
Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS

 ,
,
Abstract
1. Different Strategies
1.1. Structurally Similar Compounds
1.2. Close Eluting Compounds
1.3. Predicting Ionization Efficiency
2. Sources Decreasing the Signal of the Molecular Ion
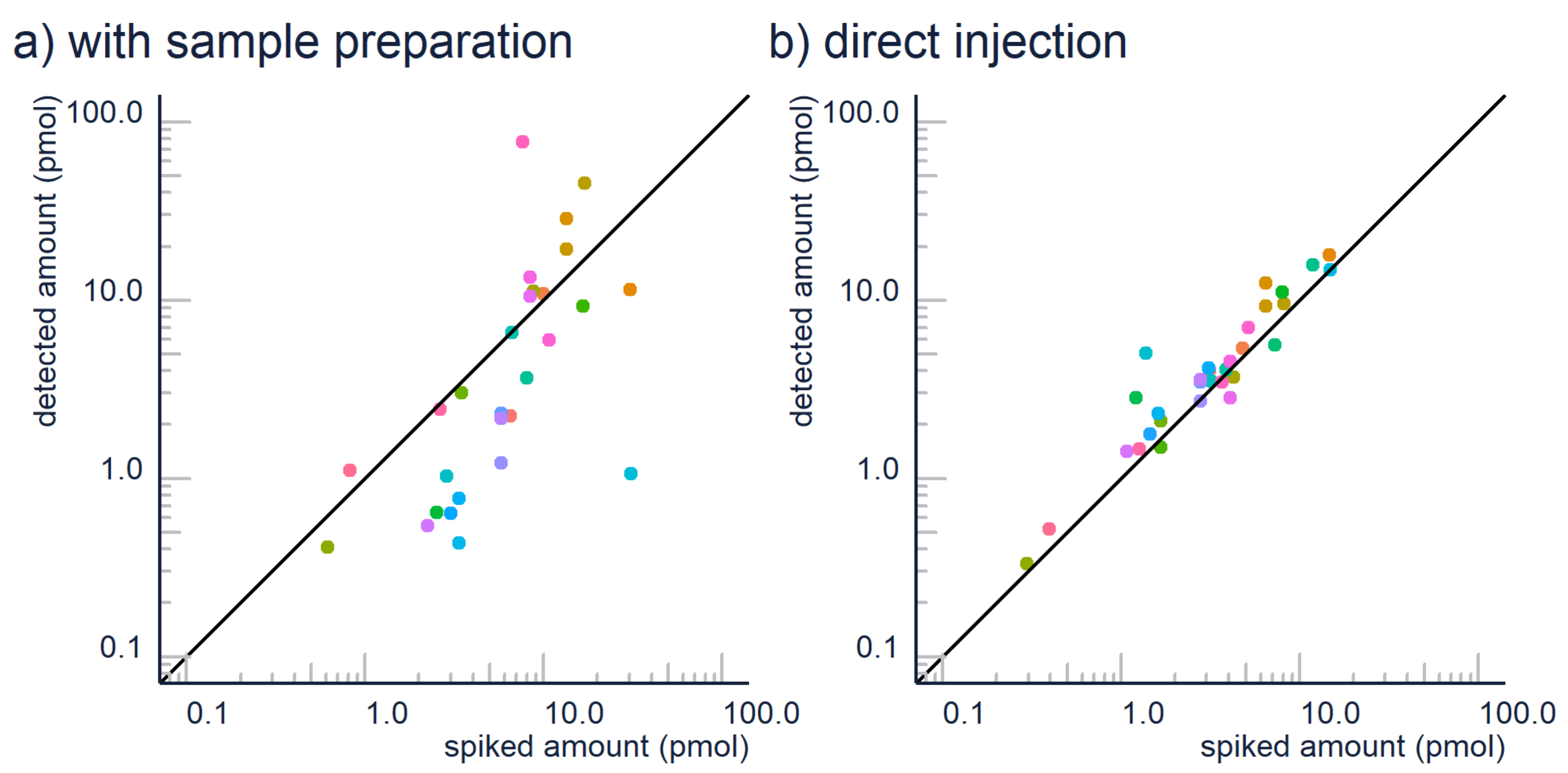
2.1. Sample Preparation
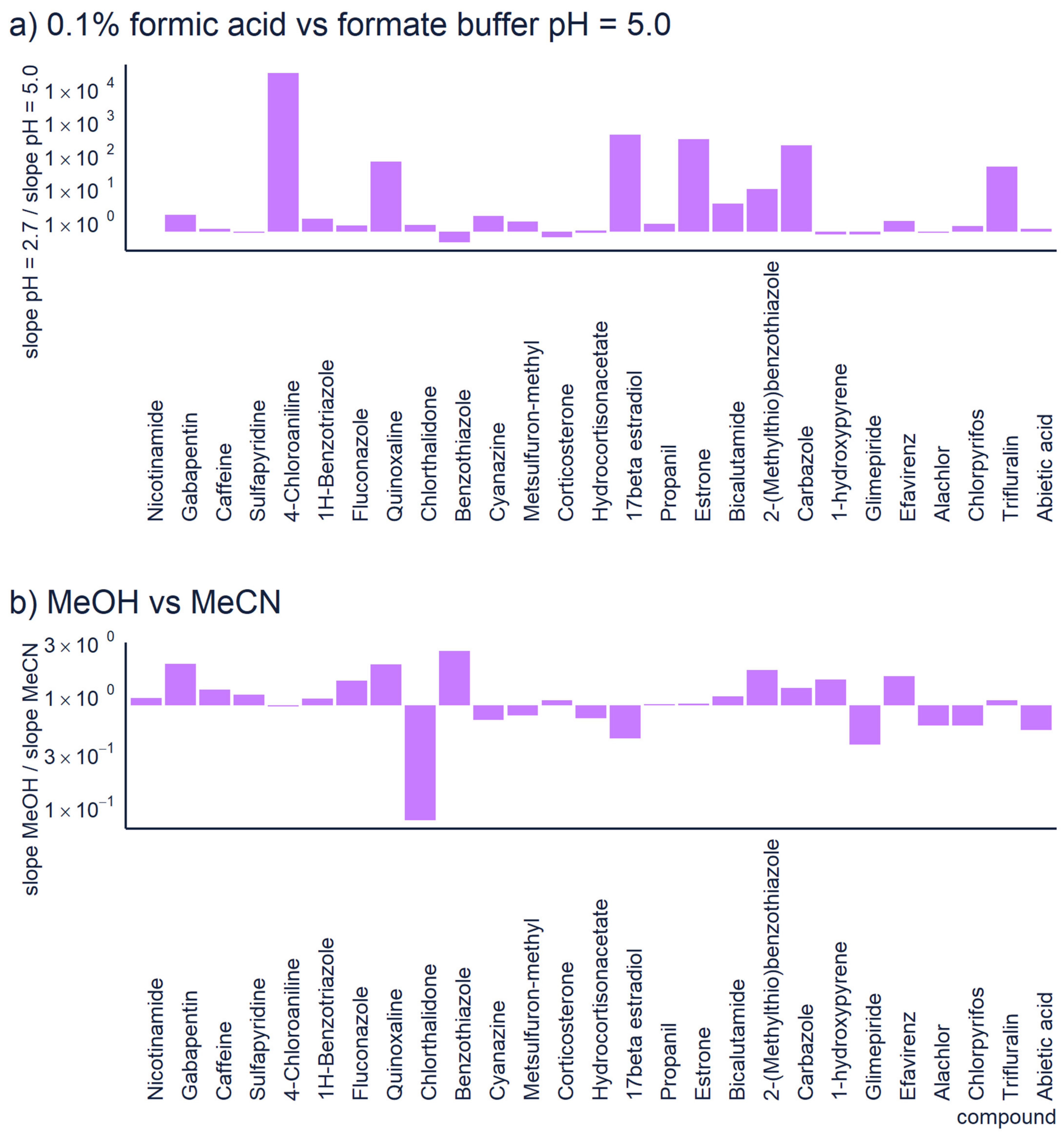
2.2. Chromatography
2.3. Ionization Conditions and Ion Transport
2.4. Data Processing
3. Improving the Performance
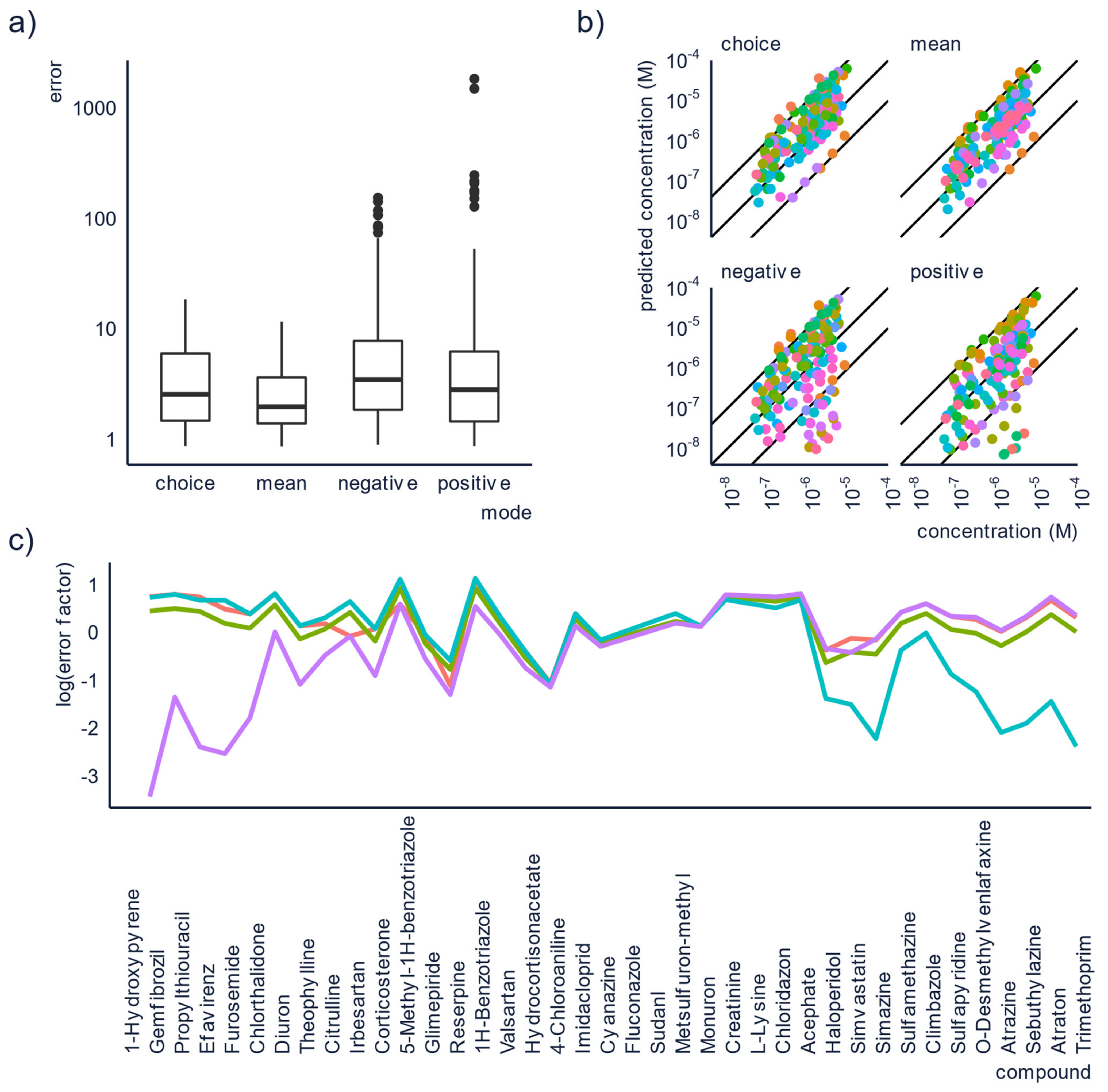
3.1. Increasing the Accuracy by Combining Positive and Negative Mode
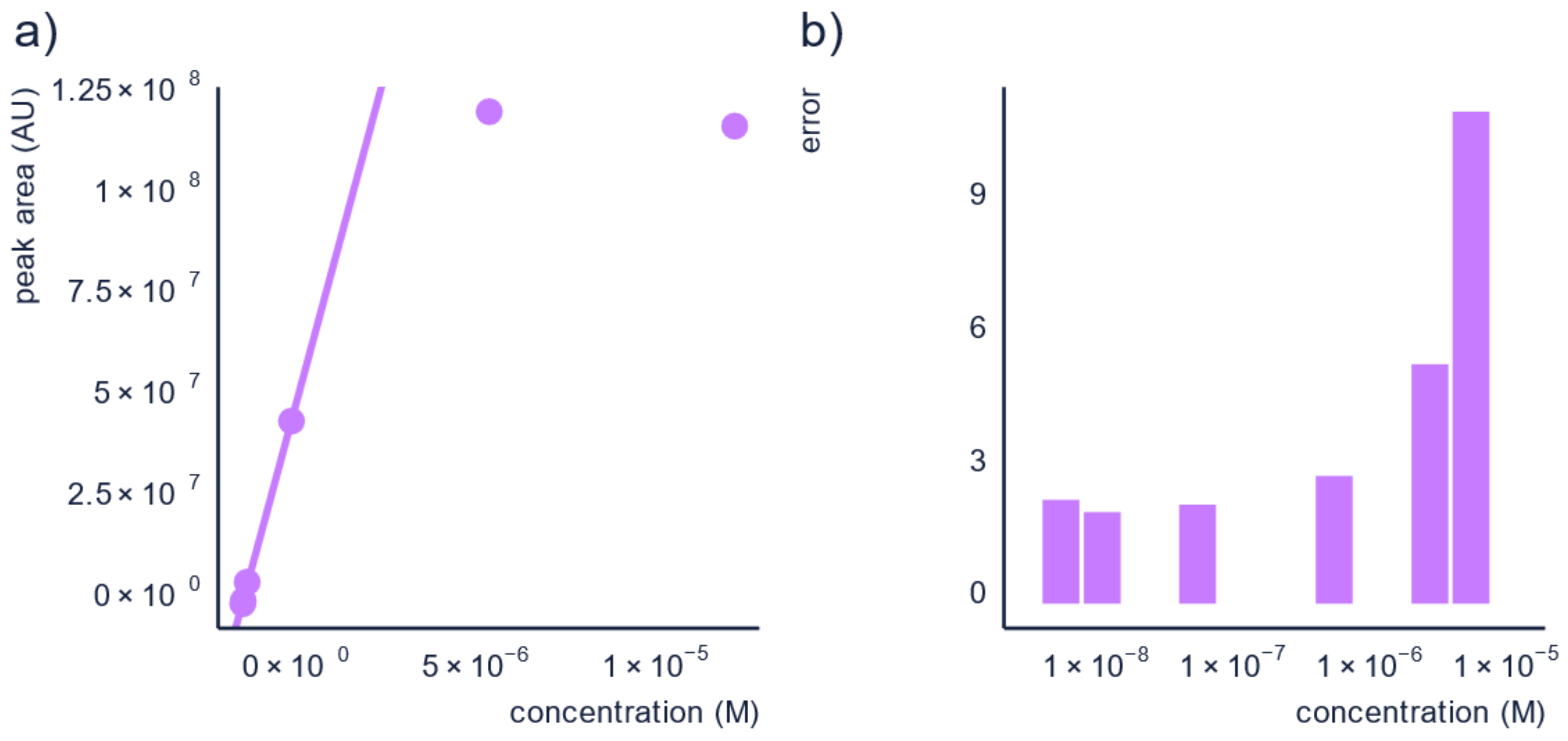
3.2. Dilution of the Sample
3.3. Quality Control
4. Opportunities Beyond Instrumental Analysis
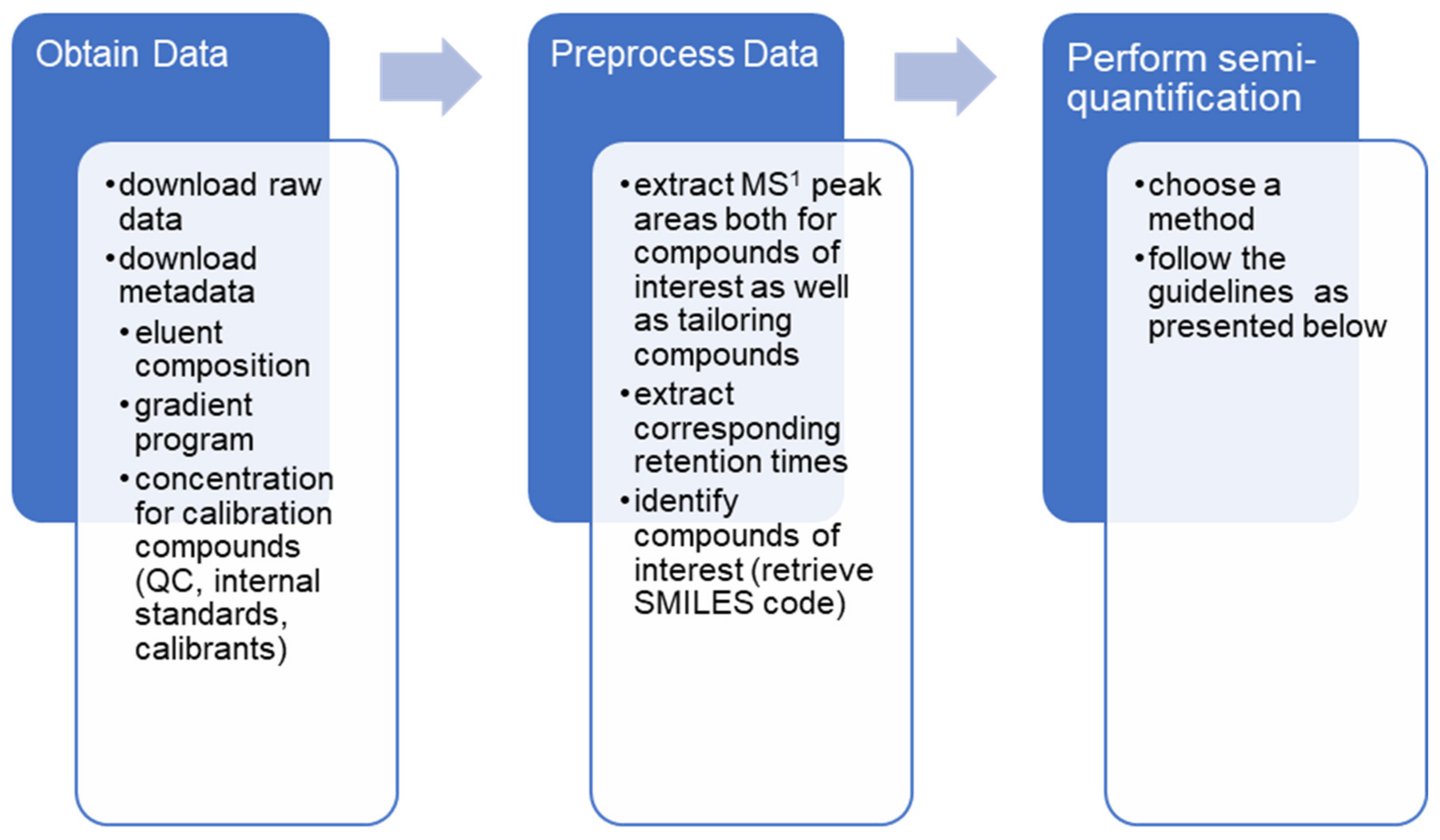
5. Carrying Out Semi-Quantitative NTS in Practice
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go? Environ. Sci. Technol. 2017, 51, 11505–11512. [Google Scholar] [CrossRef]
- Kruve, A. Semi-quantitative Non-target Analysis of Water with Liquid Chromatography/High-resolution Mass Spectrometry: How Far Are We? Rapid Commun. Mass Spectrom. 