V.; Stura, E.A. Crystallization of bi-functional ligand protein complexes. J. Struct. Biol. 2013, 182, 246–254. [Google Scholar] [CrossRef]
- Morales, R.; Perrier, S.; Florent, J.M.; Beltra, J.; Dufour, S.; De Mendez, I.; Manceau, P.; Tertre, A.; Moreau, F.; Compere, D.; et al. Crystal structures of novel non-peptidic, non-zinc chelating inhibitors bound to MMP-12. J. Mol. Biol. 2004, 341, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.P.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; Bond, B.R.; Carroll, J.; Carron, C.P.; Decrescenzo, G.A.; Easton, A.M.; Freskos, J.N.; et al. Orally active MMP-1 sparing α-tetrahydropyranyl and α-piperidinyl Sulfone matrix metalloproteinase (MMP) inhibitors with efficacy in cancer, arthritis, and cardiovascular disease. J. Med. Chem. 2010, 53, 6653–6680. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Specs.net. Available online: http://www.specs.net/ (accessed on 17 July 2021).
- OMEGA 4.1.0.4: OpenEye Scientific Software, Santa Fe, NM, USA. Available online: https://www.eyesopen.com/omega (accessed on 28 July 2021).
- McInnes, L.; Healy, J.; Astels, S. hdbscan: Hierarchical density based clustering. J. Open Source Softw. 2017, 2, 1–3. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- MarvinSketch 21.12. ChemAxon. Available online: https://www.chemaxon.com (accessed on 28 July 2021).
- Reaxys. Available online: http://www.reaxys.com (accessed on 17 July 2021).
- Qi-Zhuang, Y.; Johnson, L.L.; Nordan, I.; Hupe, D.; Hupe, L. A Recombinant Human Stromelysin Catalytic Domain Identifying Tryptophan Derivatives as Human Stromelysin Inhibitors. J. Med. Chem. 1994, 37, 206–209. [Google Scholar] [CrossRef]
- Murata, T.; Sasaki, K.; Sato, K.; Yoshizaki, F.; Yamada, H.; Mutoh, H.; Umehara, K.; Miyase, T.; Warashina, T.; Aoshima, H.; et al. Matrix metalloproteinase-2 inhibitors from Clinopodium chinense var. parviflorum. J. Nat. Prod. 2009, 72, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Albarano, T. Pharmaceutical Compositions. U.S. Patent 20200046627A1, 13 February 2020. [Google Scholar]
- Sau, S.; Iyer, A.K.; Alsaab, H. Method of Treatment for Solid Tumors Containing Hypoxia and/or Stroma Features. WO2019133914, 29 December 2019. [Google Scholar]
- Doss, J.C. Therapeutic Soap Product with UV Protection. U.S. Patent 20070071698A1, 29 March 2007. [Google Scholar]
- Wang, W.; Tang, M.; Pang, F.; Fan, X. Wrinkle-Resistant Moisturizing Cream and Preparation Method Thereof. 109330915, 15 February 2019. [Google Scholar]
- Raper, P.; Haig, C.; Ejifor, O.; Raper, R. Skin Enhancing Beverage Composition. WO/2014/114939, 23 January 2014. [Google Scholar]
- Johansen, B.; Feuerherm, A.J. Combination Therapy Comprising a Polyunsaturated Ketone and a Folic Acid Partner. WO2017207820, 7 December 2017. [Google Scholar]
- Michael, T.P. Composition for an Anti-Aging Treatment. U.S. Patent 9839604, 12 December 2017. [Google Scholar]
- Neamati, N.; Xu, S.; Tamura, S. Compositions and Methods Relating to Inhibiting Serine Hyrdoxymethyltransferase 2 Activity. WO2016085990, 24 November 2016. [Google Scholar]
- Diaz, V.H. Composition Including Superoxide Dismutase and Prickly-Pear Cactus for Minimizing and Preventing Hangovers. U.S. Patent 2008020071, 10 August 2008. [Google Scholar]
- Lin, S.; Van Reeth, I. Personal Care Compositions Containing Silicone Elastomer Gels. WO2008085360, 19 December 2008. [Google Scholar]
- Deperraz, F.; Baroth, V. Colloidal Solution. WO2008086953, 8 January 2008. [Google Scholar]
- Lee Hye, J.; Kim Mi, S.; Lee Sang, H. Composition for Improving the Skin. KR20170073308, 3 August 2017. [Google Scholar]
- Ko, E.A.; Jeon, J.H.; Hong, J.H. Cosmetic Composition Comprising Oleanolic Acid and Salvianolic Acid for Skin Whitening or Wrinkle Preventing. WO/2019/103194, 24 November 2018. [Google Scholar]
- Standardizer 16.10.10.0. ChemAxon. 2016. Available online: https://chemaxon.com/products/chemical-structure-representation-toolkit (accessed on 28 July 2021).
