2.1. Sequence Alignment
To gain insight into the definition, structure, and function of the region linking the N- and C-terminal domains of ArgRs, three ArgRs representing divergent interdomain sequences,
E. coli, B. subtilis, and
M. tuberculosis, were chosen for sequence analysis. The failure of simple sequence alignment to correctly align known secondary structural elements of the highly conserved ArgR domain structures (
Figure S1) led to an attempt to adjust the alignment manually, respecting the conserved secondary structures common to all known ArgR domain structures as judged by their overlay in three-dimensional space in structural alignments of available ArgR crystal structures. Minimal gaps were introduced between secondary structural elements as needed to preserve structure alignment and maximize sequence conservation. The resulting alignment (
Figure 1) is also fully consistent with the tertiary structures of the domains; i.e., the tertiary structure does not require any shift between the primary and secondary structures in the alignment. This finding further reinforces that the domain folds are very highly conserved despite the low overall sequence conservation.
The alignment reveals quite limited sequence conservation even within the well-aligned secondary structural elements, and it highlights significant differences in the region of the interdomain linker. BsArgR and MtArgR present irregularly structured segments of 8 and 14 divergent residues, respectively, followed by an alpha helix of 11 residues, α4. No more than one identical residue at a time can be aligned between the structurally aligned α4 helices conserved between the two organisms, in any frame of alignment. This fact suggests that α4 may have evolved independently in the two organisms, pointing to the possibility of evolutionary pressure to preserve a helix in this position. The positioning of EcArgR and VvArgR linker sequences with respect to α4 in
Figure 1 is arbitrary, and no frame of alignment would more strongly support the existence of an α4 helix equivalent in these two. For simplicity, the secondary structures in all these ArgRs are numbered as if α4 is present. Because no structure is known for intact EcArgR it is formally possible that despite very low helical propensity, its linker region adopts a helical segment similar to BsArgR and MtArgR α4. However, it seems more likely that two main groups of ArgR linker types exist, with and without helix α4. If there is covariance elsewhere in the primary structure when α4 is present vs. absent, it is hardly evident from inspection of
Figure 1. However, it may be significant that only EcArgR and VvArgR, which appear to lack α4, both have an Arg residue in α5. Arg110 in EcArgR is a key driver of trimer rotation, and BsArgR and MtArgR have no positional equivalent of EcArgR Arg110. Finally, the only completely conserved sequence of more than two residues that is common to all four ArgRs is GlyAspAsp in the tight turn between strands 5 and 6, containing Asp128 of EcArgR that interacts with Arg110 to drive rotation. The alignment of
Figure 1 makes it even more remarkable that BsArgR can substitute for EcArgR function in vivo [
6].
2.2. Phylogenetic Analysis
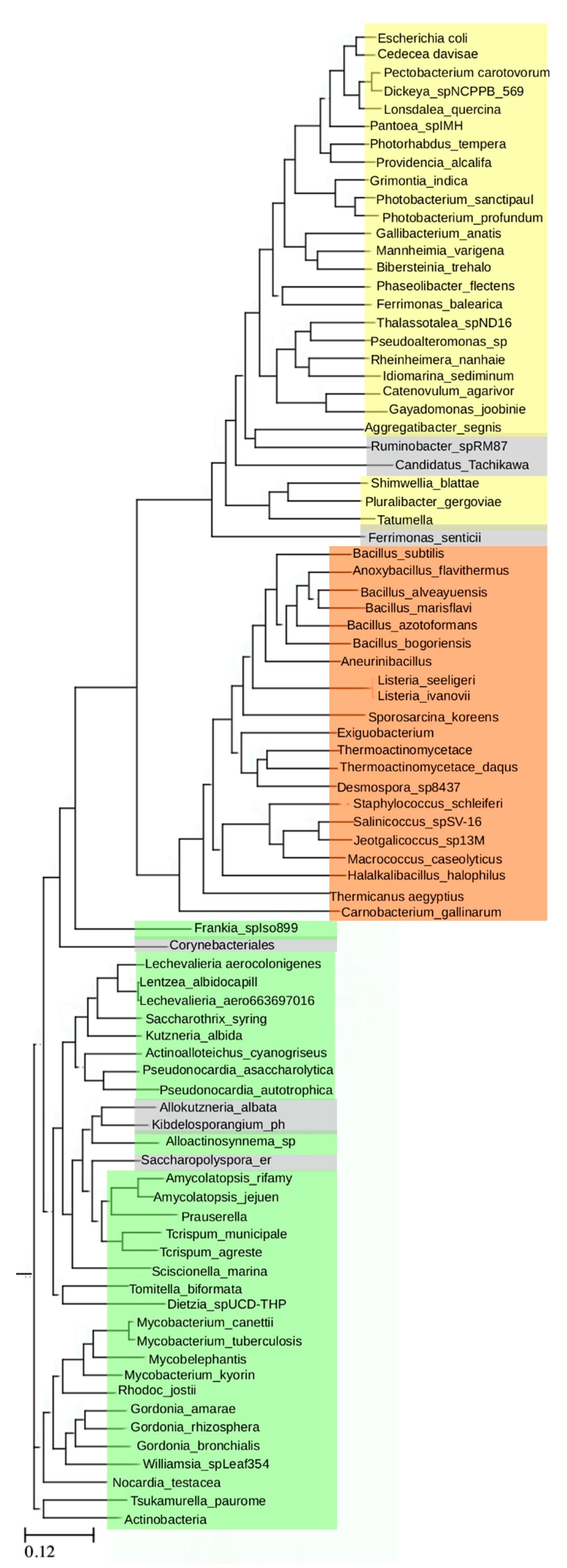
These results prompted further analysis of homologies among more distantly related bacterial ArgRs. The three ArgRs representing divergent linker regions, Ec, Bs, and Mt, were each used in a separate BLAST (Basic Local Alignment Search Tool) search to identify its homologs independently of the other two ArgRs; VvArgR was not used to avoid bias due to its high similarity to EcArgR. The query sequence in each case was the full-length ArgR, i.e., each search was conducted without definition of any motifs or domains, nor with any specific focus on the linker region itself. Each search allowed for 20,000 hits, and among these hits each search also found the other two ArgRs with e-values in the range of 10−20 to 10−30, as expected from their limited sequence identities. From each search a set of homologs was then selected comprising the most distant sequences as judged by e-value that were still annotated as ArgRs, and excluding trivial repetitions such as isoforms or mutants, resulting in 29 distant homologs for EcArgR, 21 for BsArgR, and 34 for MtArgR. The final list comprises these 84 ArgRs. When taking EcArgR as the query the list spans an e-value range from 10−100 to 10−7 and a sequence identity range from 92% to 25%, thus representing a wide range of diversity of ArgRs. This set of 84 homologs was used to construct a phylogenetic tree using standard methods.