2018, 33, 54–63. [Google Scholar] [CrossRef]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and Non-Targeted Liquid Chromatography-Mass Spectrometric Workflows for Identification of Transformation Products of Emerging Pollutants in the Aquatic Environment. TrAC Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef]
- Compound Discoverer Software; Thermo Fisher Scientific: Courtaboeuf Cedex, France.
- Schmitt, U.; Loos, M.; Singer, H. EAWAG. Available online: https://www.eawag.ch/en/department/uchem/projects/envipy/ (accessed on 10 May 2021).
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Escher, B.I.; Fenner, K. Recent Advances in Environmental Risk Assessment of Transformation Products. Environ. Sci. Technol. 2011, 45, 3835–3847. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, K.; Müller, A.; Singer, H.; Hollender, J. New Relevant Pesticide Transformation Products in Groundwater Detected Using Target and Suspect Screening for Agricultural and Urban Micropollutants with LC-HRMS. Water Res. 2019, 165, 114972. [Google Scholar] [CrossRef] [PubMed]
- Liigand, J.; Wang, T.; Kellogg, J.; Smedsgaard, J.; Cech, N.; Kruve, A. Quantification for Non-Targeted LC/MS Screening without Standard Substances. Sci. Rep. 2020, 10, 5808. [Google Scholar] [CrossRef]
- Kruve, A. Strategies for Drawing Quantitative Conclusions from Nontargeted Liquid Chromatography–High-Resolution Mass Spectrometry Analysis. Anal. Chem. 2020, 92, 4691–4699. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Practical Implications of Some Recent Studies in Electrospray Ionization Fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef]
- Liigand, J.; Kruve, A.; Leito, I.; Girod, M.; Antoine, R. Effect of Mobile Phase on Electrospray Ionization Efficiency. J. Am. Soc. Mass Spectrom. 2014, 25, 1853–1861. [Google Scholar] [CrossRef]
- Kostiainen, R.; Kauppila, T.J. Effect of Eluent on the Ionization Process in Liquid Chromatography–Mass Spectrometry. J. Chromatogr. A 2009, 1216, 685–699. [Google Scholar] [CrossRef]
- Kiontke, A.; Oliveira-Birkmeier, A.; Opitz, A.; Birkemeyer, C. Electrospray Ionization Efficiency Is Dependent on Different Molecular Descriptors with Respect to Solvent PH and Instrumental Configuration. PLoS ONE 2016, 11, e0167502. [Google Scholar] [CrossRef] [PubMed]
