
Ni Oxidation State and Ligand Saturation Impact on the Capability of Octaazamacrocyclic Complexes to Bind and Reduce CO2

 , , ,
, , ,  and
and
Abstract
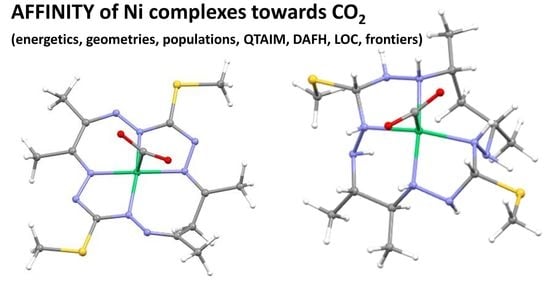
:
1. Introduction
2. Results
2.1. Energetics
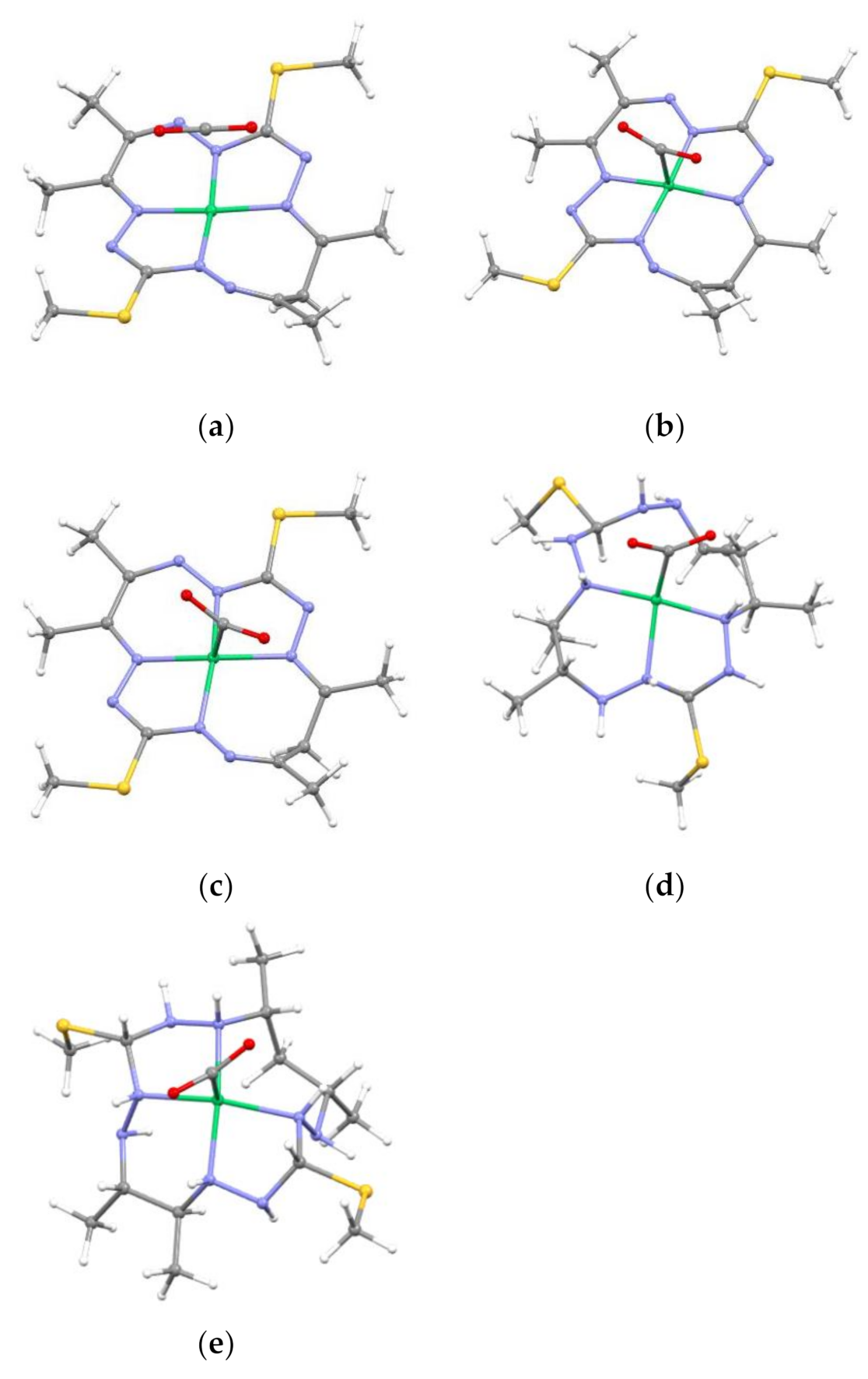
2.2. Optimized Geometries
2.3. MPA and QTAIM Charges
2.4. MPA d-Orbital Ni Populations
2.5. QTAIM BCP Analysis
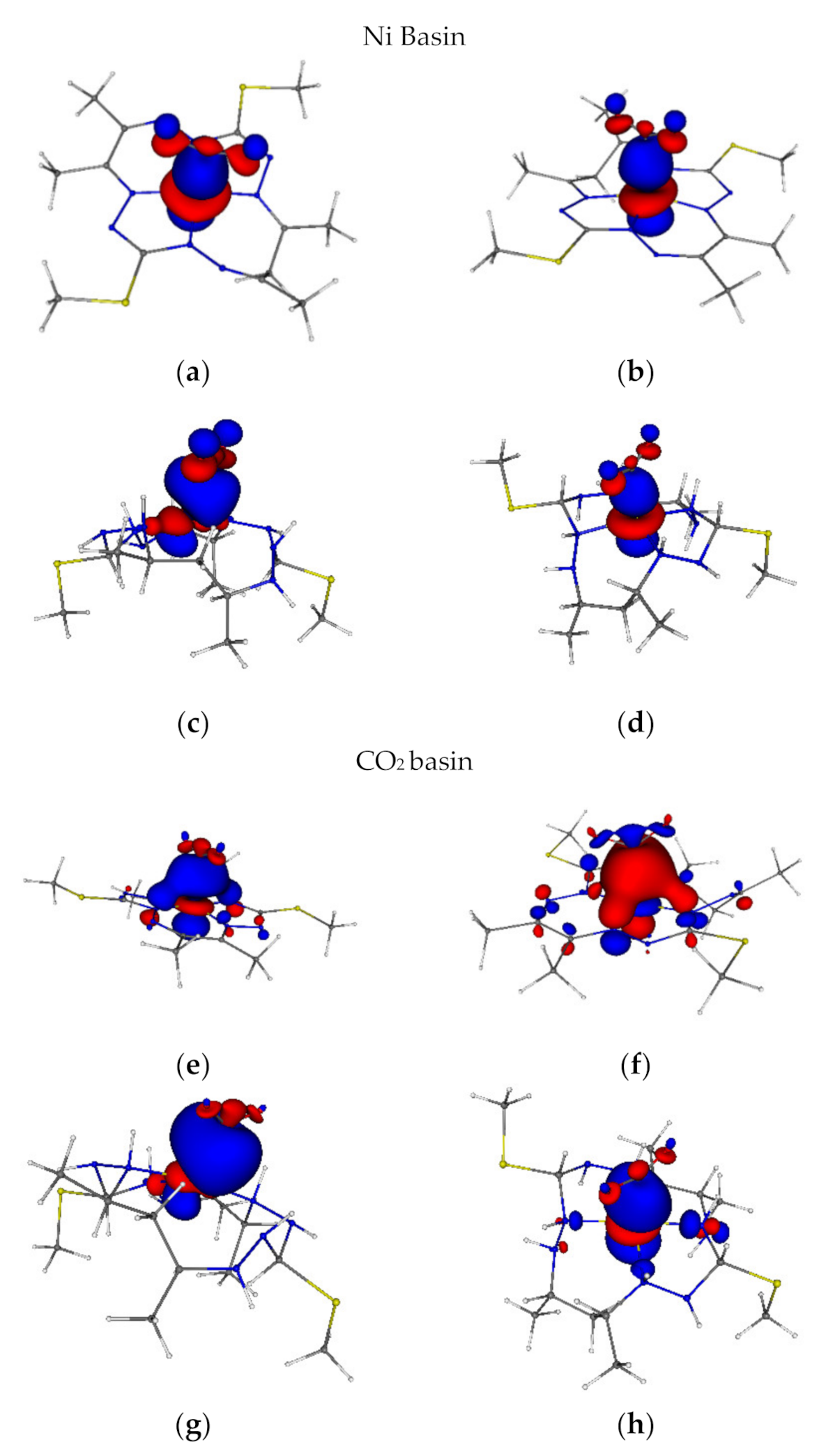
2.6. Localized Orbitals, Frontier Orbitals and DAFH Analysis
3. Methods-Computational Details
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.; Muckerman, J.T.; Fujita, E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins: Synergystic Effect of Weak Brönsted Acids. J. Am. Chem. Soc. 1996, 118, 1769–1776. [Google Scholar] [CrossRef]
- Wang, J.-W.; Liu, W.-J.; Zhong, D.-C.; Lu, T.-B. Nickel complexes as molecular catalysts for water splitting and CO2 reduction. Coord. Chem. Rev. 2019, 378, 237–261. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef] [Green Version]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabéhère-Mallart, E.; Robert, M. Molecular catalysis of CO2 reduction: Recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO-. Organometallics 1987, 6, 181–186. [Google Scholar] [CrossRef]
- Bruce, M.R.M.; Megehee, E.; Sullivan, B.P.; Thorp, H.; O’Toole, T.R.; Downard, A.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by associative activation. Organometallics 1988, 7, 238–240. [Google Scholar] [CrossRef]
- Bolinger, C.M.; Story, N.; Sullivan, B.P.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by 2,2’-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27, 4582–4587. [Google Scholar] [CrossRef]
- Slater, S.; Wagenknecht, J.H. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 1984, 106, 5367–5368. [Google Scholar] [CrossRef]
- Raebiger, J.W.; Turner, J.W.; Noll, B.C.; Curtis, C.J.; Miedaner, A.; Cox, B.; DuBois, D.L. Electrochemical Reduction of CO2 to CO Catalyzed by a Bimetallic Palladium Complex. Organometallics 2006, 25, 3345–3351. [Google Scholar] [CrossRef]
- DuBois, D.L.; Miedaner, A.; Haltiwanger, R.C. Electrochemical reduction of carbon dioxide catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: Synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113, 8753–8764. [Google Scholar] [CrossRef]