The resulting tree (
Figure 2) reveals three non-overlapping groups; these are defined by distinct sequence motifs. The EcArgR group is defined by a motif in the L-arg binding site,
107LIA
R--D
113 in α5, that includes Arg110 (bold), one of the two residues (with Asp128) driving trimer rotation in apoEcArgRC. This motif is shared by VvArgR, as shown in
Figure 1. The BsArgR and MtArgR groups share related but distinct sequence motifs in α4 of the linker region,
75K--R---D
82 in BsArgR and
86R--R---E
103 in MtArgR. The pattern of spacing of these charged residues is conserved, and the motifs align in the structure-based sequence alignment of
Figure 1. The three motif groups are mutually exclusive, i.e., linker regions in the EcArgR group have no sequences corresponding to the α4 motif, and α5 helices in the BsArgR and MtArgR groups do not have the EcArgR-group sequence motif, although the equivalent of EcArgR Asp128, but not Arg110, is present in most. A few outliers (grey in
Figure 2) in the BsArgR or EcArgR groups lack the motif according to simple sequence analysis, suggesting that more than three groups may exist, as does the fact that even an Asp128 equivalent is missing in some. The finding that the BsArgR and MtArgR groups lack an equivalent of Arg110 suggests that the trimer rotation observed in the crystal structures of their canonical members, BsArgR and MtArgR, has a different origin than the rotation observed by MD in apoEcArgRC.
2.3. Molecular Dynamics Simulations
In the earlier molecular dynamics simulations of the
E. coli protein that revealed its rotational oscillation, only the oligomerization domains (EcArgRC) could be analyzed, as no full-length intact structure of EcArgR was available then (nor even now; PDB, February, 2020). When starting this comparative work on
B. subtilis it seemed therefore logical to analyze its oligomerization domains only. However, when apoBsArgRC oligomerization domains (prepared from the intact apoBsArgR crystal structure (PDB ID: 1F9N) by removing the N-terminal domains with or without the linker region) were simulated, the hexamers soon become unstable (data not shown); thus no equilibrium state could be reached with apoBsArgRC, and all simulations were instead performed using the intact apoBsArgR structure (PDB ID: 1F9N), thus offering for the first time the possibility to also evaluate the dynamic mechanism of transcriptional activation by L-arg. To represent holoBsArgR a structural model was prepared in silico by superimposing the crystal structure of apoBsArgR (PDB ID: 1F9N) on that of holoBsArgRC (PDB ID: 2P5M) to guide placement of six molecules of L-arg manually in the six empty binding sites at the trimer-trimer interface (
Figure 3), followed by energy minimization. Docking was facilitated by the fact that the BsArgR L-arg binding site contains no resident Arg residue equivalent to EcArgR Arg110, which in apo EcArgRC blocks access to the ligand. The local structures of apoBsArgR and holoBsArgRC are essentially identical, enabling placement of L-arg ligands into the empty binding sites of apoBsArgR by superposition with holoBsArgRC. Consistent with this fact, both apo- and holoBsArgR were well-behaved in simulations of up to 2 µsec.
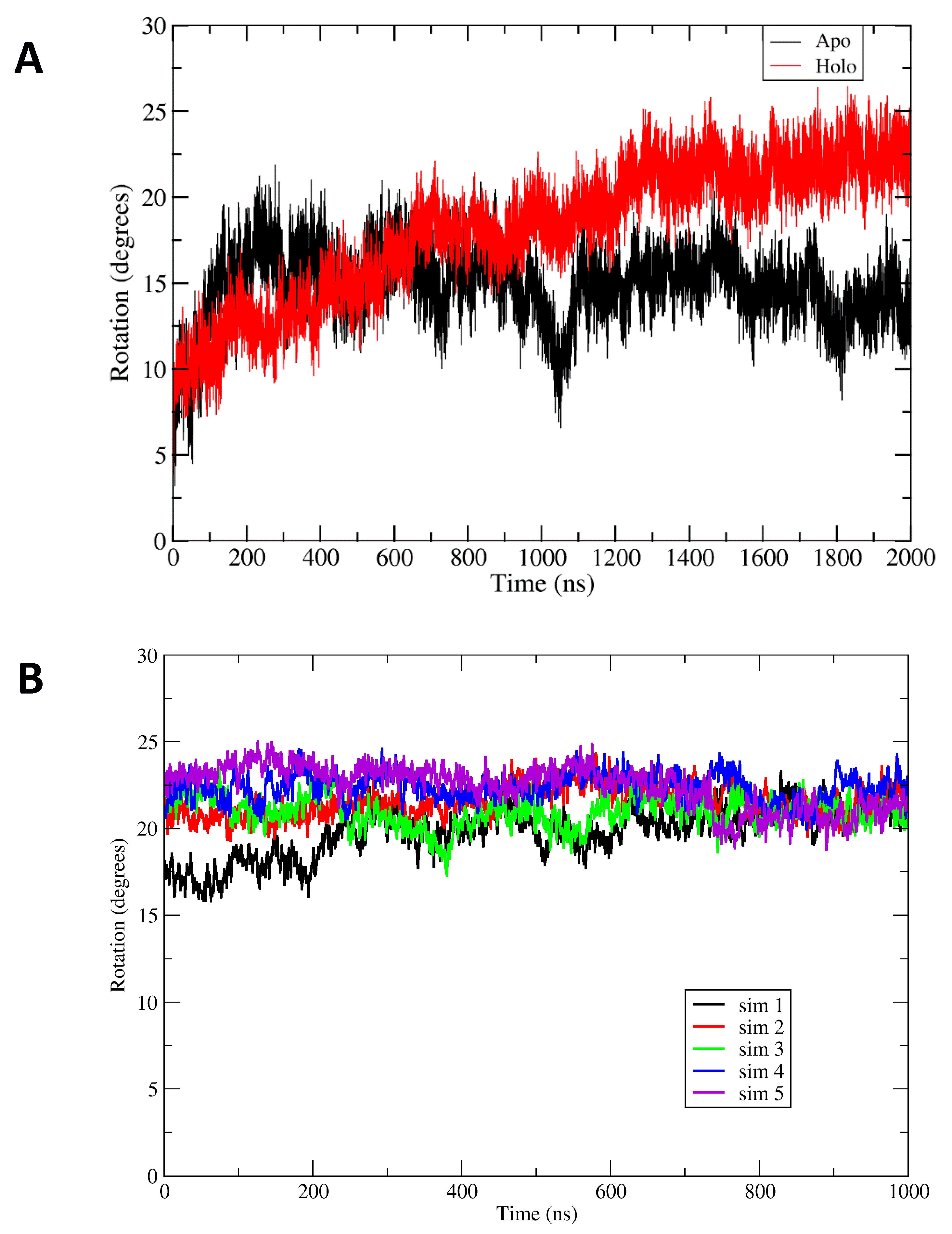
Comparison of the apoBsArgR and holoBsArgR trajectories shows that the two systems differ from each other and from the starting structures. For both proteins, a rotational shift of one trimer with respect to the other occurs very early, followed by continued evolution of the rotational angle toward an equilibrium value that persists for up to 3 µsec (
Figure 4). Trimer-trimer rotation was quantified using an in-house script [
14] as described in Methods. The initial degree of rotation in apoBsArgR crystals is 1.2° (and no crystal structure is available for holoBsArgR, which therefore has the same initial rotation as apoBsArgR from which it was prepared). During 2 µsec simulations both apo- and holoBsArgR show a clockwise rotational shift about the 3
2 axis compared to the initial structure. In the last 500 ns of the trajectories the average degree of rotation is ~14° for apoBsArgR and ~22° for holoBsArgR. The difference of ~8 degrees between apo- and holoBsArgR is similar to the difference between apo- and holoEcArgRC reported in MD simulations [
14].