- Page, J.S.; Kelly, R.T.; Tang, K.; Smith, R.D. Ionization and Transmission Efficiency in an Electrospray Ionization—Mass Spectrometry Interface. J. Am. Soc. Mass Spectrom. 2007, 18, 1582–1590. [Google Scholar] [CrossRef]
- Pieke, E.N.; Granby, K.; Trier, X.; Smedsgaard, J. A Framework to Estimate Concentrations of Potentially Unknown Substances by Semi-Quantification in Liquid Chromatography Electrospray Ionization Mass Spectrometry. Anal. Chim. Acta 2017, 975, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, M.S.; Folguera, L.; Magallanes, J.F.; Babay, P.A. Exploring Analyte Response in an ESI-MS System with Different Chemometric Tools. Chemom. Intell. Lab. Syst. 2015, 146, 120–127. [Google Scholar] [CrossRef]
- Aalizadeh, R.; Nika, M.-C.; Thomaidis, N.S. Development and Application of Retention Time Prediction Models in the Suspect and Non-Target Screening of Emerging Contaminants. J. Hazard. Mater. 2019, 363, 277–285. [Google Scholar] [CrossRef]
- Similar Compound Finder. Available online: http://dsfp.chem.uoa.gr/semiquantification/ (accessed on 10 May 2021).