- Dubois, D.L. Development of Transition Metal Phosphine Complexes as Electrocatalysts for CO2 and CO Reduction. Comments Inorg. Chem. 1997, 19, 307–325. [Google Scholar] [CrossRef]
- Meshitsuka, S.; Ichikawa, M.; Tamaru, K. Electrocatalysis by metal phthalocyanines in the reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1974, 158–159. [Google Scholar] [CrossRef]
- Fisher, B.J.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361–7363. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.P.; Ruppert, R.; Sauvage, J.P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. [Google Scholar] [CrossRef] [PubMed]
- Balazs, G.B.; Anson, F.C. Effects of CO on the electrocatalytic activity of Ni (cyclam)2+ toward the reduction of CO2. J. Electroanal. Chem. 1993, 361, 149–157. [Google Scholar] [CrossRef]
- Hammouche, M.; Lexa, D.; Momenteau, M.; Saveant, J.M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113, 8455–8466. [Google Scholar] [CrossRef]
- Grodkowski, J.; Neta, P.; Fujita, E.; Mahammed, A.; Simkhovich, L.; Gross, Z. Reduction of Cobalt and Iron Corroles and Catalyzed Reduction of CO2. J. Phys. Chem. A 2002, 106, 4772–4778. [Google Scholar] [CrossRef]
- DeLaet, D.L.; Del Rosario, R.; Fanwick, P.E.; Kubiak, C.P. Carbon dioxide chemistry and electrochemistry of a binuclear cradle complex of nickel(0), Ni2(.mu.-CNMe)(CNMe)2(PPh2CH2PPh2)2. J. Am. Chem. Soc. 1987, 109, 754–758. [Google Scholar] [CrossRef]
- Simón-Manso, E.; Kubiak, C.P. Dinuclear Nickel Complexes as Catalysts for Electrochemical Reduction of Carbon Dioxide. Organometallics 2005, 24, 96–102. [Google Scholar] [CrossRef]
- Wittrig, R.E.; Ferrence, G.M.; Washington, J.; Kubiak, C.P. Infrared spectroelectrochemical and electrochemical kinetics studies of the reaction of nickel cluster radicals [Ni3(μ2-dppm)3(μ3-L) (μ3I)]•(L = CNR, R = CH3, i-C3H7, C6H11, CH2C6H5, t-C4H9, 2,6-Me2C6H3; L = CO) with carbon dioxide. Inorg. Chim. Acta 1998, 270, 111–117. [Google Scholar] [CrossRef]
- Ferrence, G.M.; Fanwick, P.E.; Kubiak, C.P. A telluride capped trinuclear nickel cluster [Ni3(µ3-Te)2(µ-PPh2CH2PPh2)3]+ with four accessible redox states (n = −1, 0, 1, 2). Chem. Commun. 1996, 1575–1576. [Google Scholar] [CrossRef]
