
Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi


Abstract
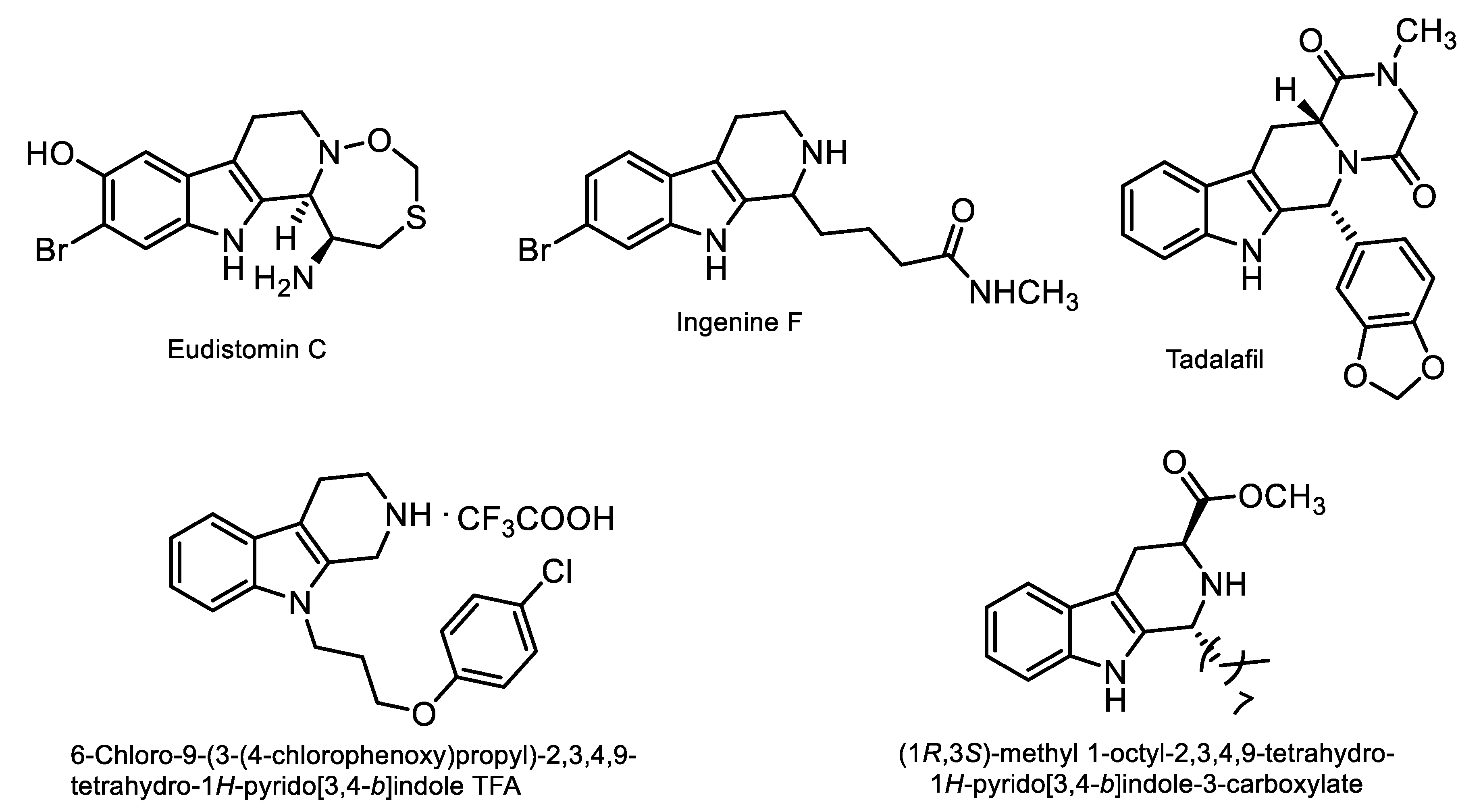
1. Introduction
2. Results and Discussion
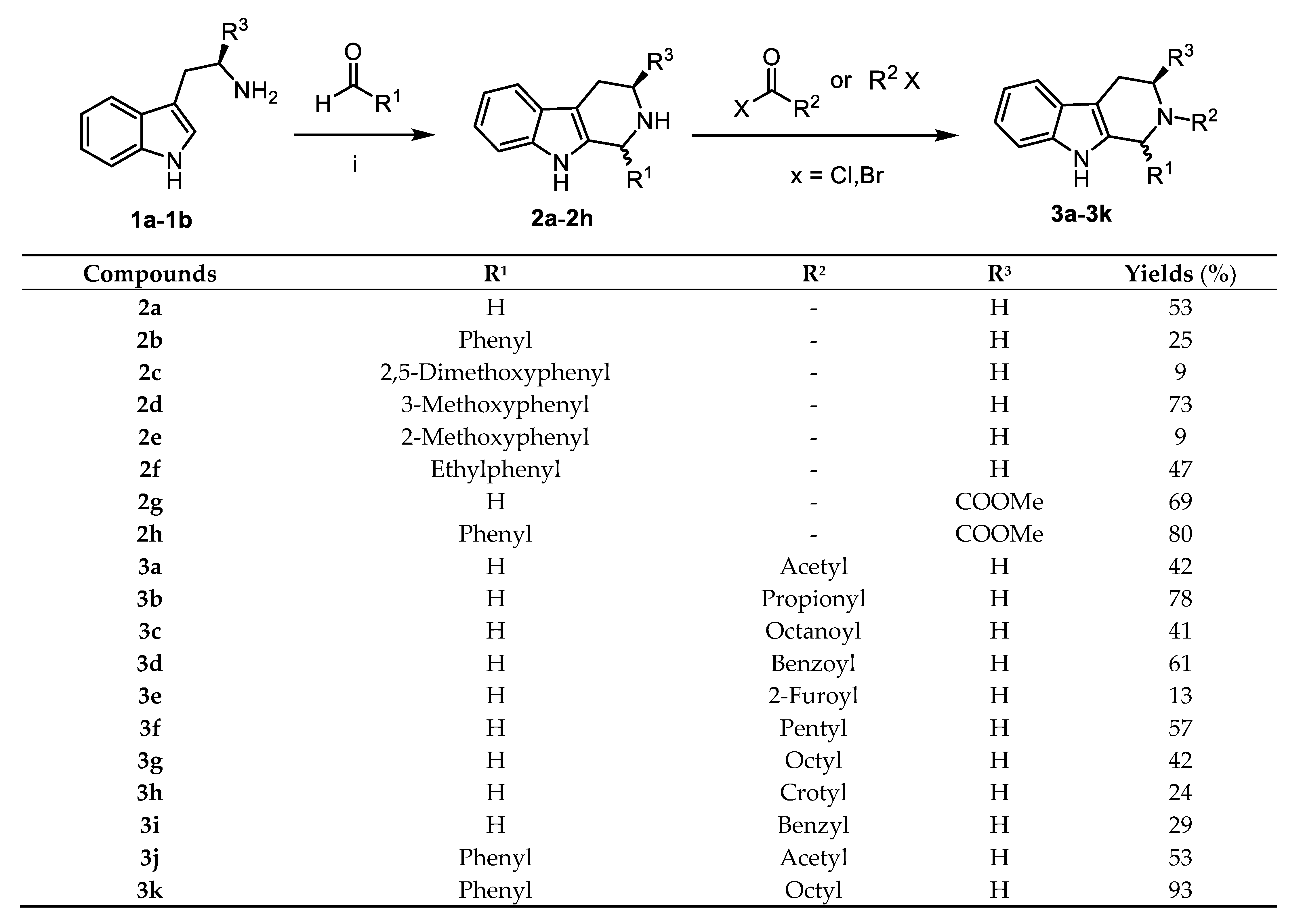
2.1. Chemistry
2.2. Antifungal Activity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of 1-substituted-tetrahydro-β-carbolines (2a–2h)
