
Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry


Abstract

1. Introduction
2. Computational Methods
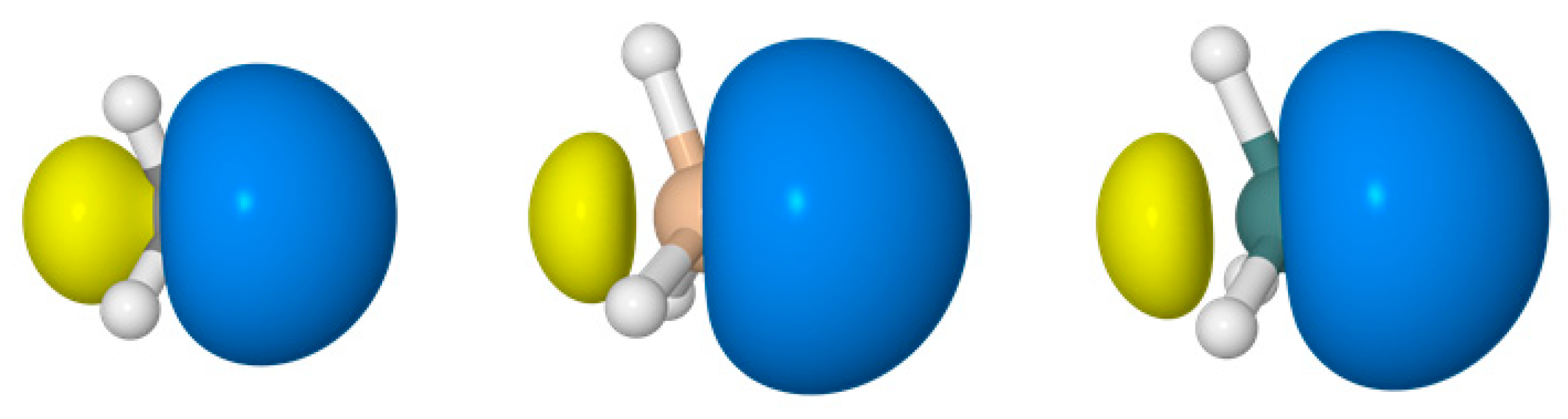
3. Directional Hybridization
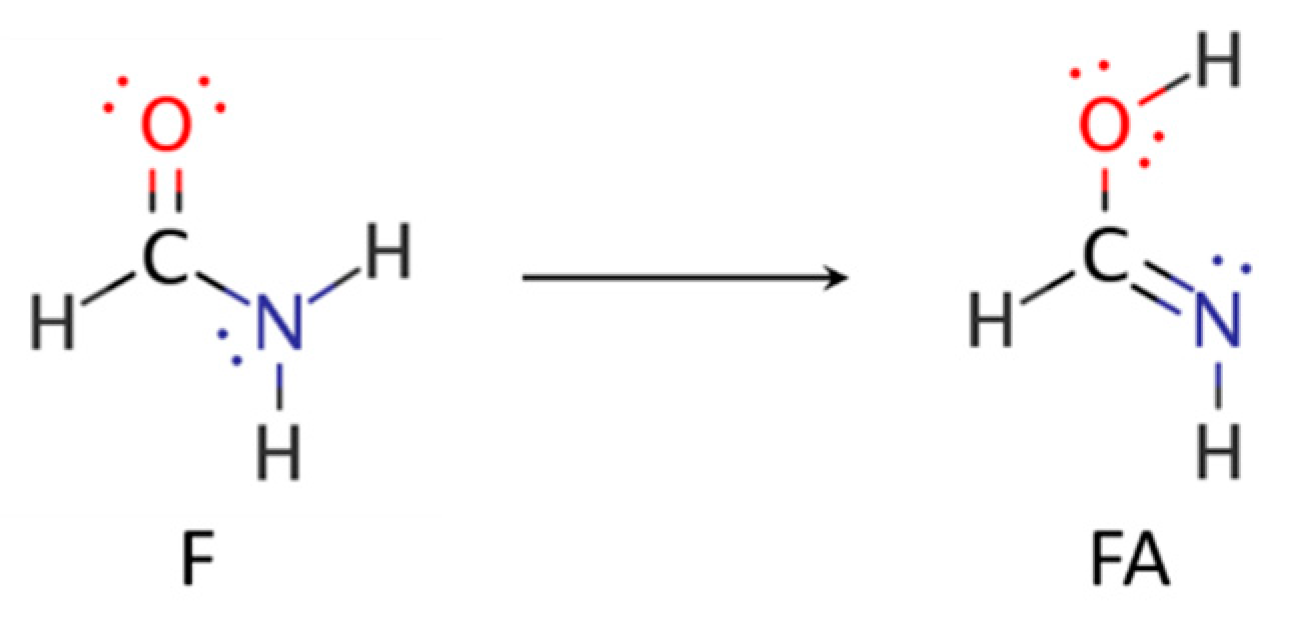
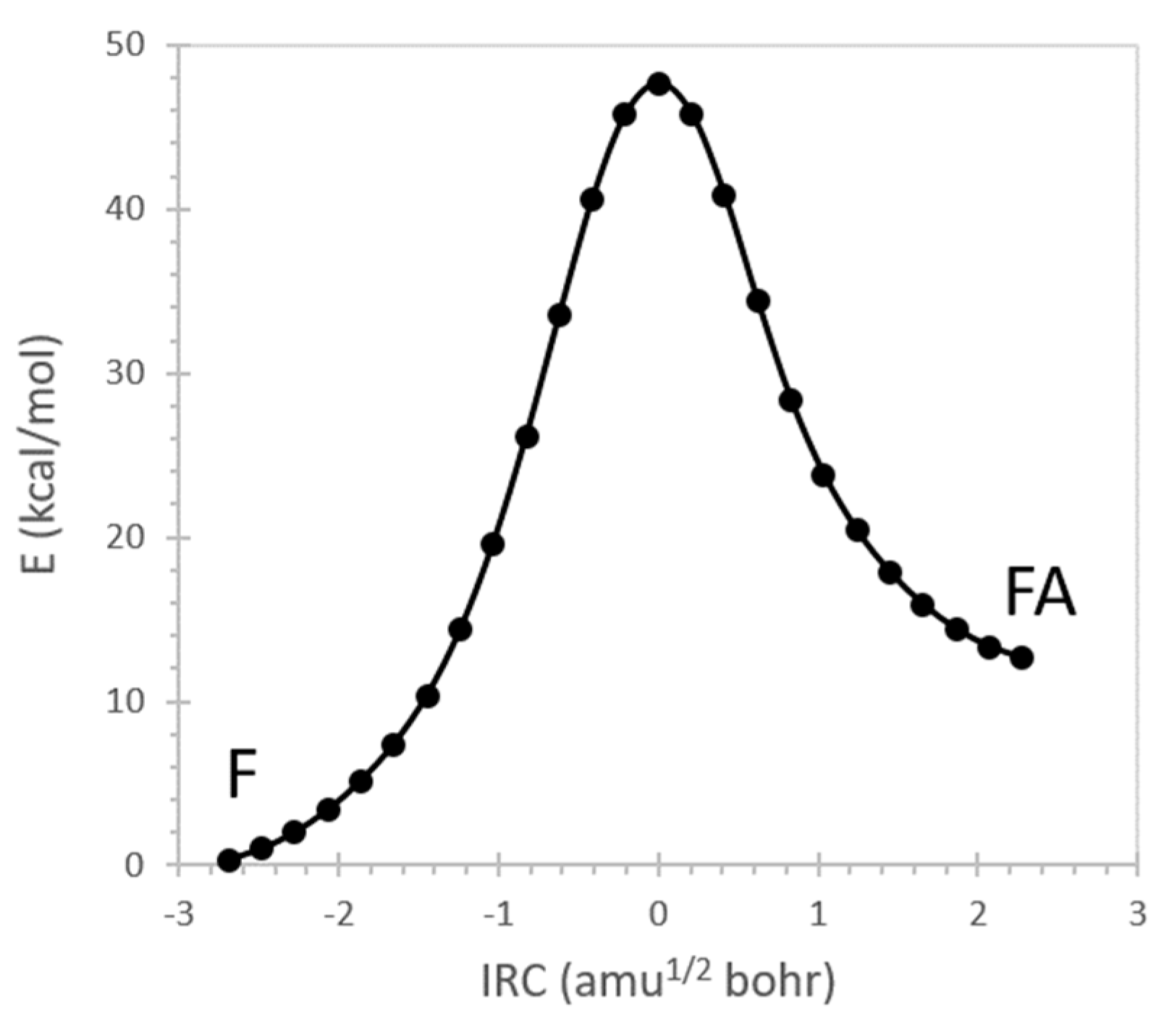
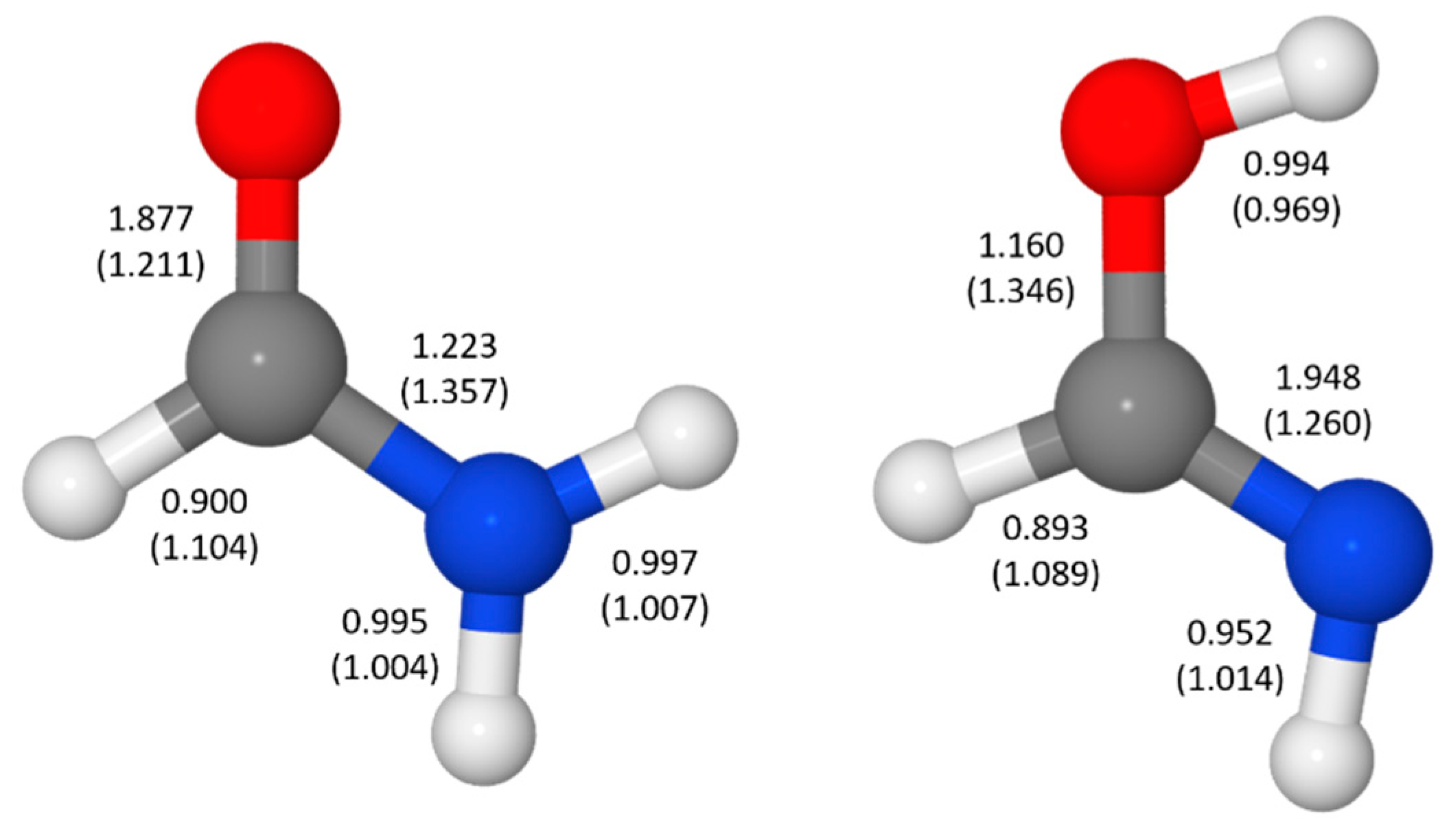
4. Resonance Delocalization
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Seiler, P.; Weisman, G.R.; Glendening, E.D.; Weinhold, F.; Johnson, V.B.; Dunitz, J.D. Observation of an Eclipsed C(sp3)-CH3 Bond in a Tricyclic Orthoamide: Experimental and Theoretical Evidence for C-H⋯O Hydrogen Bonds. Angew. Chem. Int. Ed. 1987, 26, 1175–1177. [Google Scholar] [CrossRef]
- Glendening, E.D.; Faust, R.; Streitwieser, A.; Vollhardt, K.P.C.; Weinhold, F. On the Role of Delocalization in Benzene. J. Am. Chem. Soc. 1993, 115, 10952–10957. [Google Scholar] [CrossRef][Green Version]
- Suidan, L.; Badenhoop, J.K.; Glendening, E.D.; Weinhold, F. Common Textbook and Teaching Misrepresentations of Lewis Structures. J. Chem. Educ. 1995, 72, 583–586. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory. I. General Formulation. J. Comput. Chem. 1998, 19, 593–609. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory. II. Natural Bond Order and Valency. J. Comput. Chem. 1998, 19, 610–627. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Weinhold, F. Natural Resonance Theory. III. Chemical Applications. J. Comput. Chem. 1998, 19, 628–646. [Google Scholar] [CrossRef]
- Pauling, L.; Wilson, E.B., Jr. Introduction to Quantum Mechanics; McGraw-Hill: New York, NY, USA, 1935. [Google Scholar]
- Pauling, L. Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöpolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- Pauling, L. The Application of the Quantum Mechanics to the Structure of the Hydrogen Molecule and Hydrogen Molecule-Ion and to Related Problems. Chem. Rev. 1928, 5, 173–213. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Slater, J.C. Directed valence in polyatomic molecules. Phys. Rev. 1931, 37, 481–497. [Google Scholar] [CrossRef]