- Haines, R.J.; Wittrig, R.E.; Kubiak, C.P. Electrocatalytic Reduction of Carbon Dioxide by the Binuclear Copper Complex [Cu2(6-(diphenylphosphino-2,2’-bipyridyl)2(MeCN)2][PF6]2. Inorg. Chem. 1994, 33, 4723–4728. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Ferrence, G.M.; Washington, J.; Henderson, J.I.; Rosenhein, L.; Heise, J.D.; Fanwick, P.E.; Kubiak, C.P. A Class of Halide-Supported Trinuclear Nickel Clusters [Ni3(μ3-L)(μ3-X)(μ2-dppm)3]n+ (L = I-, Br-, CO, CNR.; X = I-, Br-; n = 0, 1; dppm = Ph2PCH2PPh2): Novel Physical Properties and the Fermi Resonance of Symmetric μ3-η1 Bound Isocyanide Ligands. J. Am. Chem. Soc. 1996, 118, 2198–2207. [Google Scholar] [CrossRef]
- Dražić, B.; Antonijević-Nikolić, M.; Žižak, Ž.; Tanasković, S. Synthesis and characterization of copper(II) octaazamacrocyclic complexes with glycine derivatives. In vitro antiproliferative and antimicrobial evaluation of the Cu(II) and Co(II) analogous. J. Serb. Chem. Soc. 2020, 85, 637–649. [Google Scholar] [CrossRef]
- Antonijević-Nikolić, M.; Antić-Stanković, J.; Dražić, B.; Tanasković, S. New macrocyclic Cu(II) complex with bridge terephthalate: Synthesis, spectral properties, in vitro cytotoxic and antimicrobial activity. Comparison with related complexes. J. Mol. Struct. 2019, 1184, 41–48. [Google Scholar] [CrossRef]
- Miodragović, Z.M.; Gordana, V.; Leovac, V.M. Tetranuclear Cu(II) complex with octaazamacrocycle and bridging bicyclic dicarboxylato ligands. J. Serb. Chem. Soc. 2001, 66, 597–603. [Google Scholar] [CrossRef]
- Tanasković, S.; Antonijević-Nikolić, M.; Hollo, B.B.; Dražić, B.; Stanojković, T.; Meszaros-Szecsenyi, K.; Vucković, G. Correlations between the in vitro antiproliferative activity, structure and thermal stability of some macrocyclic dinuclear Cu(II) complexes. J. Serb. Chem. Soc. 2014, 79, 1235–1247. [Google Scholar] [CrossRef]
- Antonijević-Nikolić, M.; Antić-Stanković, J.; Tanasković, S.B.; Korabik, M.J.; Gojgić-Cvijović, G.; Vučković, G. Preparation, characterisation and study of in vitro biologically active azamacrocyclic Cu(II) dicarboxylate complexes. J. Mol. Struct. 2013, 1054–1055, 297–306. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Collin, J.P.; Jouaiti, A.; Sauvage, J.P. Electrocatalytic properties of (tetraazacyclotetradecane)nickel(2+) and Ni2(biscyclam)4+ with respect to carbon dioxide and water reduction. Inorg. Chem. 1988, 27, 1986–1990. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Miskelly, G.M.; Lewis, N.S. Effects of redox potential, steric configuration, solvent, and alkali metal cations on the binding of carbon dioxide to cobalt(I) and nickel(I) macrocycles. J. Am. Chem. Soc. 1990, 112, 3420–3426. [Google Scholar] [CrossRef]
- Fujita, E.; Creutz, C.; Sutin, N.; Szalda, D.J. Carbon dioxide activation by cobalt(I) macrocycles: Factors affecting carbon dioxide and carbon monoxide binding. J. Am. Chem. Soc. 1991, 113, 343–353. [Google Scholar] [CrossRef]
- Rudolph, M.; Dautz, S.; Jäger, E.-G. Macrocyclic [N42-] Coordinated Nickel Complexes as Catalysts for the Formation of Oxalate by Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2000, 122, 10821–10830. [Google Scholar] [CrossRef]
- Fujita, E.; Szalda, D.J.; Creutz, C.; Sutin, N. Carbon dioxide activation: Thermodynamics of carbon dioxide binding and the involvement of two cobalt centers in the reduction of carbon dioxide by a cobalt(I) macrocycle. J. Am. Chem. Soc. 1988, 110, 4870–4871. [Google Scholar] [CrossRef]
- Ogata, T.; Yanagida, S.; Brunschwig, B.S.; Fujita, E. Mechanistic and Kinetic Studies of Cobalt Macrocycles in a Photochemical CO2 Reduction System: Evidence of Co- CO2 Adducts as Intermediates. J. Am. Chem. Soc. 1995, 117, 6708–6716. [Google Scholar] [CrossRef]
- Creutz, C.; Schwarz, H.A.; Wishart, J.F.; Fujita, E.; Sutin, N. Thermodynamics and kinetics of carbon dioxide binding to two stereoisomers of a cobalt(I) macrocycle in aqueous solution. J. Am. Chem. Soc. 1991, 113, 3361–3371. [Google Scholar] [CrossRef]
- Fujita, E.; Creutz, C.; Sutin, N.; Brunschwig, B.S. Carbon dioxide activation by cobalt macrocycles: Evidence of hydrogen bonding between bound CO2 and the macrocycle in solution. Inorg. Chem. 1993, 32, 2657–2662. [Google Scholar] [CrossRef]
- Fujita, E.; Furenlid, L.R.; Renner, M.W. Direct XANES Evidence for Charge Transfer in Co−CO2 Complexes. J. Am. Chem. Soc. 1997, 119, 4549–4550. [Google Scholar] [CrossRef]
- Fujita, E.; van Eldik, R. Effect of Pressure on the Reversible Binding of Acetonitrile to the “Co(I)−CO2” Adduct To Form Cobalt(III) Carboxylate. Inorg. Chem. 1998, 37, 360–362. [Google Scholar] [CrossRef]