However, unlike EcArgRC, where the apoprotein exhibits strong rotational oscillation that is driven by formation and release of Arg110–Asp128 salt bridges across the empty ligand-binding site and is damped upon binding of L-arg, both apo- and holoBsArgR display no rotational oscillation. This finding is consistent with the fact that BsArgR has no residue corresponding to EcArgRC Arg110. The variance of ± 5–6 degrees observed in the time courses of
Figure 4 is not rotational oscillation of trimers relative to each other. Principal components analysis and calculation of the eigenvectors show that the motions contributing to the observed variances correspond to uncoordinated motions of individual subunits and not to rotation. This result comports with PCA analysis of
E. coli ArgR systems (Strawn et al., 2010). The uncoordinated motions reflect wobbling motions of monomers within trimers, a more complex and rapid multidimensional movement that contributes to apparent variation in the calculated angle of rotation; this interpretation is supported by results shown below for a mutant that does not undergo rotation. The more extreme deviations from mean rotational angle observed for apoBsArgR reflect slow motions that permit the very large system to occasionally sample states with limited rotation (~1020 ns) and states with holo-like rotational angles (~1500 ns).
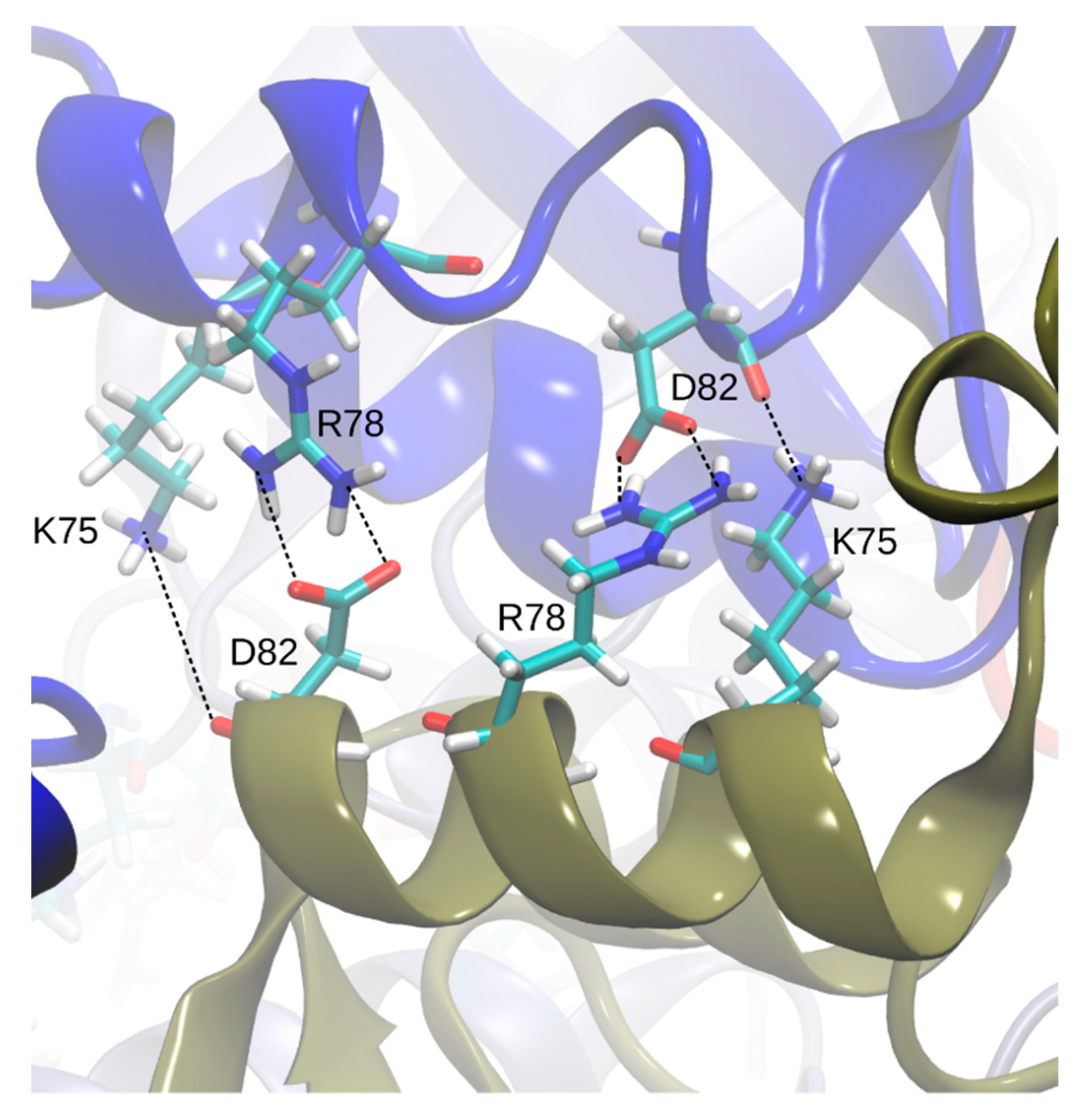
Visual screening of the apo- and holoBsArgR trajectories for potential rotation-driving interactions suggested that the linker-region sequence motif that defines the Bs phylogenetic branch (K75--R78---D82,
Table 1) plays a role in the rotational shift. Over the course of both apo- and holoBsArgR trajectories, inter-trimer interactions form between these residues located on helix α4 from a subunit on one trimer and the symmetry-equivalent residues of antiparallel helix α4′ from the closest subunit in the other trimer (