- Pauling, L. The theory of resonance in chemistry. Proc. R. Soc. Lond. Ser. A 1977, 356, 433–441. [Google Scholar]
- Todd, A.R.; Cornforth, J.W. Robert Robinson, 13 September 1886–8 February 1975. Biogr. Mem. Fellows R. Soc. Lond. 1976, 22, 415–527. [Google Scholar]
- Hund, F. Zur Deutung einiger Erscheinungen in den Molekelspektre. Z. Physik 1926, 36, 657–674. [Google Scholar] [CrossRef]
- Mulliken, R.S. The Assignment of Quantum Numbers for Electrons in Molecules. I. Phys. Rev. 1928, 32, 186–222. [Google Scholar] [CrossRef]
- Lennard-Jones, J.E. The Electronic Structure of Some Diatomic Molecules Trans. Faraday Soc. 1929, 25, 668–686. [Google Scholar] [CrossRef]
- Hartree, D.R. The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part I. Theory and Method. Proc. Camb. Philos. Soc. 1928, 24, 89–110. [Google Scholar] [CrossRef]
- Fock, V. Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems. Z. Phys. 1930, 61, 126–148. [Google Scholar] [CrossRef]
- Slater, J.C. Note on Hartree’s Method. Phys. Rev. 1930, 35, 210–211. [Google Scholar] [CrossRef]
- Coulson, C.A. Self-consistent field for molecular hydrogen. Proc. Camb. Philos. Soc. 1938, 34, 204–212. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Mulliken, R.S. Spectroscopy, molecular orbitals, and chemical bonding (Nobel Prize, 1966). Science 1967, 157, 13–24. [Google Scholar] [CrossRef]
- Norbeck, J.M.; Gallup, G.A. Valence-bond calculation of the electronic structure of benzene. J. Am. Chem. Soc. 1974, 96, 3386–3393. [Google Scholar] [CrossRef]
- Goddard, W.A., III. Improved Quantum Theory of Many-Electron Systems. I. Construction of Eigenfunctions of S2 Which Satisfy Pauli’s Principle. Phys. Rev. 1967, 157, 73–78. [Google Scholar] [CrossRef]
- Goddard, W.A., III; Dunning, T.H., Jr.; Hunt, W.J.; Hay, P.J. Generalized valence bond description of bonding in low-lying states of molecules. Acc. Chem. Res. 1973, 6, 368–376. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Beyond Molecular Orbital Theory: The Impact of Generalized Valence Bond Theory in Molecular Science. In Computational Materials, Chemistry, and Biochemistry: From Bold Initiatives to the Last Mile; Shankar, S., Muller, R., Dunning, T., Chen, G.H., Eds.; Springer: Cham, Switzerland, 2021; Volume 284, pp. 55–87. [Google Scholar]