- Kelly, C.A.; Mulazzani, Q.G.; Blinn, E.L.; Rodgers, M.A.J. Kinetics of CO Addition to Ni(cyclam)+ in Aqueous Solution. Inorg. Chem. 1996, 35, 5122–5126. [Google Scholar] [CrossRef]
- Kelly, C.A.; Blinn, E.L.; Camaioni, N.; D’Angelantonio, M.; Mulazzani, Q.G. Mechanism of CO2 and H+ Reduction by Ni(cyclam)+ in Aqueous Solution. A Pulse and Continuous Radiolysis Study. Inorg. Chem. 1999, 38, 1579–1584. [Google Scholar] [CrossRef]
- Fujita, E.; Haff, J.; Sanzenbacher, R.; Elias, H. High Electrocatalytic Activity of RRSS-[NiIIHTIM](ClO4)2 and [NiIIDMC](ClO4)2 for Carbon Dioxide Reduction (HTIM = 2,3,9,10-Tetramethyl-1,4,8,11-tetraazacyclotetradecane, DMC = C-meso-5,12-Dimethyl-1,4,8,11-tetraazacyclotetradecane). Inorg. Chem. 1994, 33, 4627–4628. [Google Scholar] [CrossRef]
- Gagne, R.R.; Ingle, D.M. One-electron-reduced nickel(II)-macrocyclic ligand complexes. Four-coordinate nickel(I) species and nickel(II)-ligand radical species which form paramagnetic, five-coordinate nickel(I) adducts. Inorg. Chem. 1981, 20, 420–425. [Google Scholar] [CrossRef]
- Furenlid, L.R.; Renner, M.W.; Szalda, D.J.; Fujita, E. EXAFS studies of nickel(II), nickel(I), and Ni(I)-CO tetraazamacrocycles and the crystal structure of (5,7,7,12,14,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-4,11-diene)nickel(I) perchlorate. J. Am. Chem. Soc. 1991, 113, 883–892. [Google Scholar] [CrossRef]
- Froehlich, J.D.; Kubiak, C.P. Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg. Chem. 2012, 51, 3932–3934. [Google Scholar] [CrossRef]
- Sakaki, S. An ab initio MO/SD-CI study of model complexes of intermediates in electrochemical reduction of carbon dioxide catalyzed by NiCl2(cyclam). J. Am. Chem. Soc. 1992, 114, 2055–2062. [Google Scholar] [CrossRef]
- Sakaki, S. Can carbon dioxide coordinate to a nickel(I) complex? An ab initio MO/SD-CI study. J. Am. Chem. Soc. 1990, 112, 7813–7814. [Google Scholar] [CrossRef]
- Dobrov, A.; Darvasiová, D.; Zalibera, M.; Bučinský, L.; Puškárová, I.; Rapta, P.; Shova, S.; Dumitrescu, D.; Martins, L.M.D.R.S.; Pombeiro, A.J.L.; et al. Nickel(II) Complexes with Redox Noninnocent Octaazamacrocycles as Catalysts in Oxidation Reactions. Inorg. Chem. 2019, 58, 11133–11145. [Google Scholar] [CrossRef] [PubMed]