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org/ (accessed on 18 July 2021).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Louppe, G.; Prettenhofer, P.; Weiss, R.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef]
- Platt, J. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. Adv. Large Margin Classif. 1999, 10, 61–74. [Google Scholar]
- Schrödinger Release 2018-1: LigPrep; Schrödinger, LLC.: New York, NY, USA, 2018.
- Schrödinger Release 2018-1: Epik; Schrödinger, LLC.: New York, NY, USA, 2018.
- Cereto-Massagué, A.; Ojeda, M.J.; Joosten, R.P.; Valls, C.; Mulero, M.; Salvado, M.J.; Arola-Arnal, A.; Arola, L.; Garcia-Vallvé, S.; Pujadas, G. The good, the bad and the dubious: VHELIBS, a validation helper for ligands and binding sites. J. Cheminform. 2013, 5, 36. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Schrödinger Suite 2018-1 Protein Preparation Wizard, Epik; Schrödinger, LLC.: New York, NY, USA; Impact, Schrödinger, LLC.: New York, NY, USA; Prime, Schrödinger, LLC.: New York, NY, USA, 2018.
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC.: New York, NY, USA, 2018.
- Schrödinger Release 2018-1: Glide; Schrödinger, LLC.: New York, NY, USA, 2018.
- Glide Fragment Library. Schrodinger. Available online: http://www.schrodinger.com/Glide/Fragment-Library (accessed on 24 April 2014).
- Salam, N.K.; Nuti, R.; Sherman, W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- EON 2.2.0.5: OpenEye Scientific Software, Santa Fe, NM, USA. Available online: https://www.eyesopen.com/eon (accessed on 28 July 2021).
- Matter, H.; Schwab, W.; Barbier, D.; Billen, G.; Haase, B.; Neises, B.; Schudok, M.; Thorwart, W.; Schreuder, H.; Brachvogel, V.; et al. Quantitative structure-activity relationship of human neutrophil collagenase (MMP-8) inhibitors using comparative molecular field analysis and X-ray structure analysis. J. Med. Chem. 1999, 42, 1908–1920. [Google Scholar] [CrossRef]
- Devel, L.; Beau, F.; Amoura, M.; Vera, L.; Cassar-Lajeunesse, E.; Garcia, S.; Czarny, B.; Stura, E.A.; Dive, V. Simple pseudo-dipeptides with a P2′ glutamate: A novel inhibitor family of matrix metalloproteases and other metzincins. J. Biol. Chem. 2012, 287, 26647–26656. [Google Scholar] [CrossRef]