- Foresman, J.B.; Frisch, A.E. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian, Inc.: Wallingford, CT, USA, 2015. [Google Scholar]
- van’t Hoff, J.H. Sur les formules de structure dans l’espace. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- LeBel, J.-A. Sur Les Relations Qui Existent Entre Les Formules Atomiques Des Corps Organiques et Le Pouvoir Rotatoire de Leurs Dissolutions. Bull. Soc. Chim. Paris 1874, 22, 337–347. [Google Scholar]
- Ramberg, P.J. Chemical Structure, Spatial Arrangement: The Early History of Stereochemistry, 1874–1914; Routledge: Abingdon, UK, 2017. [Google Scholar]
- Bragg, W.H.; Bragg, W.L. The structure of the diamond. Nature 1913, 91, 557. [Google Scholar] [CrossRef]
- Wierl, R. Elektronenbeugung und Molekülbau. Ann. Phys. 1931, 8, 521–564. [Google Scholar] [CrossRef]
- Jensen, W.P.; Palenik, G.J.; Suh, I.-H. The history of molecular structure determination viewed through the Nobel Prizes. J. Chem. Educ. 2003, 80, 753–761. [Google Scholar]
- Coulson, C.A. Valence, 2nd ed.; Oxford University Press: Oxford, UK, 1965. [Google Scholar]
- Kermack, W.O.; Robinson, R.R. An explanation of the property of induced polarity of atoms and an interpretion of the theory of partial valencies on an electronic basis. J. Chem. Soc. Trans. 1922, 121, 427–440. [Google Scholar] [CrossRef]
- Lowry, T.M. Studies of electrovalency. Part I. The polarity of double bonds. J. Chem. Soc. Trans. 1923, 123, 822–831. [Google Scholar] [CrossRef]
- Allan, J.; Oxford, A.F.; Robinson, R.; Smith, J.C. The relative directive powers of groups of the forms RO and RR’N in aromatic substitution. Part III. The nitration of some p-alkoxyanisoles. J. Chem. Soc. 1926, 129, 401–411. [Google Scholar] [CrossRef]
- Ingold, C.K.; Ingold, E.H. The nature of the alternating effect in carbon chains. Part V. A discussion of aromatic substitution with special reference to the respective roles of polar and non-polar dissociation; and a further study of the relative directive efficiencies of oxygen and nitrogen. J. Chem. Soc. 1926, 129, 1310–1328. [Google Scholar]