Figure 5). The interactions comprise salt bridges between oppositely charged residues that are brought within hydrogen-bonding distance by rotation: Lys75: Nζ with Asp82: Oδ; Lys75: Nζ with Asp82: O; and Arg78 Nη with Asp82: Oδ. In the initial state before any rotation occurs (i.e., at zero time in
Figure 4), the sidechain functional groups of these residues are too far apart to be consistent with hydrogen-bond formation, as observed in the apoBsArgR crystal structure from which both protein systems were prepared, even though the distance between their corresponding Cα atoms would permit hydrogen bonding by the extended functional groups of these long sidechains. The unexpected observation that fully charged sidechains within hydrogen-bonding distance do not interact echoes that in apoEcArgRC, where Arg110 and Asp128 sidechains are close enough to form a doubly hydrogen-bonded salt bridge across each empty L-arg binding site, but do not do so [
3], which has been suggested to reflect the very high salt concentration during crystallization [
13]. In holoBsArgRC (PDB ID: 2P5M) the sidechains of Arg78 and Asp82 are ~3 Å apart and thus can be presumed to interact, although this crystal structure was refined as a trimer, and the hexamer was prepared by a symmetry operation [
10].
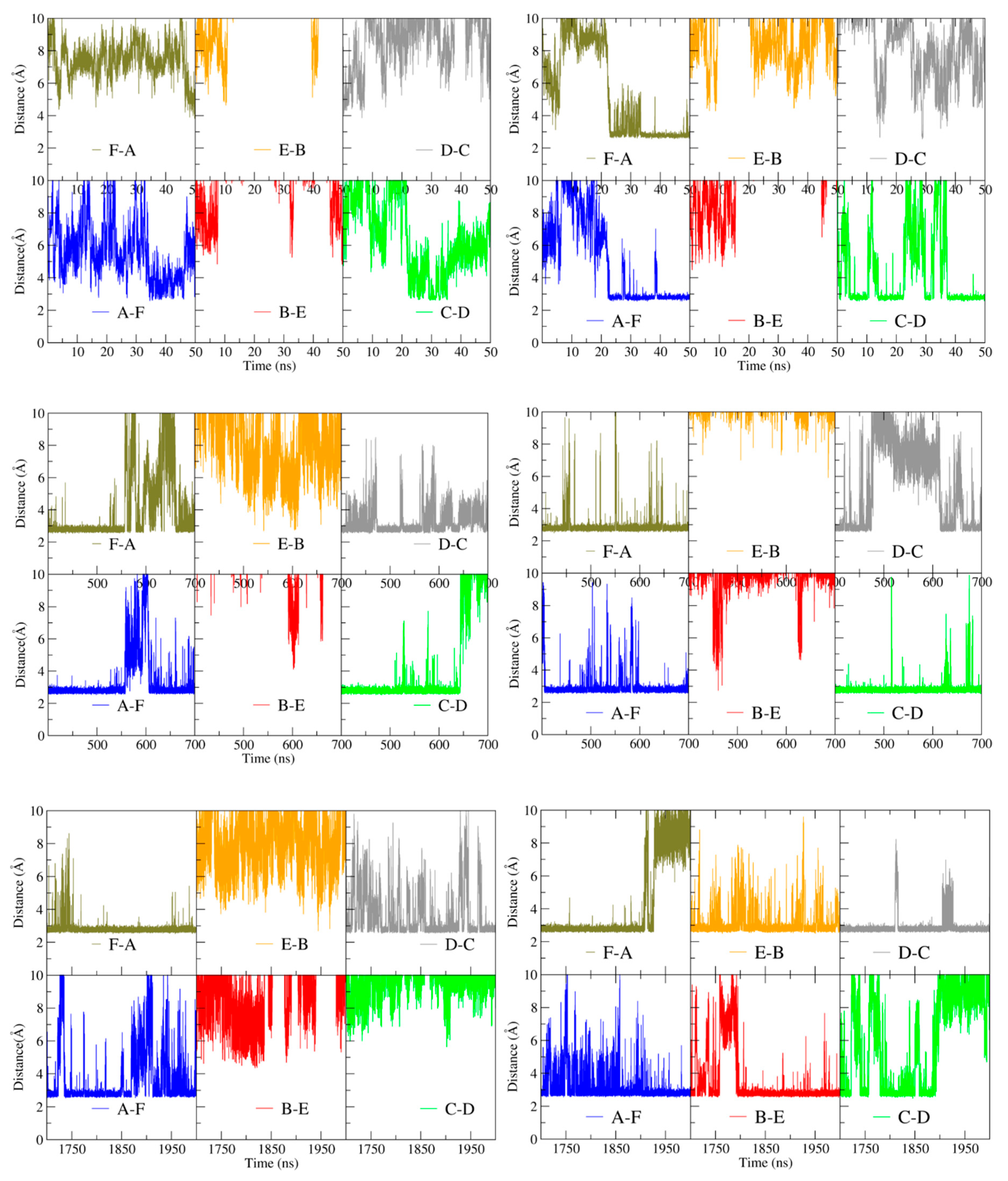
The identity of the interacting residues at the inter-trimer interface does not differ between apo- and holoBsArgR, consistent with the fact that helix α4 is far removed from the L-arg binding site (~17 Å throughout the simulations). However, the total number and persistence of inter-trimer interactions between the α4 residues increase over the course of both simulations, correlated with the degree of rotation. The interactions were quantified by monitoring the distance between the guanidino nitrogen atoms of Arg78 and the carboxylate oxygen atoms of Asp82 (
Figure 6) over the time course of
Figure 4A.