- Tochowicz, A.; Maskos, K.; Huber, R.; Oltenfreiter, R.; Dive, V.; Yiotakis, A.; Zanda, M.; Pourmotabbed, T.; Bode, W.; Goettig, P. Crystal structures of MMP-9 complexes with five inhibitors: Contribution of the flexible Arg424 side-chain to selectivity. J. Mol. Biol. 2007, 371, 989–1006. [Google Scholar] [CrossRef]
- Holmes, I.P.; Gaines, S.; Watson, S.P.; Lorthioir, O.; Walker, A.; Baddeley, S.J.; Herbert, S.; Egan, D.; Convery, M.A.; Singh, O.M.P.; et al. The identification of beta-hydroxy carboxylic acids as selective MMP-12 inhibitors. Bioorgan. Med. Chem. Lett. 2009, 19, 5760–5763. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, E.; Calderone, V.; Fragai, M.; Jaiswal, R.; Luchinat, C.; Nativi, C. Biotin-tagged probes for MMP expression and activation: Design, synthesis, and binding properties. Bioconjug. Chem. 2009, 20, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Czarny, B.; Stura, E.A.; Devel, L.; Vera, L.; Cassar-Lajeunesse, E.; Beau, F.; Calderone, V.; Fragai, M.; Luchinat, C.; Dive, V. Molecular determinants of a selective matrix metalloprotease-12 inhibitor: Insights from crystallography and thermodynamic studies. J. Med. Chem. 2013, 56, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Monovich, L.G.; Tommasi, R.A.; Fujimoto, R.A.; Blancuzzi, V.; Clark, K.; Cornell, W.D.; Doti, R.; Doughty, J.; Fang, J.; Farley, D.; et al. Discovery of potent, selective, and orally active carboxylic acid based inhibitors of matrix metalloproteinase-13. J. Med. Chem. 2009, 52, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- France, D.J.; Stepek, G.; Houston, D.R.; Williams, L.; McCormack, G.; Walkinshaw, M.D.; Page, A.P. Identification and activity of inhibitors of the essential nematode-specific metalloprotease DPY-31. Bioorgan. Med. Chem. Lett. 2015, 25, 5752–5755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robertson, E.; Harcus, Y.; Johnston, C.J.C.; Page, A.P.; Walkinshaw, M.D.; Maizels, R.M.; Houston, D. Demonstration of the anthelmintic potency of marimastat in the heligmosomoides polygyrus rodent model. J. Parasitol. 2018. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
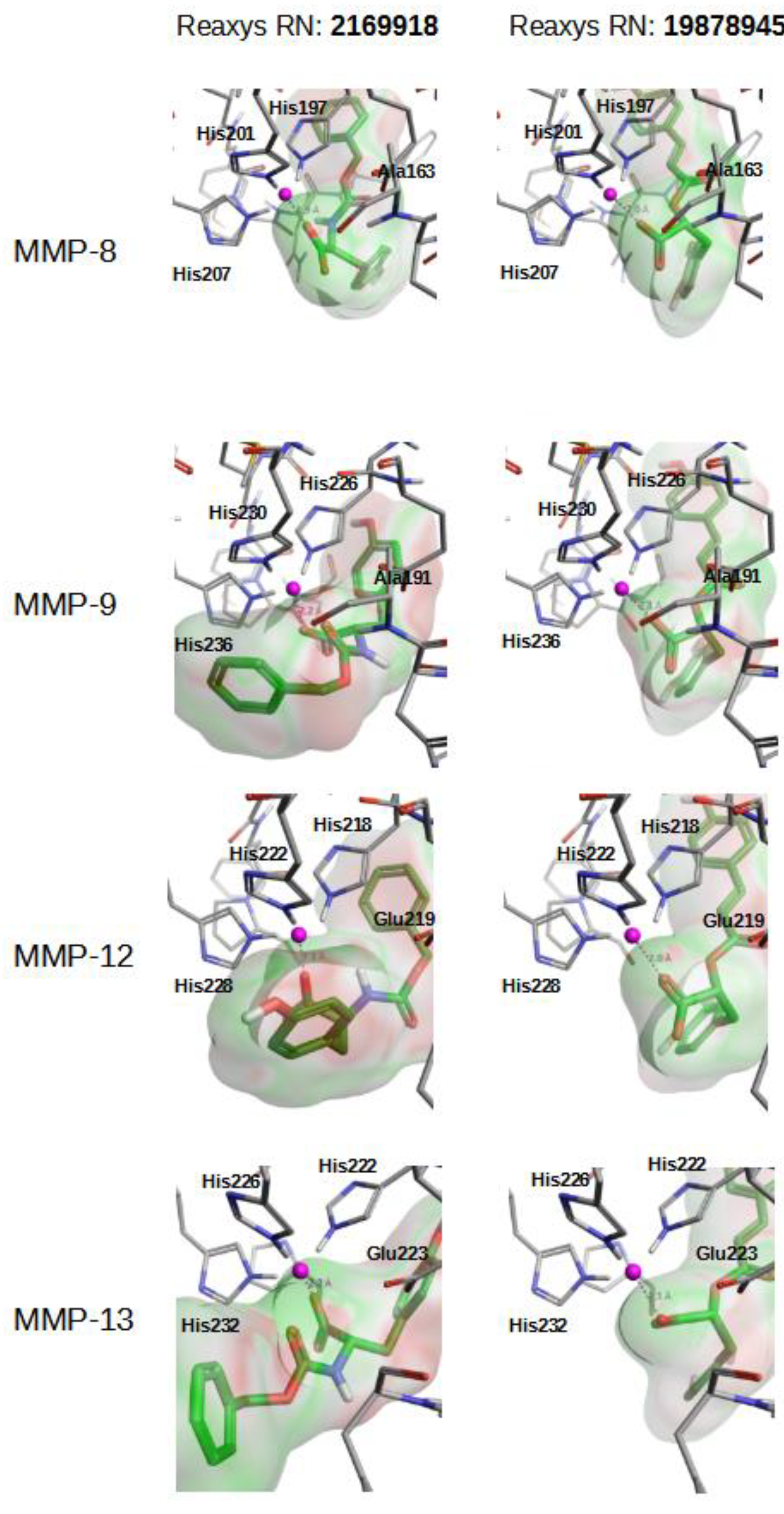{kind=link}