3.1.3. General Procedure for Preparation of 2-substituted-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (3a–3i)
3.1.4. General Procedure for the Preparation of 2-substituted-1-phenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (3j–3k)
3.2. Antifungal Activity Assay
3.2.1. Agar Well Diffusion Method
3.2.2. Minimum Inhibitory Concentration Test
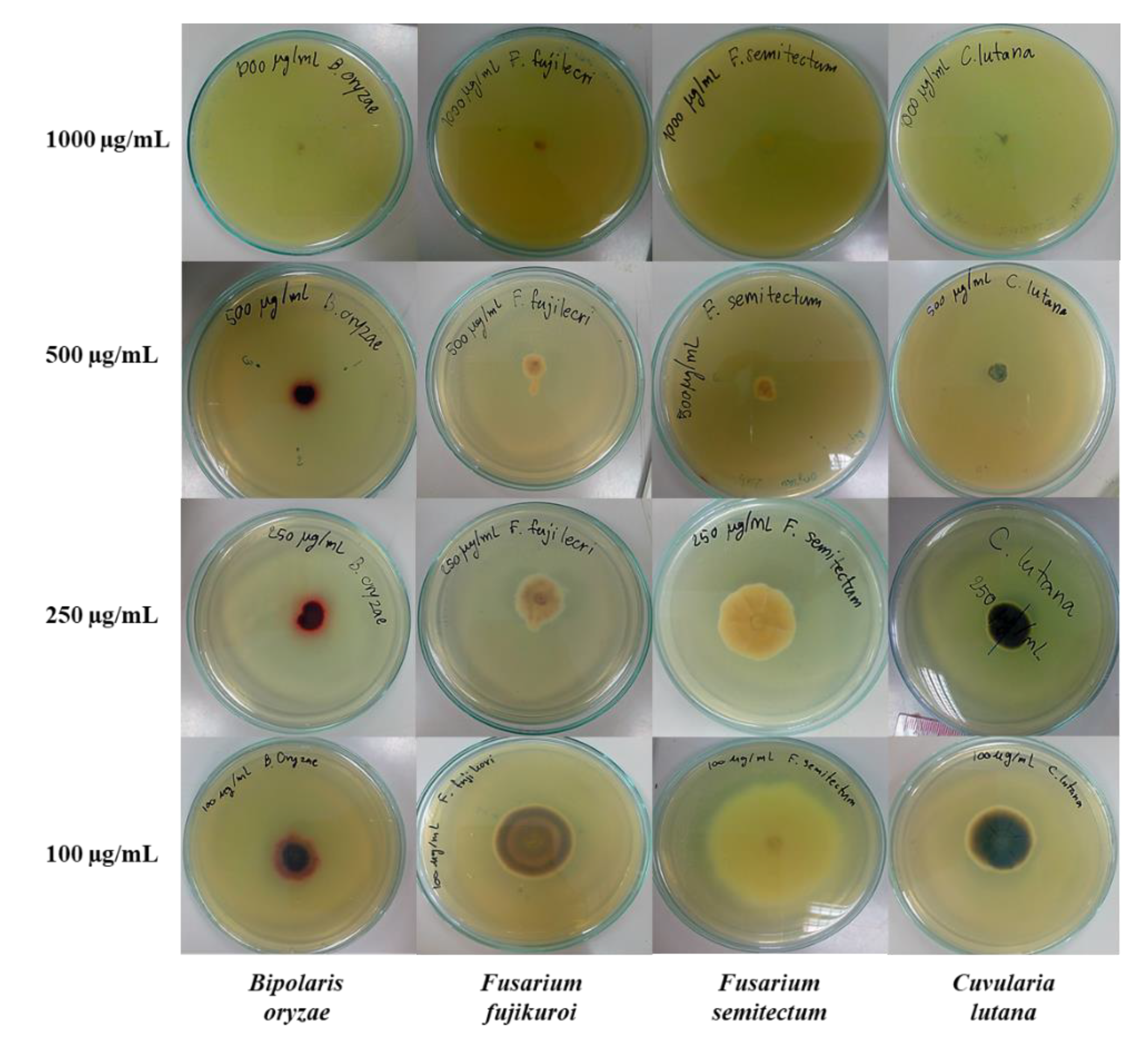
3.2.3. Test for Inhibitory Activity against Fungal Radial Growth
- IR = inhibitory activity against radial growth in percent.
- DC = diameter of fungal colony without compounds (control).