- Darvasiová, D.; Šoral, M.; Puškárová, I.; Dvoranová, D.; Vénosová, B.; Bučinský, L.; Zalibera, M.; Dujnič, V.; Dobrov, A.; Schwalbe, M.; et al. Spectroelectrochemical, photochemical and theoretical study of octaazamacrocyclic nickel(II) complexes exhibiting unusual solvent-dependent deprotonation of methylene group. Electrochim. Acta 2019, 326. [Google Scholar] [CrossRef]
- Kimura, E.; Wada, S.; Shionoya, M.; Okazaki, Y. New Series of Multifunctionalized Nickel(II)-Cyclam (Cyclam = 1,4,8,11-Tetraazacyclotetradecane) Complexes. Application to the Photoreduction of Carbon Dioxide. Inorg. Chem. 1994, 33, 770–778. [Google Scholar] [CrossRef]
- Song, J.; Klein, E.L.; Neese, F.; Ye, S. The Mechanism of Homogeneous CO2 Reduction by Ni(cyclam): Product Selectivity, Concerted Proton–Electron Transfer and C–O Bond Cleavage. Inorg. Chem. 2014, 53, 7500–7507. [Google Scholar] [CrossRef]
- Wu, Y.; Rudshteyn, B.; Zhanaidarova, A.; Froehlich, J.D.; Ding, W.; Kubiak, C.P.; Batista, V.S. Electrode-Ligand Interactions Dramatically Enhance CO2 Conversion to CO by the [Ni(cyclam)](PF6)2 Catalyst. ACS Catal. 2017, 7, 5282–5288. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle- Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density Functional Thermochemistry III The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XX. Basis Set for Correlated Wave-Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Baranov, A.I.; Ponec, R.; Kohout, M. Domain-averaged Fermi-hole analysis for solids. J. Chem. Phys. 2012, 137, 214109. [Google Scholar] [CrossRef]
- Ponec, R.; Duben, A.J. Electron pairing and chemical bonds: Bonding in hypervalent molecules from analysis of Fermi holes. J. Comput. Chem. 1999, 20, 760–771. [Google Scholar] [CrossRef]
- Cooper, D.L.; Ponec, R. Bond formation in diatomic transition metal hydrides: Insights from the analysis of domain-averaged fermi holes. Int. J. Quant. Chem. 2013, 113, 102–111. [Google Scholar] [CrossRef]
- Kohout, M. DGrid; Version 5.1; Max-Plack-Institut für Chemische Physik fester Stoffe: Dresden, Germany, 2019. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990; ISBN 0198558651. [Google Scholar]
- Keith, T.A. AIMAll; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019; Available online: http://aim.tkgristmill.com (accessed on 3 June 2021).