Figure 6 presents three selected time ranges that represent different extents of rotation; the full time course is shown in
Figure S2. The minimum possible inter-atom distance, ~3 Å, corresponds to hydrogen-bond occupancy.
In the first ~20 ns of the simulations, when only the initial rotational shift of ~10 degrees has occurred, very few close-approach distances occur for either protein, and none is persistent (upper panels). Between ~20 and ~50 ns, as rotation begins to evolve, some close-approach distances are populated, especially in holoBsArgR. Between 400 and 700 ns (middle panels) as rotation continues to evolve with a common mean rotational angle for both proteins in
Figure 4A, Arg78–Asp82 hydrogen-bond occupancies are also similar in both proteins, with four of the six possible bonding pairs formed most of the time. During the final 300 ns of each trajectory (lower panels), when each protein achieves its full extent of rotation in
Figure 4A, hydrogen-bond occupancies reach their maximum for holoBsArgR with ~22 degrees rotation and nearly full bond occupancy (seven interactions in total [data not shown], four Arg78–Asp82 sidechain and three Lys75–Asp82 backbone), and with ~14 degrees rotation and partial occupancy for apoBsArgR (three Arg78–Asp82 sidechain interactions). The much lower number of trimer-trimer interactions in apoBsArgR may explain the hexamer instability that was observed in early simulations lacking the N-terminal domains.
In addition to residues of the α4 helix, the L-arg ligand plays a direct role in enlarging the rotational angle of holoBsArgR to its fullest extent. Before the rotational angle reaches ~15 degrees the distance between the Cα carbon of Asp125 (the equivalent of EcArgR Asp128) and the position of the L-arg guanidino group is too large for interaction with the Asp125 sidechain, which faces toward the solvent (data not shown). As the rotational angle approaches ~20 degrees the Asp125 sidechain begins to point toward the position of the L-arg guanidino group in holoBsArgR simulations only. At this point the electrostatic complementarity and rotation angle begin to act in parallel in holoBsArgR, resulting ultimately in a doubly hydrogen-bonded salt bridge between the Asp125 carboxylate and the L-arg guanidino group, and with a maximum rotational angle of ~22 degrees. These results indicate that the trimer-trimer interface residues in the α4 helix, and their mutual interactions promoting rotation, are linked to L-arg binding, as expected from the fact that the ligand-binding sites span the trimer interface. Importantly, however, to reach the maximum degree of rotation and full salt-bridge occupancy, the N-terminal domains must be liberated from their interactions with the C-terminal domains, as discussed below.
If the interactions between residues of the α4 helix indeed drive the rotation observed in apo- and holoBsArgR, then eliminating the interacting residues is expected to eliminate trimer-trimer rotation. To test this prediction the triple-mutant Lys75Ala/Arg78Ala/Asp82Ala, in which all three charged sidechains are replaced by the alanine methyl group, was prepared in silico and simulations were performed for mutant apo- and holoBsArgR. Both apo and holo triple mutants show a much smaller initial rotational shift (~5 degrees) with little or no further evolution of the mean rotational angle over ~1 µs (
Figure S3). The small initial shift may reflect minor optimization of trimer-trimer interactions relative to the crystal structure. The variance observed in
Figure 4 for wildtype BsArgR is also observed for the triple mutant, affirming that it is not due to rotational oscillation, which occurs in neither mutant nor wildtype BsArgR. In the holoBsArgR triple mutant, two L-arg ligands leave the binding site within the first 100 ns of simulation, and one more ligand leaves after 200 ns (data not shown), indicating that the system is not in an L-arg binding-competent state and suggesting that in further time all six L-arg ligands may dissociate. This result can be understood by considering that when wildtype holoBsArgR is prepared by docking L-arg into the apoBsArgR structure, the ligand finds a binding-competent conformation among the conformational ensemble, and the sampled conformational space can further approach binding-compatible conformations via the rotational shift. When the rotational shift does not occur, as in the triple mutant, L-arg binding-compatible conformations remain rare.
2.4. Consequences of L-arg Binding
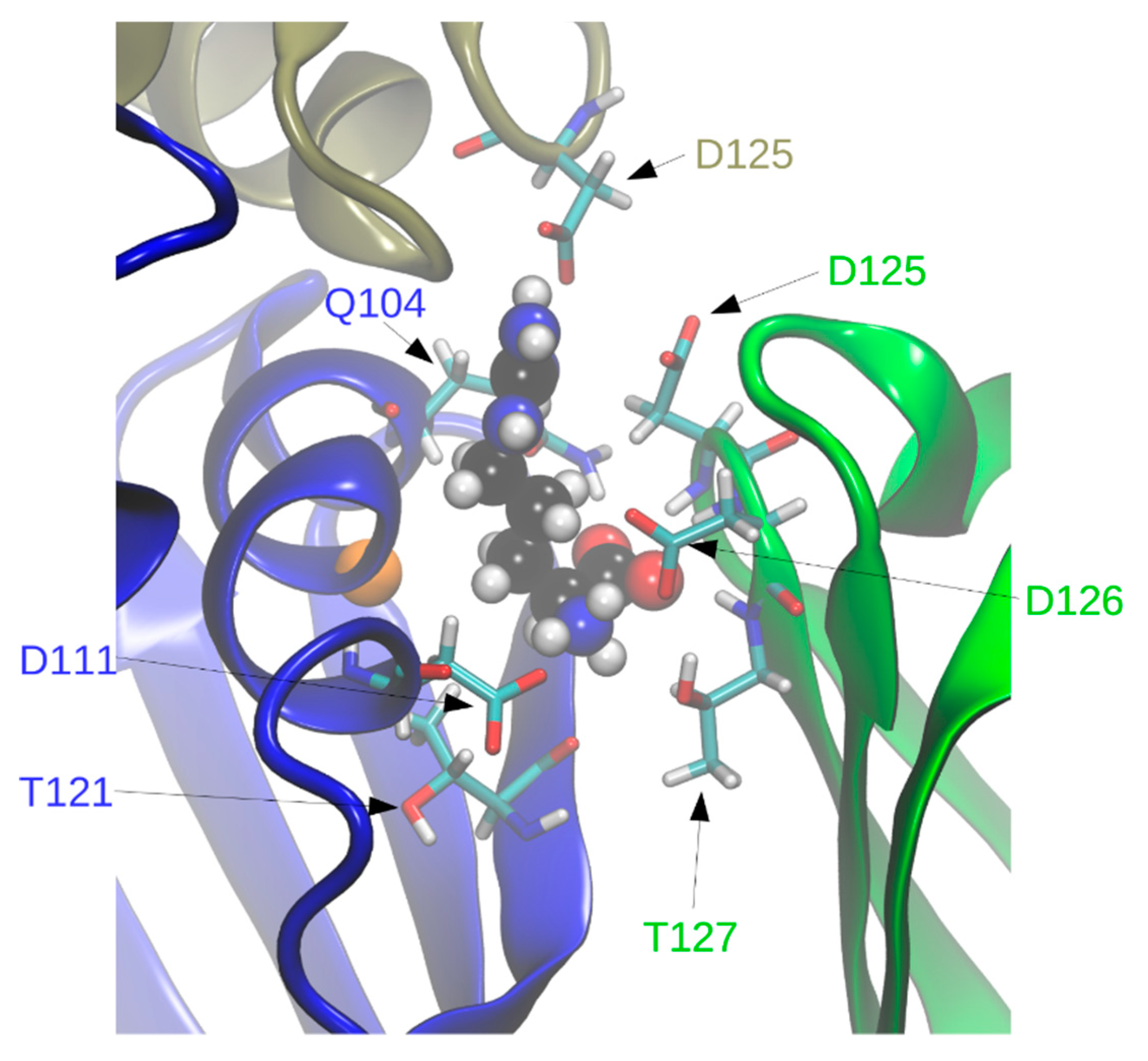
In holoBsArgR simulations each L-arginine ligand is surrounded by residues from three subunits that form each binding site (
Figure 7), consistent with the crystal structure. Seven of the surrounding residues present numerous sidechain functional groups that are appropriate for hydrogen bonding with functional groups of the L-arg ligand, and ten of these functional groups remain within hydrogen-bonding distance during much of the simulation time (