| Specs | Reaxys NP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MMP-1 | MMP-8 | MMP-9 | MMP-12 | MMP-13 | MMP-1 | MMP-8 | MMP-9 | MMP-12 | MMP-13 | |
| Initial library | 45,711 | 105,050 | ||||||||
| Random forest | 1344 | 4576 | 3334 | 3819 | 1584 | 5878 | 5811 | 11,070 | 17,684 | 2991 |
| Protein-ligand docking | - | 3775 | 2760 | 3001 | 1314 | - | 2401 | 4970 | 6896 | 1285 |
| Pharmacophore | 1064 | 2762 | 785 | 1055 | 454 | 3358 | 958 | 905 | 987 | 209 |
| Electrostatic similarity analysis | - | 79 | 60 | 32 | 27 | - | 118 | 314 | 102 | 70 |
| Hits of 2 or more VSs | 54 | 183 | ||||||||
| MMP | Sensitivity | Specificity | Precision | Fall-Out | False Negative Rate | False Discovery Rate | Accuracy | F1 Score | Matthews Correlation Coefficient |
|---|---|---|---|---|---|---|---|---|---|
| MMP-1 | 0.99 | 0.98 | 0.98 | 0.02 | 0.01 | 0.02 | 0.99 | 0.99 | 0.97 |
| MMP-8 | 0.99 | 0.97 | 0.97 | 0.03 | 0.01 | 0.03 | 0.98 | 0.98 | 0.96 |
| MMP-9 | 0.99 | 0.98 | 0.98 | 0.02 | 0.01 | 0.02 | 0.98 | 0.98 | 0.97 |
| MMP-12 | 0.99 | 0.98 | 0.98 | 0.02 | 0.01 | 0.02 | 0.98 | 0.98 | 0.96 |
| MMP-13 | 0.99 | 0.97 | 0.97 | 0.03 | 0.01 | 0.03 | 0.98 | 0.98 | 0.96 |
| MMP | Pharmacophore Hypothesis | Sensitivity | Specificity | Precision | Fall-Out | False Negative Rate | False Discovery Rate | Accuracy | F1 Score | Matthews Correlation Coefficient |
|---|---|---|---|---|---|---|---|---|---|---|
| MMP-1 | A (+) A (+) R (+) | 0.89 | 0.33 | 0.57 | 0.67 | 0.11 | 0.43 | 0.61 | 0.70 | 0.26 |
| MMP-8 | A (+) A (−) D (−) D (−) D (−) D (−) R (+) | 0.88 | 0.55 | 0.66 | 0.45 | 0.12 | 0.34 | 0.72 | 0.76 | 0.46 |
| MMP-9 | A (−) A(+) D (−) D (−) H (−) N (+) R (−) R (−) | 0.67 | 0.86 | 0.84 | 0.14 | 0.33 | 0.16 | 0.76 | 0.74 | 0.54 |
| MMP-12 | A (−) D (−) D (−) N (+) R (+) R (−) | 0.82 | 0.75 | 0.76 | 0.25 | 0.18 | 0.24 | 0.78 | 0.79 | 0.57 |
| MMP-13 | A (−) D (−) D (−) D (−) N (+) R (−) R (+) | 0.82 | 0.74 | 0.76 | 0.26 | 0.18 | 0.24 | 0.78 | 0.79 | 0.56 |
| Compound | MMP-1 | MMP-8 | MMP-9 | MMP-12 | MMP-13 |
|---|---|---|---|---|---|
| 1 | 10.5% | 19.6% | 18.2% | 16.1% | 21.3% |
| 2 b | ND | ND | ND | ND | ND |
| 3 | 84.2% | 80.9% | 80.1% | 79.7% | 69.5% |
| 4 | 17.5% | 19.8% | 24.8% | 17.8% | 17.8% |