- Ingold, C.K. Mesomerism and tautomerism. Nature 1934, 133, 946–947. [Google Scholar] [CrossRef]
- Ingold, C.K. Principles of an electronic theory of organic reactions. Chem. Rev. 1934, 15, 225–274. [Google Scholar] [CrossRef]
- Saltzman, M.D. CK Ingold’s development of the concept of mesomerism. Bull. Hist. Chem. 1996, 19, 25–32. [Google Scholar]
- Kerber, R.C. If it’s resonance, what is resonating? J. Chem. Educ. 2006, 83, 223–227. [Google Scholar] [CrossRef]
- Coulson, C.A.; Fischer, I. Notes on the Molecular Orbital Treatment of the Hydrogen Molecule. Phil. Mag. 1949, 40, 386–393. [Google Scholar] [CrossRef]
- Grushow, A. Is it time to retire the hybrid orbital? J. Chem. Educ. 2011, 88, 860–862. [Google Scholar] [CrossRef]
- Tro, N.J. Retire the Hybrid Atomic Orbital? Not So Fast. J. Chem. Educ. 2012, 89, 567–568. [Google Scholar] [CrossRef]
- DeKock, R.L.; Strikwerda, J.R. Retire Hybrid Atomic Orbitals? J. Chem. Educ. 2012, 89, 569. [Google Scholar] [CrossRef]
- Landis, C.R.; Weinhold, F. Comments on ‘Is It Time To Retire the Hybrid Atomic Orbital?’. J. Chem. Educ. 2012, 89, 570–572. [Google Scholar] [CrossRef]
- Truhlar, D.G. Are Molecular Orbitals Delocalized? J. Chem. Educ. 2012, 89, 573–574. [Google Scholar] [CrossRef]
- Hiberty, P.C.; Volatron, F.; Shaik, S. In Defense of the Hybrid Atomic Orbitals. J. Chem. Educ. 2012, 89, 575–577. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Hiberty, P.C.; Shaik, S.; Gordon, M.S.; Danovich, D. Orbitals and the Interpretation of Photoelectron Spectroscopy and (e,2e) Ionization Experiments. Angew. Chem. Int. Ed. 2019, 58, 12332–12338. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.R. A Soviet Marxist view of structural chemistry: The theory of resonance controversy. Isis 1964, 55, 20–31. [Google Scholar] [CrossRef]
- The Soviet Resonance Controversy. Available online: https://paulingblog.wordpress.com/2009/03/26/paulings-theory-of-resonance-a-soviet-controversy/ (accessed on 28 May 2021).