- Varetto, U. Molekel, Version 5.4. Available online: https://ugovaretto.github.io/molekel/wiki/pmwiki.php/Main/DownloadBinary.html (accessed on 3 June 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆ER [kJ mol−1] | ∆EI [kJ mol−1] | ∆EICPC [kJ mol−1] | ∆HR [kJ mol−1] | d(Ni-CCO2) [Å] | |
|---|---|---|---|---|---|
| 3[NiL]2− | −27.16 | −210.59 | −159.49 | −88.13 | 2.051 |
| 1[NiL]2− | −7.70 | −1.28 | 3.689 | ||
| 2[NiL]− | −5.27 | −1.55 | 3.511 | ||
| 1[NiL]0 | −4.37 | −11.08 | 1.21 | −0.33 | 3.576 |
| 2[NiL]+ | −2.68 | −1.11 | 3.603 | ||
| 1[NiL]2+ | −1.86 | 2.83 | 3.629 | ||
| 1[NiLH]0 | −160.655 | −451.90 | −343.55 | −150.26 | 1.887 |
| 2[NiLH]+ | −7.906 | −106.86 | −66.32 | −3.24 | 2.079 |
| Ni | C | O * | ||||
|---|---|---|---|---|---|---|
| MPA | QTAIM | MPA | QTAIM | MPA | QTAIM | |
| 3[NiL]2− | 0.73 | 0.74 | - | - | - | - |
| 1[NiL]2− | 0.89 | 0.89 | - | - | - | - |
| 2[NiL]- | 0.96 | 0.94 | - | - | - | - |
| 1[NiL]0 | 1.00 | 0.96 | - | - | - | - |
| 2[NiL]+ | 1.05 | 0.99 | - | - | - | - |
| 1[NiL]2+ | 1.10 | 1.03 | - | - | - | - |
| 3[NiL-CO2]2− | 1.06 | 0.94 | 0.11 | 1.66 | −0.41 | −1.16 |
| 1[NiLfrCO2]0 | 1.10 | 1.00 | 0.21 | 1.81 | −0.32 | −1.10 |
| 1[NiL-CO2]0 | 1.00 | 0.96 | 0.48 | 2.15 | −0.25 | −1.08 |
| 1[NiLH]0 | 0.04 | 0.15 | - | - | - | - |
| 2[NiLH]+ | 0.76 | 0.66 | - | - | - | - |
| 1[NiLH]2+ | 1.05 | 0.94 | - | - | - | - |
| 1[NiLH-CO2]0 | 0.84 | 0.68 | 0.03 | 1.44 | −0.48 | −1.21 |
| 2[NiLH-CO2]+ | 1.15 | 0.89 | 0.19 | −0.10 | −0.33 | −0.08 |
| CO2 | - | - | 0.53 | 2.15 | −0.26 | −1.08 |
| Formal ox. State of Ni | dxz | dyz | dxy | sσ | dtotal | |||
|---|---|---|---|---|---|---|---|---|
| 3[NiL]2− | I | 1.745 | 1.878 | 1.830 | 1.146 | 1.886 | 0.693 | 8.487 |
| (0.043) | (0.009) | (0.032) | (0.768) | (0.006) | (−0.055) | (0.858) | ||
| 1[NiL]2− | II | 1.797 | 1.925 | 1.903 | 0.846 | 1.910 | 0.684 | 8.381 |
| 2[NiL]- | II | 1.820 | 1.924 | 1.908 | 0.796 | 1.912 | 0.639 | 8.360 |
| 1[NiL]0 | II | 1.835 | 1.920 | 1.906 | 0.791 | 1.911 | 0.599 | 8.363 |
| 2[NiL]+ | II | 1.851 | 1.903 | 1.908 | 0.790 | 1.913 | 0.562 | 8.365 |
| 1[NiL]2+ | II | 1.868 | 1.893 | 1.886 | 0.803 | 1.915 | 0.527 | 8.365 |
| 3[NiL-CO2]2− | II | 1.420 | 1.918 | 1.915 | 1.205 | 1.911 | 0.553 | 8.343 |
| (0.273) | (0.009) | (0.012) | (0.774) | (0.009) | (−0.012) | (1.077) | ||