| 5 | 25.2% | 27.8% | 32.0% | 26.3% | 22.4% |
| 6 | 71.2% | 68.5% | 78.1% | 70.4% | 77.8% |
| 7 | 74.0% | 71.1% | 72.7% | 73.5% | 60.0% |
| 8 | 50.9% | 53.1% | 59.3% | 56.5% | 48.8% |
| 9 | 17.6% | 8.7% | 14.9% | 18.9% | 16.8% |
| 10 | 19.1% | 21.7% | 25.6% | 31.5% | 19.4% |
| 11 | 5.5% | 6.0% | 27.8% | 19.3% | 18.8% |
| 12 | 23.7% | 10.8% | 28.4% | 31.2% | 20.9% |
| 13 c | ND | ND | ND | ND | ND |
| 14 | 18.9% | 25.6% | 26.1% | 30.4% | 31.2% |
| 15 | 50.3% | 66.5% | 60.2% | 51.8% | 71.3% |
| 16 | 15.3% | 5.6% | 20.5% | 58.4% | 20.0% |
| 17 | 20.4% | 22.6% | 76.8% | 35.5% | 27.3% |
| 18 | 10.0% | 20.4% | 32.3% | 52.6% | 28.4% |
| 19 | 32.0% | 30.3% | 69.7% | 32.6% | 37.8% |
| 20 | 6.4% | 29.2% | 27.2% | 32.2% | 26.9% |
| Compound | MMP-1 | MMP-8 | MMP-9 | MMP-12 | MMP-13 |
|---|---|---|---|---|---|
| 3 | 21 ± 2 | 23 ± 2 | 23 ± 1 | 24 ± 1 | 35 ± 3 |
| 6 | 32 ± 4 | 31 ± 5 | 26 ± 2 | 33 ± 5 | 33 ± 4 |
| 7 | 41 ± 2 | 41 ± 5 | 31 ± 1 | 30 ± 2 | 62 ± 4 |
| 8 | 92 ± 9 | 103 ± 17 | 80 ± 4 | 107 ± 9 | 108 ± 10 |
| 15b | 70 ± 9 | 77 ± 10 | 47 ± 6 | 111 ± 1 | 46 ± 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimeno, A.; Cuffaro, D.; Nuti, E.; Ojeda-Montes, M.J.; Beltrán-Debón, R.; Mulero, M.; Rossello, A.; Pujadas, G.; Garcia-Vallvé, S. Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening. Molecules 2021, 26, 4553. https://doi.org/10.3390/molecules26154553
Gimeno A, Cuffaro D, Nuti E, Ojeda-Montes MJ, Beltrán-Debón R, Mulero M, Rossello A, Pujadas G, Garcia-Vallvé S. Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening. Molecules. 2021; 26(15):4553. https://doi.org/10.3390/molecules26154553
Chicago/Turabian StyleGimeno, Aleix, Doretta Cuffaro, Elisa Nuti, María José Ojeda-Montes, Raúl Beltrán-Debón, Miquel Mulero, Armando Rossello, Gerard Pujadas, and Santiago Garcia-Vallvé. 2021. "Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening" Molecules 26, no. 15: 4553. https://doi.org/10.3390/molecules26154553
APA StyleGimeno, A., Cuffaro, D., Nuti, E., Ojeda-Montes, M. J., Beltrán-Debón, R., Mulero, M., Rossello, A., Pujadas, G., & Garcia-Vallvé, S. (2021). Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening. Molecules, 26(15), 4553. https://doi.org/10.3390/molecules26154553