- Frenking, G.; Krapp, A. Unicorns in the world of chemical bonding models. J. Comput. Chem. 2007, 28, 15–24. [Google Scholar] [CrossRef]
- Landis, C.R.; Hughes, R.P.; Weinhold, F. Bonding Analysis of TM(cAAC)2 (TM = Cu, Ag, Au) and the Importance of Reference State. Organometallics 2015, 34, 3442–3449. [Google Scholar] [CrossRef]
- Landis, C.R.; Hughes, R.P.; Weinhold, F. Comment on ‘Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr. or Ba) that mimic transition metals’. Science 2019, 365, 553. [Google Scholar] [CrossRef]
- Xu, L.T.; Thompson, J.V.K.; Dunning, T.H., Jr. Spin-coupled generalized valence bond description of group 14 species: The carbon, silicon and germanium hydrides, XHn (n = 1–4). J. Phys. Chem. A 2019, 123, 2401–2419. [Google Scholar] [CrossRef]
- Xu, L.T.; Dunning, T.H., Jr. Orbital hybridization in modern valence bond wave functions: Methane, ethylene, and acetylene. J. Phys. Chem. A 2020, 124, 204–214. [Google Scholar] [CrossRef]
- Foster, J.M.; Boys, S.F. Canonical configurational interaction procedure. Rev. Mod. Phys. 1960, 32, 300–302. [Google Scholar] [CrossRef]
- Edmiston, C.; Ruedenberg, K. Localized atomic and molecular orbitals. Rev. Mod. Phys. 1963, 35, 457–464. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- Khaliullin, R.Z.; Head-Gordon, M.; Bell, A.T. An efficient self-consistent field method for large systems of weakly interacting components. J. Chm. Phys. 2006, 124, 204105. [Google Scholar] [CrossRef]
- Jansík, B.; Høst, S.; Kristensen, K.; Jørgensen, P. Local orbitals by minimizing powers of the orbital variance. J. Chem. Phys. 2011, 134, 194104. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO Analysis and How is it Useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Theory: Discovering Chemistry with NBO7. In Complementary Bonding Analysis; Grabowsky, S., Ed.; De Gruyer: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Resonance Theory Reboot. J. Am. Chem. Soc. 2019, 141, 4156–4166. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef] [PubMed]