Table 2). The fact that L-arg forms multi-dentate contacts with multiple subunits is similar to the case of the
E. coli protein, although the identity of contacting residues is different except for Asp125 and Asp126, the aligned sequence equivalents of EcArgR Asp128 and Asp129 (compare
Figure 1). Unlike the
E. coli case, no resident Arg residue equivalent to EcArgR Arg110 is displaced by ligand binding to BsArgR. In holoEcArgR the very extensive network of interactions surrounding the alpha-amino and alpha-carboxylate groups of the L-arg ligand enables the free amino acid to compete effectively with resident residue Arg110 despite the far greater local effective concentration of a covalently attached residue. For both Bs and EcArgR, multi-subunit contacts provide a direct structural means for signaling among subunits upon L-arg binding.
The fact that each ligand has extensive contacts with three subunits spanning both trimers suggests that the binding of L-arg contributes to the stability of the hexameric assembly. Indeed, not only L-arg but also the N-terminal domains and α4 helix appear to be involved in hexamer stabilization, as suggested by the following results with BsArgRC. HoloBsArgRC trimers stay associated, and hexamers remain symmetric, during the entire simulation periods, whether or not the α4 helix is present (data not shown). In contrast, the apoBsArgRC hexamer including α4 does not equilibrate, and although trimers do not separate, the hexamer becomes increasingly asymmetric due to the wobbling motions described above. In apoBsArgRC without the α4 helix, hexamers become asymmetric even more rapidly, and trimers separate within the first 100 ns. These findings may be related to previously published reports that some ArgRs crystallize, and even bind to DNA in solution, as trimers [
4,
7,
15,
16,
17,
18].
Interestingly, in BsArgR the position structurally equivalent to
E. coli Arg110 in helix α5 is occupied by Gly107 (
Figure 7). This observation is consistent with the speculation advanced previously for ArgR [
13], and supported experimentally for the
E. coli tryptophan repressor [
19,
20], that binding sites for amino acid ligands can develop when mutations replace protein residues corresponding to the free amino acid. Such a step in the early evolution of ArgR as the master feedback regulator of the arginine regulon might thus be captured in the comparison of Bs, Ec, and Mt ArgRs presented here. However, the fact that all three ArgRs respond functionally to L-arg binding, even though EcArgR retains residue Arg110 and the others do not, makes it difficult to evaluate which organism may be more similar to the evolutionary progenitor, and which is more similar to the progeny.
2.5. Interactions between N- and C-Terminal Domains
Visual analysis of trajectories reveals that by the end of the simulations the N-terminal domains occupy positions that differ between apo- and holoBsArgR and from each starting structure, and with little or no internal rearrangement within either N-terminal or C-terminal domains. To quantify the domain interactions, distances were measured between potential hydrogen-bonding pairs that can span the N- and C-terminal domains, with distances less than 3 Å during the final 500 ns of the simulations and populated more than 50% of the time scored as interdomain hydrogen-bonding interactions. The identities of the residues and their atoms that come within hydrogen-bonding distance are listed in
Table 3. Of the eight hydrogen-bond pairs that are sampled in apoBsArgR, only three are also sampled in holoBsArgR, and in fewer subunits in all cases. One hydrogen-bond pair, between Arg78 and His49, is sampled uniquely in holoBsArgR, but in only one subunit per hexamer. None of the interactions comprise hydrogen-bonded salt-bridge pairs; rather, each involves at least one uncharged polar backbone atom. Thus, the orientation of N- and C-terminal domains observed in apoBsArgR is enforced by the collective effect of many weak interactions rather than by a few dominant ones. Consequently, once the angle of rotation grows large enough upon L-arg binding, all the weak N- to C-terminal domain bonds are compromised together, releasing the N-terminal domains. It is notable that none of the residues involved in interactions between the N- and C-terminal domains of BsArgR is conserved in EcArgR, which is also consistent with the predominance of backbone atoms in the interactions. Even between BsArgR and Mt ArgR, which share a similar pattern of charged residues in helix α4, some of which are involved in interdomain interactions, no homologous residues are found among the hydrogen-bonding pairs in
Table 3 (data not shown).
These results are consistent with visual inspection of the trajectories, which indicates that when L-arg is bound the N-terminal domains are more mobile and have moved away from the C-terminal domains.