- DT = diameter of fungal colony treated with compound.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laine, A.E.; Lood, C.; Koskinen, A.M.P. Pharmacological importance of optically active tetrahydro-β-carbolines and synthetic approaches to create the C1 stereocenter. Molecules 2014, 19, 1544–1567. [Google Scholar] [CrossRef] [PubMed]
- Manda, S.; Khan, S.I.; Jain, S.K.; Mohammed, S.; Tekwani, B.L.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-β-carbolines. Bioorganic Med. Chem. Lett. 2014, 24, 3247–3250. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.R.; Wilson, J.; Emmerson, D.; Garcia, M.D.; Smith, M.R.; Gray, S.J.; Britton, R.G.; Mahale, S.; Chaudhuri, B. Design, synthesis and biological evaluation of new tryptamine and tetrahydro-β-carboline-based selective inhibitors of CDK4. Bioorg. Med. Chem. 2008, 16, 7728–7739. [Google Scholar] [CrossRef] [PubMed]
- Foderaro, T.A.; Barrows, L.R.; Lassota, P.; Ireland, C.M. Bengacarboline, a New β-Carboline from a Marine Ascidian Didemnum sp. J. Org. Chem. 1997, 62, 6064–6065. [Google Scholar] [CrossRef]
- Peng, J.; Hu, J.; Kazi, A.B.; Li, Z.; Avery, M.; Peraud, O.; Hill, R.T.; Franzblau, S.G.; Zhang, F.; Schinazi, R.F.; et al. Manadomanzamines A and B: A Novel Alkaloid Ring System with Potent Activity against Mycobacteria and HIV-1. J. Am. Chem. Soc. 2003, 125, 13382–13386. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.D.; Freyer, A.J.; Carte, B.; Taylor, P.B.; Johnson, R.K.; Faulkner, D.J. Haploscleridamine, a Novel Tryptamine-Derived Alkaloid from a Sponge of the Order Haplosclerida: An Inhibitor of Cathepsin, K.J. Nat. Prod. 2002, 65, 628–629. [Google Scholar] [CrossRef]
- Davis, R.A.; Duffy, S.; Avery, V.M.; Camp, D.; Hooper, J.N.A.; Quinn, R.J. (+)-7-Bromotrypargine: An antimalarial β-carboline from the Australian marine sponge Ancorina sp. Tetrahedron Lett. 2010, 51, 583–585. [Google Scholar] [CrossRef]
- Maity, P.; Adhikari, D.; Jana, K.A. An Overview on Synthetic Entries to Tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A-Q, beta.-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Youssef, D.T.A. Hyrtioerectines A-C, Cytotoxic Alkaloids from the Red Sea Sponge Hyrtios erectus. J. Nat. Prod. 2005, 68, 1416–1419. [Google Scholar] [CrossRef]
- Shen, Y.C.; Chen, C.Y.; Hsieh, P.W.; Duh, C.Y.; Lin, Y.M.; Ko, C.L. The Preparation and Evaluation of 1-Substituted 1,2,3,4-Tetrahydro- and 3,4-Dihydro-β-carboline Derivatives as Potential Antitumor Agents. Chem. Pharm. Bull. 2005, 53, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kubota, T.; Kobayashi, J.I. Eudistomidins H–K, new β-carboline alkaloids from the Okinawan marine tunicate Eudistoma glaucus. Bioorganic Med. Chem. Lett. 2011, 21, 4220–4223. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Mohamed, G.; Al Haidari, R.; El-Kholy, A.; Zayed, M.; Ingenine, F. A New Cytotoxic Tetrahydro Carboline Alkaloid from the Indonesian Marine Sponge Acanthostrongylophora ingens. Pharmacogn. Mag. 2018, 14, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Zhu, L.F.; Yu, X.C.; Sun, M.Q.; Miao, F.; Zhou, L. Design, Synthesis, and Structure–Activity Relationship of New 2-Aryl-3,4-dihydro-β-carbolin-2-ium Salts as Antifungal Agents. J. Agric. Food Chem. 2016, 64, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Sarma, B.K.; Mishra, P.K.; Ray, A.B. Antifungal activity of venenatine, an indole alkaloid isolated from Alstonia venenata. Folia Microbiol. 