- Kruve, A.; Aalizadeh, R.; Malm, L.; Alygizakis, N.; Thomaidis, N.S. Interlaboratory Comparison on Strategies for Semi-Quantitative Non-Targeted LC-ESI-HRMS. Available online: https://www.norman-network.net/sites/default/files/files/QA-QC%20Issues/Invitation%20letter%20JPA%202020%20semi-quant%20inter%20lab%20%28002%29.pdf (accessed on 21 July 2020).
- NORMAN Network; Aalizadeh, R.; Alygizakis, N.; Schymanski, E.; Slobodnik, J.; Fischer, S.; Cirka, L. S0|SUSDAT|Merged NORMAN Suspect List: SusDat (Version NORMAN-SLE-S0.0.3.2) [Data set]. Zenodo 2021. [Google Scholar] [CrossRef]
- Kruve, A.; Kiefer, K.; Hollender, J. Benchmarking of the Quantification Approaches for the Non-Targeted Screening of Micropollutants and Their Transformation Products in Groundwater. Anal. Bioanal. Chem. 2021, 413, 1549–1559. [Google Scholar] [CrossRef]
- Richardson, S.D.; Kimura, S.Y. Water Analysis: Emerging Contaminants and Current Issues. Anal. Chem. 2020, 92, 473–505. [Google Scholar] [CrossRef]
- Chalcraft, K.R.; Lee, R.; Mills, C.; Britz-McKibbin, P. Virtual Quantification of Metabolites by Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry: Predicting Ionization Efficiency Without Chemical Standards. Anal. Chem. 2009, 81, 2506–2515. [Google Scholar] [CrossRef]
- ChemAxon. Available online: https://chemicalize.com/ (accessed on 7 July 2020).
- Dahal, U.P.; Jones, J.P.; Davis, J.A.; Rock, D.A. Small Molecule Quantification by Liquid Chromatography-Mass Spectrometry for Metabolites of Drugs and Drug Candidates. Drug Metab. Dispos. 2011, 39, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Oss, M.; Kruve, A.; Herodes, K.; Leito, I. Electrospray Ionization Efficiency Scale of Organic Compounds. Anal. Chem. 2010, 82, 2865–2872. [Google Scholar] [CrossRef]
- Huffman, B.A.; Poltash, M.L.; Hughey, C.A. Effect of Polar Protic and Polar Aprotic Solvents on Negative-Ion Electrospray Ionization and Chromatographic Separation of Small Acidic Molecules. Anal. Chem. 2012, 84, 9942–9950. [Google Scholar] [CrossRef]
- Panagopoulos Abrahamsson, D.; Park, J.-S.; Singh, R.R.; Sirota, M.; Woodruff, T.J. Applications of Machine Learning to In Silico Quantification of Chemicals without Analytical Standards. J. Chem. Inf. Modeling 2020, 60, 2718–2727. [Google Scholar] [CrossRef]
- Mayhew, A.W.; Topping, D.O.; Hamilton, J.F. New Approach Combining Molecular Fingerprints and Machine Learning to Estimate Relative Ionization Efficiency in Electrospray Ionization. ACS Omega 2020, 5, 9510–9516. [Google Scholar] [CrossRef] [PubMed]
- Liigand, P.; Liigand, J.; Kaupmees, K.; Kruve, A. 30 Years of Research on ESI/MS Response: Trends, Contradictions and Applications. Anal. Chim. Acta 2021, 1152, 238117. [Google Scholar] [CrossRef]
- Quantem Analytics. Available online: https://app.quantem.co/ (accessed on 10 May 2021).