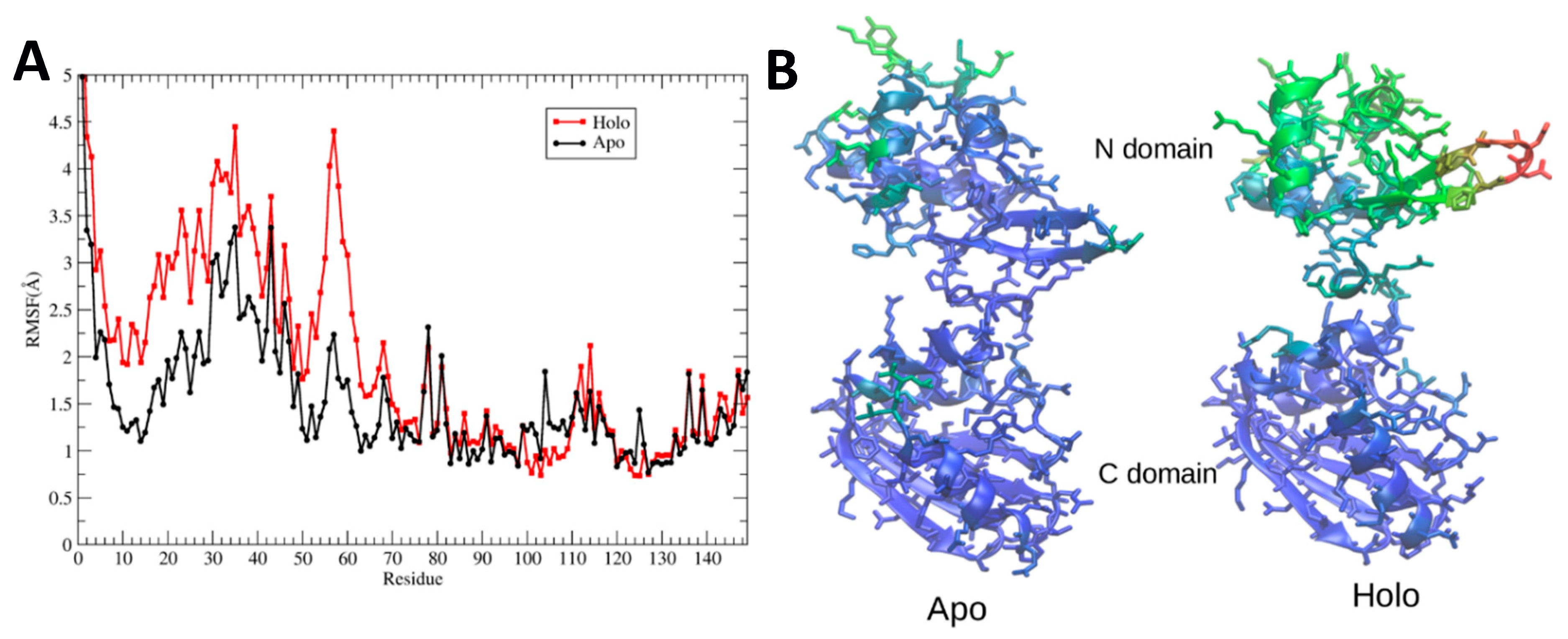
Figure 8A quantifies this behavior by plotting the maximum root-mean-square fluctuations of heavy atoms of apo- and holoBsArgR during the final 500 ns of each simulation. The results clearly show large fluctuations N-terminal to linker residue 66, and very limited fluctuations beyond this point. Root mean square fluctuation (RMSF) values are larger overall for the N-terminal domains of holoBsArgR by approximately 1 Å, and largely parallel to those of apoBsArgR except in the turn between strands 1 and 2, where the RMSF difference is approximately 2.5 Å. The fact that RMSFs for apo- and holoproteins change in parallel supports the view that the N-terminal domains move as units, because parallel RMSFs are not expected if internal structural change occurs.
Figure 8B displays on apo- and holoBsArgR structures the B-factors calculated from the corresponding RMSF values. This comparison shows that in addition to the overall increase in their mobility, the N-terminal domains of holoBsArgR shift slightly away from the C-terminal domains. These results, together with the finding that the triple mutant does not rotate, strongly suggest that rotation is indeed the cause of the smaller number of interactions in holoBsArgR that permits greater mobility of its N-terminal domains compared to apoBsArgR.
This interpretation is also supported by analysis of root mean square deviation (RMSD) values calculated for the whole hexamer and for the individual domains (
Figure S4).
Figure S4A shows that although RMSD values for the C-terminal domain are in the range 1–1.5 Å typically expected in an equilibrated protein system, global domain movements cause RMSD values of ~3 Å for apoBsArgR, and values above 5 Å for holoBsArgR. Thus, RMSD does not reflect fluctuations of internal secondary or tertiary structure within the domains. However the result does suggest that the N-terminal domains of apoBsArgR either are much less mobile than those of holoBsArgR, or that if they are not more mobile then they must have other changes to account for the RMSD. These two possibilities are resolved in
Figure S4B, where superposition of individual domains removes translational and rotational domain movements. Increased RMSD values in the N-terminal domains are instead correlated with fluctuations in the turn between helices α2 and α3, as can be seen by comparing with the RMSF values in
Figure 8. Repositioning of these helices accounts for the RMSD value of ~3Å. Because there is clearly no global unfolding or other changes to the internal domain structure during the simulations, the RMSD results confirm that in holoBsArgR the N-terminal domains move as units, and more freely than in apoBsArgR.
Liberation of the N-terminal domains increases the entropy contribution to the free energy of the system, further contributing to the stability of holoBsArgR. To quantify the entropic differences, the contribution of configurational entropy to the total free energy of each system was calculated from the trajectories as described in Methods. The configurational entropy contribution is increased for holoBsArgR (81,094 ± 3087 J/mol K) compared to apoBsArgR (72,693 J/mol K). This result presumably reflects the higher flexibility observed for the N-terminal domains of holoBsArgR even though lower entropy contributions are expected from the C-terminal domains due to the extensive bonding between each L-arg ligand and three of the six the subunits. Indeed, in holoBsArgR the contribution of the N-terminal domains is higher than in apoBsArgR, 46378 ± 1718 J/mol K vs. 40,445 J/mol K. However, the entropy contribution of the C-terminal domains of holoBsArgR is also increased relative to apoBsArgR, 38,787 ± 1705 J/mol K vs. 35521 J/mol K, although the difference between apo- and holoBsArgR C-terminal domain contributions is smaller than for their N-terminal domains (a gain of 3266 J/mol K for the C-terminal domains vs. 5933 J/mol K for the N-terminal domains). Thus, the increase in entropy of holoBsArgR compared to apoBsArgR is not evenly distributed throughout the protein; rather, the N-terminal domains contribute disproportionately, ~60% of the total entropy gain, although the C-terminal domains also contribute ~40% of the total entropy gain. Please note that the method for calculating entropy contributions of domains and intact proteins does not necessarily result in additive values.