2000, 45, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.M.F.; Queiroz, E.F.; Zeraik, M.L.; Marti, G.; Favre-Godal, Q.; Simões-Pires, C.; Marcourt, L.; Carrupt, P.A.; Cuendet, M.; Paulo, M.Q.; et al. Antifungals and acetylcholinesterase inhibitors from the stem bark of Croton heliotropiifolius. Phytochem. Lett. 2014, 10, lxxxviii–xciii. [Google Scholar] [CrossRef]
- Sheng, C.; Che, X.; Wang, W.; Wang, S.; Cao, Y.; Yao, J.; Miao, Z.; Zhang, W. Design and synthesis of antifungal benzoheterocyclic derivatives by scaffold hopping. Eur. J. Med. Chem. 2011, 46, 1706–1712. [Google Scholar] [CrossRef]
- Tu, J.; Li, Z.; Jiang, Y.; Ji, C.; Han, G.; Wang, Y.; Liu, N.; Sheng, C. Discovery of Carboline Derivatives as Potent Antifungal Agents for the Treatment of Cryptococcal Meningitis. J. Med. Chem. 2019, 62, 2376–2389. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Liu, W.; Liu, N.; Zhang, Y.; Dong, G.; Liu, Y.; Li, Z.; He, X.; Miao, Z.; et al. Novel Carboline Derivatives as Potent Antifungal Lead Compounds: Design, Synthesis, and Biological Evaluation. ACS Med. Chem. Lett. 2014, 5, 506–511. [Google Scholar] [CrossRef]
- Singh, R.; Jaisingh, A.; Maurya, I.K.; Salunke, D.B. Design, synthesis and bio-evaluation of C-1 alkylated tetrahydro-β-carboline derivatives as novel antifungal lead compounds. Bioorganic Med. Chem. Lett. 2020, 30, 126869. [Google Scholar] [CrossRef]
- Li, H.; Dang, H.T.; Li, J.; Sim, C.J.; Hong, J.; Kim, D.K.; Jung, J.H. Pyroglutamyl dipeptides and tetrahydro-β-carboline alkaloids from a marine sponge Asteropus sp. Biochem. Syst. Ecol. 2010, 38, 1049–1051. [Google Scholar] [CrossRef]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.C.; van Soest, R.W.M. 5-Hydroxytryptamine-Derived Alkaloids from Two Marine Sponges of the Genus Hyrtios. J. Nat. Prod. 2002, 65, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kubota, T.; Fromont, J.; Kobayashi, J.I. Zamamidines A and B, New Manzamine Alkaloids from the Sponge Amphimedon Species. Org. Lett. 2009, 11, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Pilli, R.A.; Rawal, V.H. Enantioselective Total Syntheses of (+)-Arborescidine A, (−)-Arborescidine B, and (−)-Arborescidine, C.J. Org. Chem. 2004, 69, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, R.M.; Jompa, J.; Tahir, A.; Ireland, C.M. Trypargine Alkaloids from a Previously Undescribed Eudistoma sp. Ascidian. J. Nat. Prod. 1999, 62, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Pénez, N.; Culioli, G.; Pérez, T.; Briand, J.F.; Thomas, O.P.; Blache, Y. Antifouling Properties of Simple Indole and Purine Alkaloids from the Mediterranean Gorgonian Paramuricea clavata. J. Nat. Prod. 2011, 74, 2304–2308. [Google Scholar]
- Ovenden, P.B.S.; Nielson, J.L.; Liptrol, H.C.; Willis, H.R.; Tapiolas, M.D.; Wright, D.A.; Motti, A.C. Callophycin A, a cytotoxic tetrahydro-β-carboline from the red alga Callophycus oppositifolius. Phytochem. Lett. 2011, 4, 69–71. [Google Scholar] [CrossRef]
- Badre, A.; Boulanger, A.; Abou-Mansour, E.; Banaigs, B.; Combaut, G.; Francisco, C. Eudistomin U and Isoeudistomin U, New Alkaloids from the Carribean Ascidian Lissoclinum fragile. J. Nat. Prod. 1994, 57, 528–533. [Google Scholar] [CrossRef]