- Dührkop, K.; Nothias, L.-F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic Classification of Unknown Metabolites Using High-Resolution Fragmentation Mass Spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Bertelkamp, C.; Dingemans, M.M.L.; Kolkman, A.; Wols, B.; Harmsen, D.; Siegers, W.; Martijn, B.J.; Oorthuizen, W.A.; ter Laak, T.L. Integration of Target Analyses, Non-Target Screening and Effect-Based Monitoring to Assess OMP Related Water Quality Changes in Drinking Water Treatment. Sci. Total Environ. 2020, 705, 135779. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.P.; Longrée, P.; Loos, M.; Ruff, M.; Stravs, M.A.; Ripollés Vidal, C.; Hollender, J. Strategies to Characterize Polar Organic Contamination in Wastewater: Exploring the Capability of High Resolution Mass Spectrometry. Environ. Sci. Technol. 2014, 48, 1811–1818. [Google Scholar] [CrossRef]
- Sørensen, L.; McCormack, P.; Altin, D.; Robson, W.J.; Booth, A.M.; Faksness, L.-G.; Rowland, S.J.; Størseth, T.R. Establishing a Link between Composition and Toxicity of Offshore Produced Waters Using Comprehensive Analysis Techniques—A Way Forward for Discharge Monitoring? Sci. Total Environ. 2019, 694, 133682. [Google Scholar] [CrossRef]
- Blum, K.M.; Andersson, P.L.; Renman, G.; Ahrens, L.; Gros, M.; Wiberg, K.; Haglund, P. Non-Target Screening and Prioritization of Potentially Persistent, Bioaccumulating and Toxic Domestic Wastewater Contaminants and Their Removal in on-Site and Large-Scale Sewage Treatment Plants. Sci. Total Environ. 2017, 575, 265–275. [Google Scholar] [CrossRef]
- Baz-Lomba, J.A.; Salvatore, S.; Gracia-Lor, E.; Bade, R.; Castiglioni, S.; Castrignanò, E.; Causanilles, A.; Hernandez, F.; Kasprzyk-Hordern, B.; Kinyua, J.; et al. Comparison of Pharmaceutical, Illicit Drug, Alcohol, Nicotine and Caffeine Levels in Wastewater with Sale, Seizure and Consumption Data for 8 European Cities. BMC Public Health 2016, 16, 1035. [Google Scholar] [CrossRef]
- González-Mariño, I.; Gracia-Lor, E.; Rousis, N.I.; Castrignanò, E.; Thomas, K.V.; Quintana, J.B.; Kasprzyk-Hordern, B.; Zuccato, E.; Castiglioni, S. Wastewater-Based Epidemiology To Monitor Synthetic Cathinones Use in Different European Countries. Environ. Sci. Technol. 2016, 50, 10089–10096. [Google Scholar] [CrossRef] [PubMed]
- Schulze, T.; Ahel, M.; Ahlheim, J.; Aït-Aïssa, S.; Brion, F.; Di Paolo, C.; Froment, J.; Hidasi, A.O.; Hollender, J.; Hollert, H.; et al. Assessment of a Novel Device for Onsite Integrative Large-Volume Solid Phase Extraction of Water Samples to Enable a Comprehensive Chemical and Effect-Based Analysis. Sci. Total Environ. 2017, 581–582, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Veprikova, Z.; Godula, M.; Hajslova, J. Multi-Analyte High Performance Liquid Chromatography Coupled to High Resolution Tandem Mass Spectrometry Method for Control of Pesticide Residues, Mycotoxins, and Pyrrolizidine Alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef]
- Kafeenah, H.I.S.; Osman, R.; Bakar, N.K.A. Effect of Mobile Phase PH on the Electrospray Ionization Efficiency and Qualitative Analysis of Pharmaceuticals in ESI + LC-MS/MS. J. Chromatogr. Sci. 2019, 57, 847–854. [Google Scholar] [CrossRef]
- Zhou, S.; Cook, K.D. Protonation in Electrospray Mass Spectrometry: Wrong-Way-Round or Right-Way-Round? J. Am. Soc. Mass Spectrom. 2000, 11, 961–966. [Google Scholar] [CrossRef]
- Shou, W.; Naidong, W. Simple Means to Alleviate Sensitivity Loss by Trifluoroacetic Acid (TFA) Mobile Phases in the Hydrophilic Interaction Chromatography–Electrospray Tandem Mass Spectrometric (HILIC–ESI/MS/MS) Bioanalysis of Basic Compounds. J. Chromatogr. B 2005, 825, 186–192. [Google Scholar] [CrossRef]
- Mallet, C.R.; Lu, Z.; Mazzeo, J.R. A Study of Ion Suppression Effects in Electrospray Ionization from Mobile Phase Additives and Solid-Phase Extracts. Rapid Commun. Mass Spectrom. 2004, 18, 49–58. [Google Scholar] [CrossRef]
- Snyder, L.R.; Kirkland, J.J.; Glajch, J.L. Practical HPLC Method Development, 2nd ed.; Wiley: New York, NY, USA, 1997; ISBN 978-0-471-00703-6. [Google Scholar]
- Ojakivi, M.; Liigand, J.; Kruve, A. Modifying the Acidity of Charged Droplets. ChemistrySelect 2018, 3, 335–338. [Google Scholar] [CrossRef]
- Rebane, R.; Kruve, A.; Liigand, J.; Liigand, P.; Gornischeff, A.; Leito, I. Ionization Efficiency Ladders as Tools for Choosing Ionization Mode and Solvent in Liquid Chromatography/Mass Spectrometry. Rapid Commun. Mass Spectrom. 2019, 33, 1834–1843. [Google Scholar] [CrossRef]
- Colizza, K.; Mahoney, K.E.; Yevdokimov, A.V.; Smith, J.L.; Oxley, J.C. Acetonitrile Ion Suppression in Atmospheric Pressure Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1796–1804. [Google Scholar] [CrossRef]
- Buszewski, B.; Bocian, S.; Felinger, A. Artifacts in Liquid-Phase Separations–System, Solvent, and Impurity Peaks. Chem. Rev. 2012, 112, 2629–2641. [Google Scholar] [CrossRef] [PubMed]
- Srbek, J.; Coufal, P.; Bosáková, Z.; Tesařová, E. System Peaks and Their Positive and Negative Aspects in Chromatographic Techniques. J. Sep. Sci. 2005, 28, 1263–1270. [Google Scholar] [CrossRef]
- Oberacher, H.; Sasse, M.; Antignac, J.-P.; Guitton, Y.; Debrauwer, L.; Jamin, E.L.; Schulze, T.; Krauss, M.; Covaci, A.; Caballero-Casero, N.; et al. A European Proposal for Quality Control and Quality Assurance of Tandem Mass Spectral Libraries. Environ. Sci. Eur. 2020, 32, 43. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN Small Molecule Dataset for Machine Learning-Based Retention Time Prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef]
- Minkus, S.; Bieber, S.; Moser, S.; Letzel, T. Optimization of Electrospray Ionization Parameters in a RPLC-HILIC-MS/MS Coupling by Design of Experiment. Available online: http://afin-ts.de/literature/?lang=en (accessed on 11 May 2021).