2.6. Approaching DNA-Binding-Competent Conformations
The conformational differences observed between holoBsArgR and apoBsArgR in the positions of their DNA-binding domains may be related to the mechanism of activation of DNA by L-arg binding. To evaluate this possibility the structure of holoBsArgR in the final 500 ns of the simulation was compared with known requirements for DNA binding based on biochemical evidence for EcArgR [
9,
21,
22] and crystal structures of other ArgR-DNA complexes [
7,
10,
11,
12]. In a crystal structure of two isolated N-terminal domains of BsArgR bound to a short palindromic DNA duplex (PDB ID: 2P5L) the major groove in successive openings on one “face” of the DNA is occupied by two N-terminal domains, with key residue Arg43 of each domain contacting symmetry-equivalent guanine residues of the palindromic sequence. In this structure the distance between C-alpha carbons of the two Arg43 residues is ~26 Å, consistent with footprinting and stoichiometric binding data indicating that one ArgR N-terminal domain binds to each half-palindrome, and that pairs of domains bind to each full palindrome [
9,
21,
22]. Those biochemical data also indicate that the tandem palindromes of typical operators in the arg regulon engage four of the six subunits of an ArgR hexamer, requiring positioning of two pairs of N-terminal domains to allow contact with four successive openings of the DNA major groove on one face. The distance between C-alpha carbons of Arg43 residues in N-terminal domain pairs of ArgR was therefore measured over the course of the simulations for apoBsArgR and holoBsArgR.
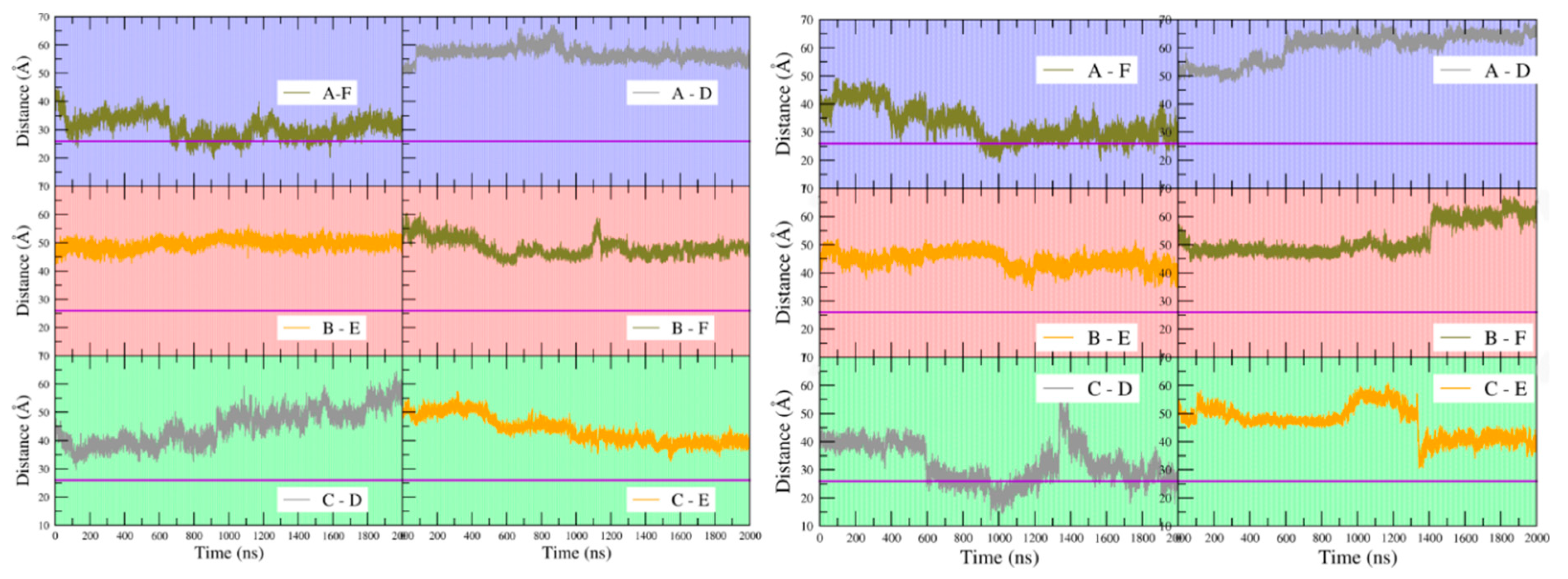
Figure 9 shows that at the beginning of the simulations the distance between Arg43 C-alpha carbons on any pair of subunits is in the range of ~40 to ~50 Å for both apo- and holoBsArgR, consistent with the crystal structure of apoBsArgR from which holoBsArgR was prepared, where the initial distance is ~40 Å. This result shows that the multiple bonding interactions between the N- and C-terminal domains of apoBsArgR (
Table 3) position the domains too far apart for DNA binding. The interdomain distances become smaller or larger over time, and with no obvious covariance between domain pairs for either apo- or holoBsArgR. In apoBsArgR the distance approaches ~26 Å occasionally only for monomers A and F (
Figure 9 left). This finding suggests that apoBsArgR may be on its way toward a DNA-binding-competent state that would be constitutively active for binding to a single palindrome. In holoBsArgR the distance between Arg43 C-alpha carbons on any pair of subunits approaches ~26 Å for subunit pairs A–F and C–D (
Figure 9 right), indicating structures that may be on the way toward binding-competent states for the tandem palindromes of natural Arg operators. In neither apo- nor holoBsArgR is the position of the N-terminal domains fully optimal for DNA binding, however, as helix α3 must also be oriented approximately parallel to the base pair planes in the DNA major groove opening according to known crystal structures. Whole domain movements are expected to be slow (on a μs–ms rather than ns–μs timescale), and on a longer timescale the conformations sampled by holoBsArgR might progress toward DNA-binding-competent states. Whether or not they do so, proximity to a DNA segment bearing suitably spaced palindromic sequences is expected to enforce the distances and orientations required for binding.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}