- NBO. Affiliated Electronic Structure Systems and Protected Interfaces. Available online: https://nbo7.chem.wisc.edu/affil_css.htm (accessed on 2 July 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian-16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Cooper, D.L.; Thorsteinsson, T.; Gerratt, J. Fully variational optimization of modern VB wave functions using the CASVB strategy. Int. J. Quant. Chem. 1997, 65, 439–451. [Google Scholar] [CrossRef]
- Cooper, D.L.; Thorsteinsson, T.; Gerratt, J. Modern VB representations of CASSCF wave functions and the fully-variational optimization of modern VB wave functions using the CASVB strategy. Adv. Quant. Chem. 1998, 32, 51–67. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, 2021.1, A Package of Ab Initio Programs; Technologie-Transfer-Initiative GmbH an der Universität Stuttgart: Stuttgart, Germany, 2021. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Bent, H.A. Distribution of atomic s character in molecules and its chemical implications. J. Chem. Educ. 1960, 37, 616–624. [Google Scholar] [CrossRef]
- Bent, H.A. An appraisal of valence-bond structures and hybridization in compounds of the first-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Bresch, S.; Manoharan, M. Hybridization trends for main group elements and the Bent’s rule beyond carbon: More than electronegativity. J. Phys. Chem. A 2014, 118, 3663–3677. [Google Scholar] [CrossRef]
- Eisenberg, D. The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 2003, 30, 11207–11210. [Google Scholar] [CrossRef]
- Hazra, M.K.; Chakraborty, T. Formamide tautomerization: Catalytic role of formic acid. J. Phys. Chem. A 2005, 109, 7621–7625. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. NBO Program Manual; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 1996; p. B-82ff. Available online: https://nbo7.chem.wisc.edu/nboman.pdf (accessed on 2 July 2021).
- Jiao, Y.; Weinhold, F. NBO/NRT two-state theory of bond-shift spectral excitation. Molecules 2020, 25, 4052. [Google Scholar] [CrossRef]
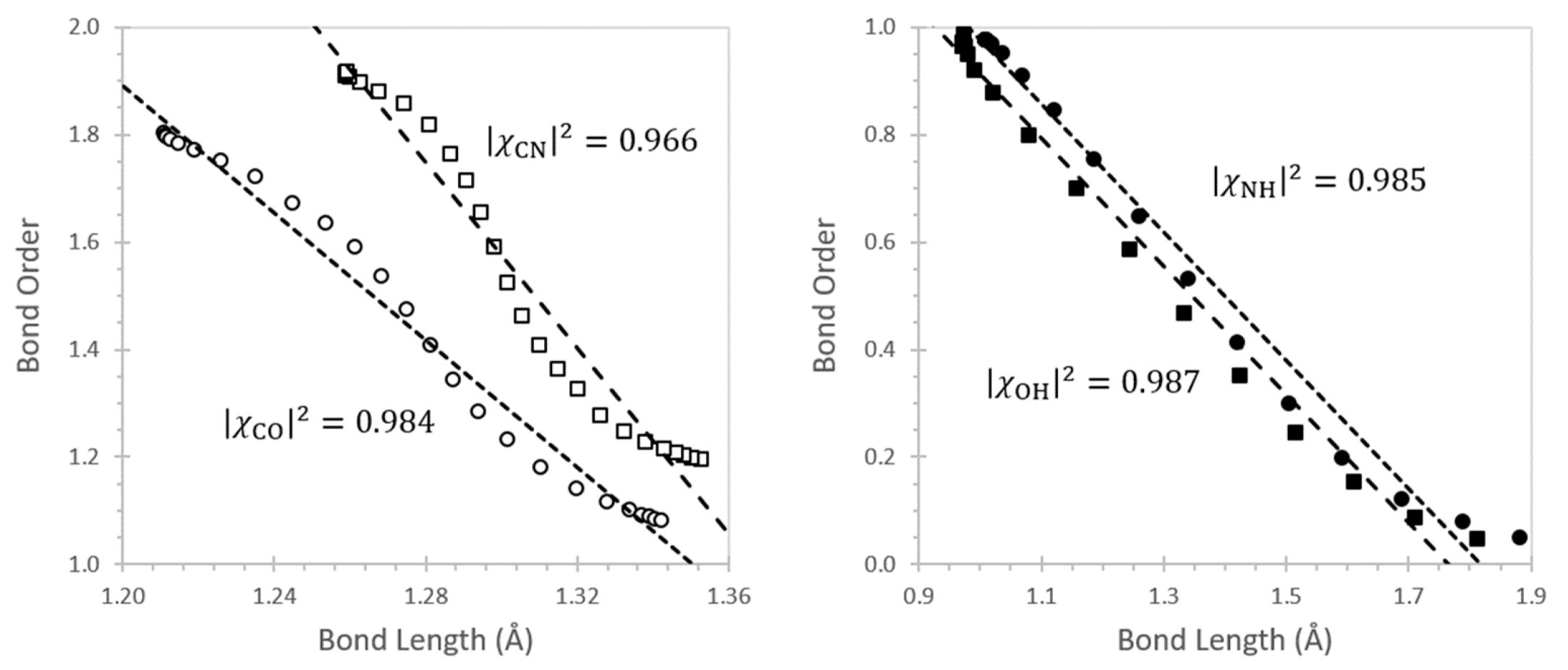