| 1[NiLfrCO2]0 | II | 1.562 | 1.922 | 1.938 | 0.951 | 1.923 | 0.490 | 8.296 |
| 1[NiL-CO2]0 | II | 1.833 | 1.919 | 1.907 | 0.794 | 1.911 | 0.602 | 8.364 |
| 1[NiLH]0 | 0 | 1.649 | 1.861 | 1.890 | 1.527 | 1.829 | 1.135 | 8.756 |
| 2[NiLH]+ | I | 1.736 | 1.919 | 1.908 | 1.123 | 1.897 | 0.662 | 8.583 |
| 1[NiLH]2+ | II | 1.836 | 1.941 | 1.933 | 0.762 | 1.928 | 0.588 | 8.400 |
| 1[NiLH-CO2]0 | I | 1.329 | 1.892 | 1.870 | 1.636 | 1.890 | 0.577 | 8.617 |
| 2[NiLH-CO2]+ | II | 1.504 | 1.928 | 1.929 | 1.165 | 1.923 | 0.426 | 8.448 |
| AO Populations | Total | |||||
|---|---|---|---|---|---|---|
| s(Ni) | s(C) | pz(C) | Ni | C | ||
| 1[NiLfrCO2]0 | 0.18 | 1.38 | 0.17 | 0.27 | 1.56 | 0.44 |
| 3[NiL-CO2]2− α | 0.08 | 0.74 | 0.04 | 0.07 | 0.82 | 0.11 |
| 3[NiL-CO2]2− β | 0.12 | 0.36 | 0.32 | 0.30 | 0.48 | 0.62 |
| 2[NiLH-CO2]+ α | 0.09 | 0.81 | 0.02 | 0.05 | 0.93 | 0.07 |
| 2[Ni LH-CO2]+ β | 0.14 | 0.53 | 0.17 | 0.23 | 0.66 | 0.40 |
| 1[Ni LH-CO2]0 | 0.18 | 0.62 | 0.62 | 0.52 | 0.80 | 1.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vénosová, B.; Jelemenská, I.; Kožíšek, J.; Rapta, P.; Zalibera, M.; Novotný, M.; Arion, V.B.; Bučinský, L. Ni Oxidation State and Ligand Saturation Impact on the Capability of Octaazamacrocyclic Complexes to Bind and Reduce CO2. Molecules 2021, 26, 4139. https://doi.org/10.3390/molecules26144139
Vénosová B, Jelemenská I, Kožíšek J, Rapta P, Zalibera M, Novotný M, Arion VB, Bučinský L. Ni Oxidation State and Ligand Saturation Impact on the Capability of Octaazamacrocyclic Complexes to Bind and Reduce CO2. Molecules. 2021; 26(14):4139. https://doi.org/10.3390/molecules26144139
Chicago/Turabian StyleVénosová, Barbora, Ingrid Jelemenská, Jozef Kožíšek, Peter Rapta, Michal Zalibera, Michal Novotný, Vladimir B. Arion, and Lukáš Bučinský. 2021. "Ni Oxidation State and Ligand Saturation Impact on the Capability of Octaazamacrocyclic Complexes to Bind and Reduce CO2" Molecules 26, no. 14: 4139. https://doi.org/10.3390/molecules26144139
APA StyleVénosová, B., Jelemenská, I., Kožíšek, J., Rapta, P., Zalibera, M., Novotný, M., Arion, V. B., & Bučinský, L. (2021). Ni Oxidation State and Ligand Saturation Impact on the Capability of Octaazamacrocyclic Complexes to Bind and Reduce CO2. Molecules, 26(14), 4139. https://doi.org/10.3390/molecules26144139