- Lake, R.J.; Blunt, J.W.; Munro, M.H.G. Eudistomins from the New Zealand Ascidian Ritterella sigillinoides. Aust. J. Chem. 1989, 42, 1201–1206. [Google Scholar] [CrossRef]
- Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Rinehart, K.L., Jr. Eudistomins A, D, G, H, I, J, M, N, O, P, and Q, bromo, hydroxy, pyrrolyl and iminoazepino beta-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1526–1528. [Google Scholar] [CrossRef]
- Schupp, P.; Poehner, T.; Edrada, R.; Ebel, R.; Berg, A.; Wray, V.; Proksch, P. Eudistomins W and X, Two New β-Carbolines from the Micronesian Tunicate Eudistoma sp. J. Nat. Prod. 2003, 66, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Chinen, T.; Yoshida, K.; Kudo, S.; Nagumo, Y.; Shiwa, Y.; Yamada, R.; Umihara, H.; Iwasaki, K.; Masumoto, H.; et al. Eudistomin C, an Antitumor and Antiviral Natural Product, Targets 40S Ribosome and Inhibits Protein Translation. Chembiochem 2016, 17, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Mohamed, G.A. Ingenine E, a new cytotoxic beta-carboline alkaloid from the Indonesian sponge Acanthostrongylophora ingens. J. Asian. Nat. Prod. Res. 2017, 19, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.; Zayed, M.F.; Sayed, H.M. Ingenines A and B, Two New Alkaloids from the Indonesian Sponge Acanthostrongylophora ingens. Drug Res. 2015, 65, 361–365. [Google Scholar] [CrossRef][Green Version]
- Gorki, V.; Singh, R.; Walter, N.S.; Bagai, U.; Salunke, D.B. Synthesis and Evaluation of Antiplasmodial Efficacy of β-Carboline Derivatives against Murine Malaria. ACS Omega 2018, 3, 13200–13210. [Google Scholar] [CrossRef]
- Eardley, I.; Cartledge, J. Tadalafil (Cialis) for men with erectile dysfunction. Int. J. Clin. Pract. 2002, 56, 300–304. [Google Scholar]
- Levin, Y.D.; White, R.J. Novel therapeutic approaches in pulmonary arterial hypertension: Focus on tadalafil. Drugs Today 2011, 47, 145–156. [Google Scholar] [CrossRef]
- Shrestha, S.K.; Garzan, A.; Garneau-Tsodikova, S. Novel alkylated azoles as potent antifungals. Eur. J. Med. Chem. 2017, 133, 309–318. [Google Scholar] [CrossRef]
- Ji, H.; Zhang, W.; Zhang, M.; Kudo, M.; Aoyama, Y.; Yoshida, Y.; Sheng, C.; Song, Y.; Yang, S.; Zhou, Y.; et al. Structure-Based de Novo Design, Synthesis, and Biological Evaluation of Non-Azole Inhibitors Specific for Lanosterol 14α-Demethylase of Fungi. J. Med. Chem. 2003, 46, 474–485. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, J.; Zhou, Y.; Li, Y.; Cheng, J.; Zheng, C. Design, synthesis, and antifungal activities in vitro of novel tetrahydroisoquinoline compounds based on the structure of lanosterol 14α-demethylase (CYP51) of fungi. Bioorganic Med. Chem. Lett. 2006, 16, 5285–5289. [Google Scholar] [CrossRef]
- Yao, B.; Ji, H.; Cao, Y.; Zhou, Y.; Zhu, J.; Lü, J.; Li, Y.; Chen, J.; Zheng, C.; Jiang, Y.; et al. Synthesis and Antifungal Activities of Novel 2-Aminotetralin Derivatives. J. Med. Chem. 2007, 50, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Steinbuch, K.B.; Benhamou, R.I.; Levin, L.; Stein, R.; Fridman, M. Increased Degree of Unsaturation in the Lipid of Antifungal Cationic Amphiphiles Facilitates Selective Fungal Cell Disruption. ACS Infect. Dis. 2018, 4, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Neto, V.; Voisin, A.; Héroguez, V.; Grelier, S.; Coma, V. Influence of the Variation of the Alkyl Chain Length of N-Alkyl-β-d-glycosylamine Derivatives on Antifungal Properties. J. Agric. Food Chem. 2012, 60, 10516–10522. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, M.; Rai, N.N.; Sawant, D. Mild and efficient cyanuric chloride catalyzed Pictet–Spengler reaction. Beilstein J. Org. Chem. 2013, 9, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Dóra, L.; Clauder, O. The reduction of 1,2,3,4-tetrahydro-1-oxo-beta-carboline. Acta Pharm. Hung. 1968, 38, 78–83. [Google Scholar]
- Qi, S.H.; Zhang, S.; Yang, L.H.; Qian, P.Y. Antifouling and antibacterial compounds from the gorgonians Subergorgia suberosa and Scripearia gracillis. Nat. Prod. Res. 2008, 22, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Water as an efficient medium for the synthesis of tetrahydro-β-carbolines via Pictet-Spengler reactions. Tetrahedron Lett. 2007, 48, 1379–1383. [Google Scholar] [CrossRef]
- Cheve, G.; Duriez, P.; Fruchart, J.; Teissier, E.; Poupaert, J.; Lesieur, D. Antioxidant activity of pinoline analogues in the LDL oxidation model. J. Mad. Chem. 2002, 11, 361–379. [Google Scholar]
- Barbero, M.; Bazzi, S.; Cadamuro, S.; Dughera, S. o-Benzenedisulfonimide as a reusable acid catalyst for an easy, efficient, and green synthesis of tetrahydroisoquinolines and tetrahydro-β-carbolines through Pictet–Spengler reaction. Tetrahedron Lett. 2010, 51, 6356–6359. [Google Scholar] [CrossRef]
- Ikeda, R.; Iwaki, T.; Iidaa, T.; Okabayashi, T.; Nishi, E.; Kurosawa, M.; Sakai, N.; Konakahara, T. 3-Benzylamino-ß-carboline derivatives induce apoptosis through G2/M arrest in human carcinoma cells HeLa S-3. Eur. J. Med. Chem. 2011, 46, 636–646. [Google Scholar] [CrossRef]
- Ding, L.; Dahse, H.-M.; Hertweck, C. Cytotoxic Alkaloids from Fusarium incarnatum Associated with the Mangrove Tree Aegiceras corniculatum. J. Nat. Prod. 2012, 75, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.B.; Mclsaac, W.M.; Walker, E.K. Inhibitors of monoamine oxidase II. Syntheses of some N-2(9)-Substituted tetrahydro-β-carbolines and evaluation of their inhibitory a ctivities. J. Pharm. Sci. 1968, 57, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Wilson, A.J.; Jenkins, R.P.; Mahale, S.; Chaudhuri, B.; Maréchalc, J.-D.; Sutcliffe, J.M. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org. Biomol. Chem. 2006, 4, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, F.; Stephensen, H. (Cyanomethyl)trialkylphosphonium Iodides: Efficient Reagents for the Intermolecular Alkylation of Amines with Alcohols in Solution and on Solid Phase. J. Org. Chem. 2001, 66, 2518–2521. [Google Scholar] [CrossRef] [PubMed]
- Freter, K.; HugoHübner, H.; Metz, H.; Schroeder, H.D.; Zeile, K. Reactions of 1-aryl-1,2,3,4-tetrahydro-β-carbolines in acid solution. Justus Liebigs Ann. Chem. 1965, 684, 159–187. [Google Scholar] [CrossRef]
- Taechowisan, T.; Chaiseang, S.; Phutdhawong, S.W. Antifungal activity of biphenyls from Streptomyces sp. bo07 against selective phytopathogenic fungi of rice. Int. J. Curr. Res. 2018, 10, 66239–66244. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Tests for Filamentous Fungi; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2002. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Compounds | B. oryzae | F. fujikuroi | F. semitectum | C. lutana | ||||