- Seo, J.; Warnke, S.; Gewinner, S.; Schöllkopf, W.; Bowers, M.T.; Pagel, K.; von Helden, G. The Impact of Environment and Resonance Effects on the Site of Protonation of Aminobenzoic Acid Derivatives. Phys. Chem. Chem. Phys. 2016, 18, 25474–25482. [Google Scholar] [CrossRef]
- Liigand, P.; Kaupmees, K.; Haav, K.; Liigand, J.; Leito, I.; Girod, M.; Antoine, R.; Kruve, A. Think Negative: Finding the Best Electrospray Ionization/MS Mode for Your Analyte. Anal. Chem. 2017, 89, 5665–5668. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.B. Electrospray and MALDI Mass Spectrometry Fundamentals, Instrumentation, Practicalities, and Biological Applications; Wiley: Hoboken, NJ, USA, 2011; ISBN 978-1-118-21155-7. [Google Scholar]
- Kruve, A.; Kaupmees, K. Predicting ESI/MS Signal Change for Anions in Different Solvents. Anal. Chem. 2017, 89, 5079–5086. [Google Scholar] [CrossRef]
- Wang, T.; Liigand, J.; Frandsen, H.L.; Smedsgaard, J.; Kruve, A. Standard Substances Free Quantification Makes LC/ESI/MS Non-Targeted Screening of Pesticides in Cereals Comparable between Labs. Food Chem. 2020, 318, 126460. [Google Scholar] [CrossRef]
- Lagerwerf, F.M.; van Dongen, W.D.; Steenvoorden, R.J.J.M.; Honing, M.; Jonkman, J.H.G. Exploring the Boundaries of Bioanalytical Quantitative LC–MS–MS. TrAC Trends Anal. Chem. 2000, 19, 418–427. [Google Scholar] [CrossRef]
- Kirwan, J.A.; Broadhurst, D.I.; Davidson, R.L.; Viant, M.R. Characterising and Correcting Batch Variation in an Automated Direct Infusion Mass Spectrometry (DIMS) Metabolomics Workflow. Anal. Bioanal. Chem. 2013, 405, 5147–5157. [Google Scholar] [CrossRef] [PubMed]
- Brunius, C.; Shi, L.; Landberg, R. Large-Scale Untargeted LC-MS Metabolomics Data Correction Using between-Batch Feature Alignment and Cluster-Based within-Batch Signal Intensity Drift Correction. Metabolomics 2016, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, Q.; Li, Q.; Zhang, S.; Qu, X.; Zhu, J.; Zhong, G.; Huang, M. Signal Drift in Liquid Chromatography Tandem Mass Spectrometry and Its Internal Standard Calibration Strategy for Quantitative Analysis. Anal. Chem. 2020, 92, 7690–7698. [Google Scholar] [CrossRef] [PubMed]
- Knolhoff, A.M.; Premo, J.H.; Fisher, C.M. A Proposed Quality Control Standard Mixture and Its Uses for Evaluating Nontargeted and Suspect Screening LC/HR-MS Method Performance. Anal. Chem. 2021, 93, 1596–1603. [Google Scholar] [CrossRef]
- Schulze, B.; Jeon, Y.; Kaserzon, S.; Heffernan, A.L.; Dewapriya, P.; O’Brien, J.; Gomez Ramos, M.J.; Ghorbani Gorji, S.; Mueller, J.F.; Thomas, K.V.; et al. An Assessment of Quality Assurance/Quality Control Efforts in High Resolution Mass Spectrometry Non-Target Workflows for Analysis of Environmental Samples. TrAC Trends Anal. Chem. 2020, 133, 116063. [Google Scholar] [CrossRef]
- Alygizakis, N.A.; Oswald, P.; Thomaidis, N.S.; Schymanski, E.L.; Aalizadeh, R.; Schulze, T.; Oswaldova, M.; Slobodnik, J. NORMAN Digital Sample Freezing Platform: A European Virtual Platform to Exchange Liquid Chromatography High Resolution-Mass Spectrometry Data and Screen Suspects in “Digitally Frozen” Environmental Samples. TrAC Trends Anal. Chem. 2019, 115, 129–137. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A Resource Evolving in Response to the Needs of Its Scientific Community. Nucleic Acids Res. 2019, gkz1019. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An International Repository for Metabolomics Data and Metadata, Metabolite Standards, Protocols, Tutorials and Training, and Analysis Tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Kellogg, J.J.; Graf, T.N.; Paine, M.F.; McCune, J.S.; Kvalheim, O.M.; Oberlies, N.H.; Cech, N.B. Comparison of Metabolomics Approaches for Evaluating the Variability of Complex Botanical Preparations: Green Tea ( Camellia Sinensis ) as a Case Study. J. Nat. Prod. 2017, 80, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Hernando, M.D.; Suárez-Barcena, J.M.; Bueno, M.J.M.; Garcia-Reyes, J.F.; Fernández-Alba, A.R. Fast Separation Liquid Chromatography–Tandem Mass Spectrometry for the Confirmation and Quantitative Analysis of Avermectin Residues in Food. J. Chromatogr. A 2007, 1155, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Liigand, J.; Kruve, A.; Liigand, P.; Laaniste, A.; Girod, M.; Antoine, R.; Leito, I. Transferability of the Electrospray Ionization Efficiency Scale between Different Instruments. J. Am. Soc. Mass Spectrom. 2015, 26, 1923–1930. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Similarity Score | pKa | logP | RF |
|---|---|---|---|---|
Atrazine | 100% | 4.2 | 2.2 | 7.3 × 1017 |
Atrazine-2-hydroxy | 58.3% | 3.6 | −3.1 | 1.0 × 1017 |
Atrazine-desethyl | 53.9% | 4.4 | 0.8 | 4.1 × 1016 |
Atrazine-desethyl-2-hydroxy | 38.9% | 3.0 | −3.5 | 3.0 × 1016 |
Atrazine-desisopropyl | 44.6% | 4.4 | 0.4 | 2.7 × 1016 |
Atrazine-desisopropyl-2-hydroxy | 31.5% | 3.1 | −3.9 | 4.6 × 1016 |
Atrazine-desethyl-desisopropyl | 19.8% | 4.6 | −0.2 | 4.9 × 1015 |