- Coulson, C.A. The electronic structure of some polyenes and aromatic molecules. VII. Bonds of fractional order by the molecular orbital method. Proc. R. Soc. Lond. A 1939, 169, 413–428. [Google Scholar]
- Coulson, C.A. Bond lengths in conjugated molecules: The present position. Proc. R. Soc. Lond. A 1951, 207, 91–100. [Google Scholar]
- Mishra, P.C.; Rai, D.K. Bond order-bond length relationship in all-valence-electron molecular orbital theory. Mol. Phys. 1972, 23, 631–634. [Google Scholar] [CrossRef]
- Johnston, H.S.; Parr, C. Activation energies from bond energies. I. Hydrogen transfer reactions. J. Am. Chem. Soc. 1963, 85, 2544–2551. [Google Scholar] [CrossRef]
- Bent, H.A. Structural chemistry of donor-acceptor interactions. Chem. Rev. 1968, 68, 587–648. [Google Scholar] [CrossRef]
- Bürgi, H.; Dunitz, J.D. Fractional bonds: Relations among their lengths, strengths, and stretching force constants. J. Am. Chem. Soc. 1987, 109, 2924–2926. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Loureiro, R.M.S.; Lurdes, M.; Cristiano, S.; Labat, G. Bond energy/bond order relationships for N-O linkages and a quantitative measure of ionicitiy: The role of nitro groups in hydrogen-bonding. ARKIVOC 2010, 2010, 142–169. [Google Scholar] [CrossRef]
- Badger, R.M. A relation between internuclear distances and bond force constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef]
- Boyer, M.A.; Marsalek, O.; Heindel, J.P.; Markland, T.E.; McCoy, A.B.; Xantheas, S.S. Beyond Badger’s rule: The origins and generality of the structure-spectra relationship of aqueous hydrogen bonds. J. Phys. Chem. Lett. 2019, 10, 918–924. [Google Scholar] [CrossRef]
- Gründemann, S.; Limbach, H.-H.; Buntkowsky, G.; Sabo-Etienne, S.; Chaudret, B. Distance and scalar HH-coupling correlations in transition metal dihydrides and dihydrogen complexes. J. Phys. Chem. A 1999, 103, 4752–4754. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RHF | B3LYP | SCGVB | CAS | MP2 | CCSD | |
|---|---|---|---|---|---|---|
| BH3 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 |
| CH4 | 2.99 | 3.00 | 2.99 | 2.99 | 2.99 | 2.99 |