|---|---|---|---|---|---|---|---|---|
| Zone of Inhibition | MIC 1 | Zone of Inhibition | MIC | Zone of Inhibition | MIC | Zone of Inhibition | MIC | |
| 2a | 0.00 | NT 2 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2b | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | NT |
| 2c | 0.10 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2d | 0.60 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2e | 0.25 ± 0.3 | NT | 0.00 | NT | 0.00 | NT | 0.40 ± 0.0 | NT |
| 2f | 0.40 ± 0.0 | NT | 0.10 ± 0.0 | NT | 0.00 | NT | 0.20 ± 0.0 | NT |
| 2g | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2h | 0.15 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.20 ± 0.1 | NT |
| 3a | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | 870 ± 0.0 | 0.00 | NT |
| 3b | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3c | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3d | 0.30 ± 0.1 | 200 ± 0.1 | 0.1 ± 0.0 | >512 | 0.20 ± 0.0 | >512 | 0.30 ± 0.0 | >512 |
| 3e | 0.60 ± 0.0 | 400 ± 0.0 | 0.20 ± 0.1 | >512 | 0.25 ± 0.1 | >512 | 0.25 ± 0.2 | 250 ± 0.0 |
| 3f | 0.10 ± 0.0 | 110 ± 0.0 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3g | 0.10 ± 0.0 | 28 ± 0.0 | 0.1 ± 0.0 | >512 | 0.1 ± 0.1 | >512 | 0.15 ± 0.1 | 200 ± 0.0 |
| 3h | 0.10 ± 0.0 | 220 ± 0.1 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3i | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | 250 ± 0.0 |
| 3j | 0.00 | NT | 0.00 | NT | 0.20 ± 0.1 | >512 | 0.00 | NT |
| 3k | 0.35 ± 0.1 | 210 ± 0.0 | 0.00 | NT | 0.20 ± 0.0 | 1700 ± 0.0 | 0.10 ± 0.0 | 270 ± 0.0 |
| Amphotericin B | 0.21 ± 0.1 | 0.78 ± 0.0 | 0.00 | >512 | 0.13 ± 0.1 | >512 | 0.15 ± 0.1 | 0.33 ± 0.1 |
| Compound | Concentration (µg/mL) | Diameter of Colony (cm) of Tested Fungi | |||
|---|---|---|---|---|---|
| B. oryzae | F. fujikuroi | F. semitectum | C. lutana | ||
| None (control) | - | 7.77 ± 0.15 | 5.48 ± 0.08 | 5.17 ± 0.23 | 5.00 ± 0.27 |
| 3g | 1000 | NG 1 (100%) | NG (100%) | NG (100%) | NG (100%) |
| 500 | 0.85 ± 0.05 (89.1%) | 1.30 ± 0.35 (76.3%) | 0.90 ± 0.07 (82.6%) | 0.90 ± 0.12 (82.0%) | |
| 250 | 1.43 ± 0.11 (81.7%) | 2.23 ± 0.08 (59.4%) | 3.08 ± 0.04 (40.6%) | 1.98 ± 0.11 (60.5%) | |
| 100 | 2.48 ± 0.13 (68.2%) | 3.43 ± 0.04 (37.4%) | 5.90 ± 0.21 (−14.0%) | 3.00 ± 0.07 (40%) | |
| Amphotericin B | 0.6 | 3.30 ± 0.14 (57.6%) | 5.68 ± 0.04 (−3.7%) | 2.45 ± 0.17 (52.7%) | 4.03 ± 0.08 (19.5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buaban, K.; Phutdhawong, W.; Taechowisan, T.; Phutdhawong, W.S. Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi. Molecules 2021, 26, 207. https://doi.org/10.3390/molecules26010207
Buaban K, Phutdhawong W, Taechowisan T, Phutdhawong WS. Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi. Molecules. 2021; 26(1):207. https://doi.org/10.3390/molecules26010207
Chicago/Turabian StyleBuaban, Koonchira, Weerachai Phutdhawong, Thongchai Taechowisan, and Waya S. Phutdhawong. 2021. "Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi" Molecules 26, no. 1: 207. https://doi.org/10.3390/molecules26010207
APA StyleBuaban, K., Phutdhawong, W., Taechowisan, T., & Phutdhawong, W. S. (2021). Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi. Molecules, 26(1), 207. https://doi.org/10.3390/molecules26010207