Atrazine-desethyl-desisopropyl-2-hydroxy | 13.4% | 3.1 | −4.5 | 4.0 × 1014 |
| Compound | pH | Neighbour | RTcompound (min) | RTneighbour (min) | RFcompound | RFneighbour | |
Valsartan | 2.7 | Estrone | 12.58 | 12.58 | 5.99 × 1011 | 1.11 × 1012 | 0.54 |
| 5.0 | Chlorthalidone | 7.30 | 7.41 | 1.52 × 1011 | 9.42 × 1010 | 1.61 | |
| 8.0 | Sulfamethazine | 5.23 | 5.27 | 7.93 × 1011 | 4.77 × 1012 | 0.166 | |
Metsulfuron-methyl | 2.7 | Prometryn | 10.32 | 10.26 | 4.31 × 1012 | 6.23 × 1012 | 0.692 |
| 5.0 | 2-napthoic acid | 9.62 | 9.62 | 2.17 × 1012 | 2.53 × 109 | 857 | |
| 8.0 | Gabapentin | 3.78 | 3.75 | 5.80 × 1012 | 2.52 × 1012 | 2.30 |
| NTS Step | Suggested Procedure | Reasoning |
|---|---|---|
| Sample Preparation | Avoid extensive sample preparation where possible. | If you analyze fairly simple liquids like water samples, urine, beverages, etc., direct injection of the sample is suggested over sample clean-up to avoid losses of analyte. Matrix effect can be evaluated by comparing the results from different dilutions, see below. |
| Standards | Prepare a solution with a set of compounds with known concentrations and analyze this set together with your samples. A suggested set of compounds could be tetrahexylammonium salt, haloperidol, diphenyl phthalate, tetraethylammonium salt, phenylalanine, dimethyl phthalate, progesterone, alanine, uracil, and saccharin for positive ESI mode. In negative mode we suggest 4-aminobutyric acid, sorbic acid, vanillin, benzoic acid, salicylic acid, p-nitrophenol, 3-nitrobenzenesulphonamide, perfluorobutyric acid, tetradecanoic acid, 3,5-diiodosalicylic acid, perfluorooctanesulfonic acid [32]. | Make sure that these compounds cover a wide ionization efficiencies range and elute over the full chromatographic run. These compounds will be used to transfer the ionization efficiency predictions to your instrument scale. |
| Sample Analysis | Analyze samples on at least two dilutions. | Additional to running duplicates or triplicates, you can also analyze your samples at different dilutions or with different injection volumes. This will allow you to assure that all measurements are in the linear range as well as account for possible matrix effect. Matrix effect [72] is known to be less severe for more diluted samples. |
| Chromatography | Use generic chromatographic parameters. Avoid exotic additives and organic solvents. To generalize these methods, we recommend using 0.1% formic acid in ESI positive mode and ammonium hydroxide or ammonium formiate pH = 8.0 in ESI negative mode. In both cases a linear gradient from 5 to 100% of acetonitrile over 15 min for a 10 cm C18 column with 3 µm particle size has proven generic. | The predictions of any model are applicable only to the conditions used in the training/validation of the model. Therefore, rare LC conditions are likely not to be covered by the quantification model used. |
| Ionization Conditions | Choose soft ionization conditions, use the default parameters of the vendor as guide. If possible, do not alter these parameters too much. Run in both positive and negative ESI mode. | Soft conditions are likely to cause less fragmentation. The units and range of values of source parameters depend on the vendor, so exact parameters cannot be transferred between instruments. However, using vendor recommendations across instruments yields similar relative ionization efficiency values, and therefore, semi-quantification results [73]. Running analysis in both positive and negative mode enables combining and comparing results for polyfunctional compounds ionizing in both modes. |
| MS Parameters | Use a wide scan range, e.g., from 100 to 1000 Da. | Wide scan range will enable pinpointing fragments formed in the ionization source. |
| Data Processing | Combine the signal of the precursor ion and fragments together. Check that the integrations is acceptable. | Fragmentation occurs separately from ionization and is not accounted in the prediction algorithms. Poor integration of tailing or split peaks may significantly decrease semi-quantification accuracy. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malm, L.; Palm, E.; Souihi, A.; Plassmann, M.; Liigand, J.; Kruve, A. Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS. Molecules 2021, 26, 3524. https://doi.org/10.3390/molecules26123524
Malm L, Palm E, Souihi A, Plassmann M, Liigand J, Kruve A. Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS. Molecules. 2021; 26(12):3524. https://doi.org/10.3390/molecules26123524
Chicago/Turabian StyleMalm, Louise, Emma Palm, Amina Souihi, Merle Plassmann, Jaanus Liigand, and Anneli Kruve. 2021. "Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS" Molecules 26, no. 12: 3524. https://doi.org/10.3390/molecules26123524
APA StyleMalm, L., Palm, E., Souihi, A., Plassmann, M., Liigand, J., & Kruve, A. (2021). Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS. Molecules, 26(12), 3524. https://doi.org/10.3390/molecules26123524