| NH3 | 2.89 | 2.90 | 2.96 | 2.99 | 2.93 | 2.94 |
| H2O | 3.24 | 3.31 | 3.42 | 3.47 | 3.45 | 3.44 |
| HF | 3.69 | 3.86 | 4.05 | 4.09 | 4.10 | 4.08 |
| AlH3 | 1.97 | 1.98 | 1.98 | 1.98 | 1.98 | 1.98 |
| SiH4 | 2.96 | 2.98 | 2.96 | 2.96 | 2.96 | 2.96 |
| PH3 | 5.38 | 5.72 | 5.49 | 5.60 | 5.57 | 5.67 |
| H2S | 5.55 | 5.83 | 5.88 | 6.00 | 5.83 | 5.93 |
| HCl | 5.79 | 5.93 | 6.46 | 6.55 | 6.17 | 6.24 |
| GaH3 | 1.99 | 2.00 | 1.99 | 1.99 | 1.99 | 1.99 |
| GeH4 | 2.99 | 3.00 | 2.99 | 2.99 | 2.99 | 2.99 |
| AsH3 | 6.14 | 6.91 | 6.37 | 6.47 | 6.54 | 6.64 |
| H2Se | 6.43 | 7.11 | 7.00 | 7.05 | 6.95 | 7.06 |
| HBr | 6.82 | 7.27 | 7.81 | 7.80 | 7.43 | 7.52 |
| ∠HXH | α | β | |
|---|---|---|---|
| NH3 | 106.8 | 110.0 | 107.9 |
| H2O | 104.1 | 106.8 | 104.7 |
| PH3 | 93.6 | 100.3 | 98.0 |
| H2S | 92.2 | 99.9 | 95.6 |
| AsH3 | 92.5 | 98.8 | 98.8 |
| H2Se | 91.1 | 98.3 | 96.8 |
| # | Structure | Weight (%) | Donor–Acceptor Interaction b | |
|---|---|---|---|---|
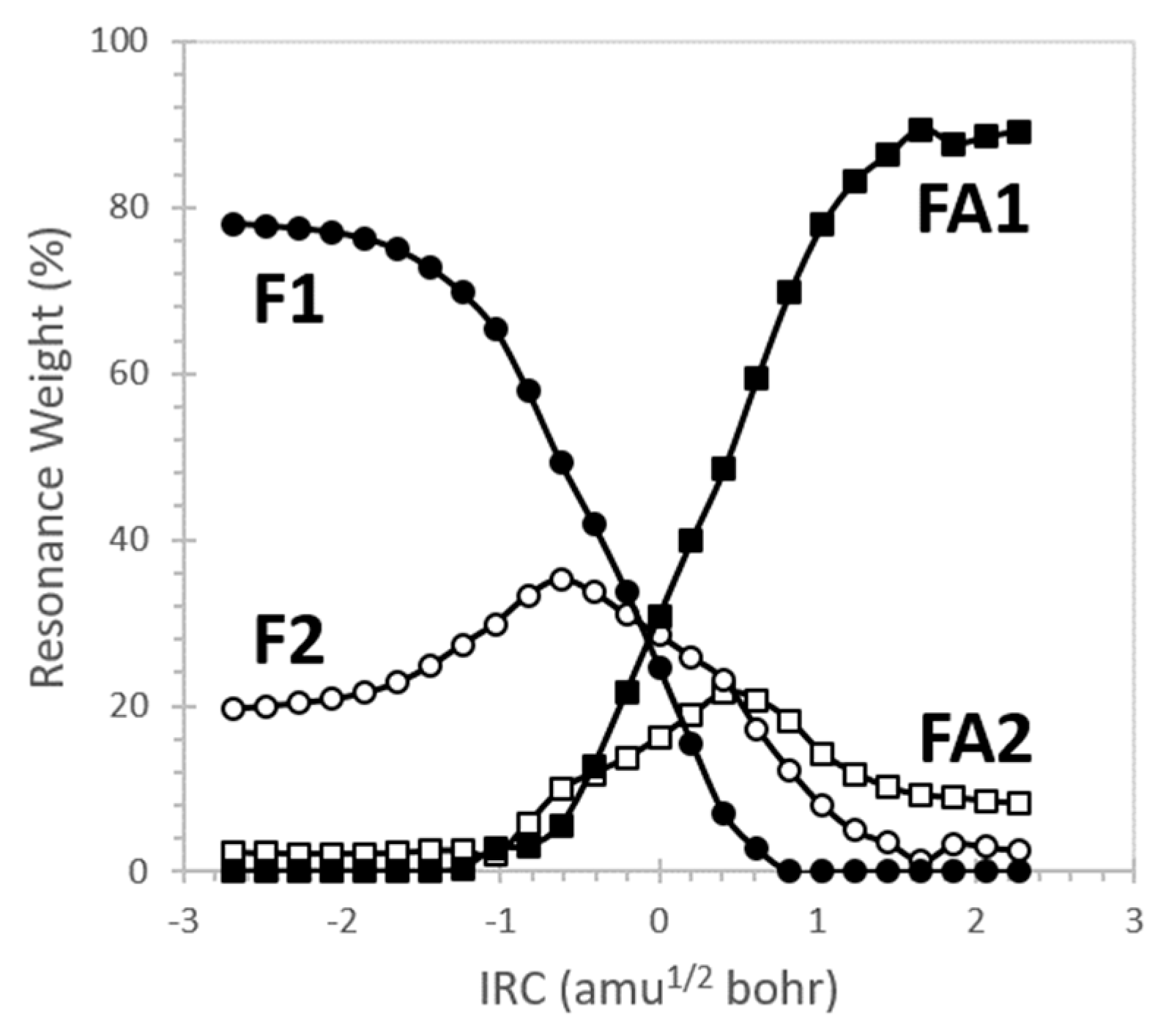
| F1 |  | 43.8 | ||
| F2 |  | 33.7 | nN→πCO * (61.9) |  |
| F3 |  | 12.1 | nO→σCN * (26.2) |  |
| F4 |  | 9.7 | nO→σCH * (22.8) |  |
| # | Structure | Weight (%) | Donor–Acceptor Interaction b | |
|---|---|---|---|---|
| FA1 |  | 64.2 | ||
| FA2 |  | 20.2 | nO→πCN * (40.4) |  |
| FA3 |  | 10.1 | nN→σCH * (10.7) |  |
| FA4 |  | 3.7 | σNH→σCO * (10.6) |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glendening, E.D.; Weinhold, F. Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry. Molecules 2021, 26, 4110. https://doi.org/10.3390/molecules26144110
Glendening ED, Weinhold F. Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry. Molecules. 2021; 26(14):4110. https://doi.org/10.3390/molecules26144110
Chicago/Turabian StyleGlendening, Eric D., and Frank Weinhold. 2021. "Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry" Molecules 26, no. 14: 4110. https://doi.org/10.3390/molecules26144110
APA StyleGlendening, E. D., & Weinhold, F. (2021). Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry. Molecules, 26(14), 4110. https://doi.org/10.3390/molecules26144110