
Bar Adsorptive Microextraction Coated with Carbon-Based Phase Mixtures for Performance-Enhancement to Monitor Selected Benzotriazoles, Benzothiazoles, and Benzenesulfonamides in Environmental Water Matrices

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Instrumental Operating Conditions
2.2. Optimization of the BAµE-µLD Efficiency
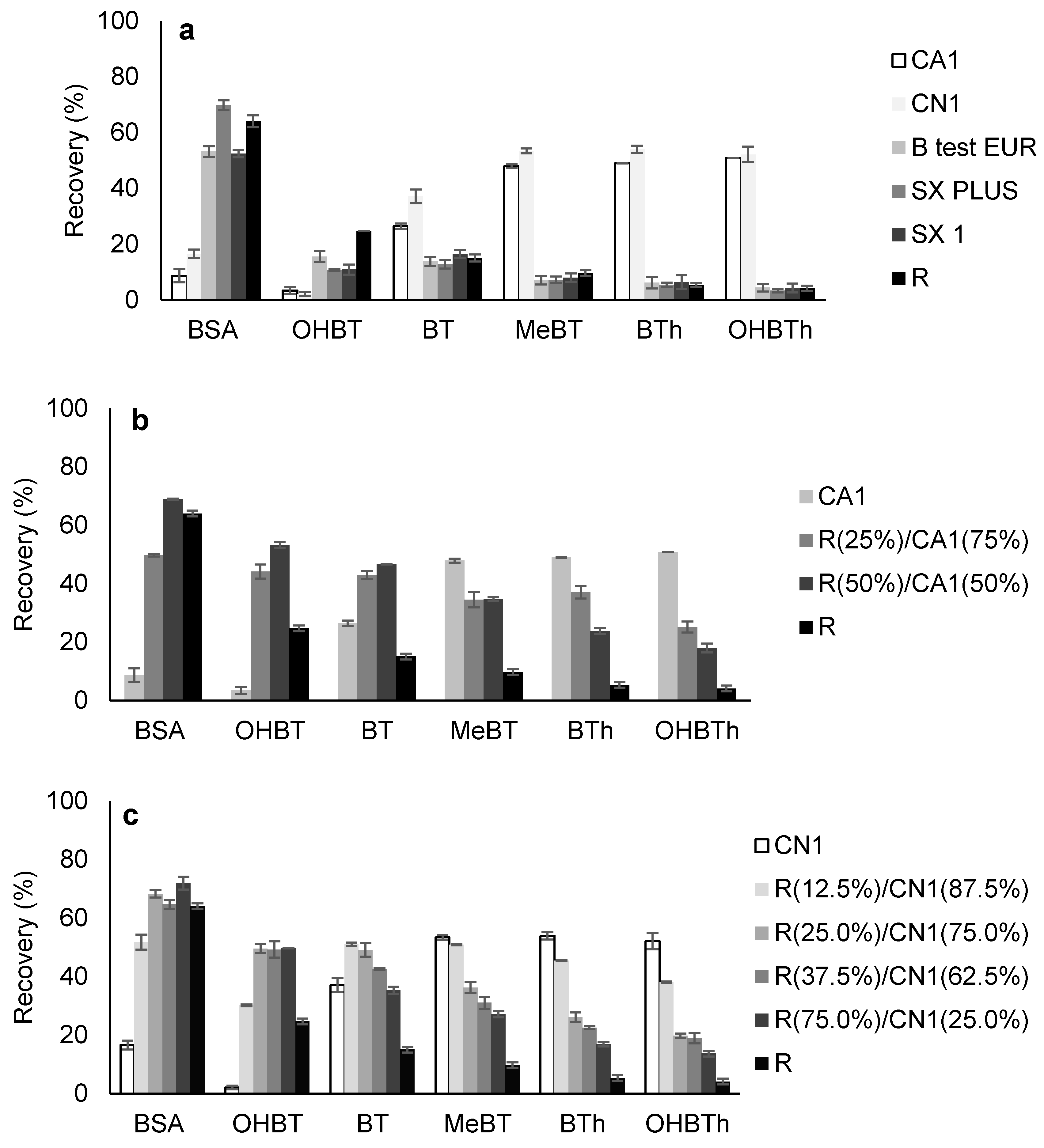
2.2.1. Selection of the Carbon-Based Phase
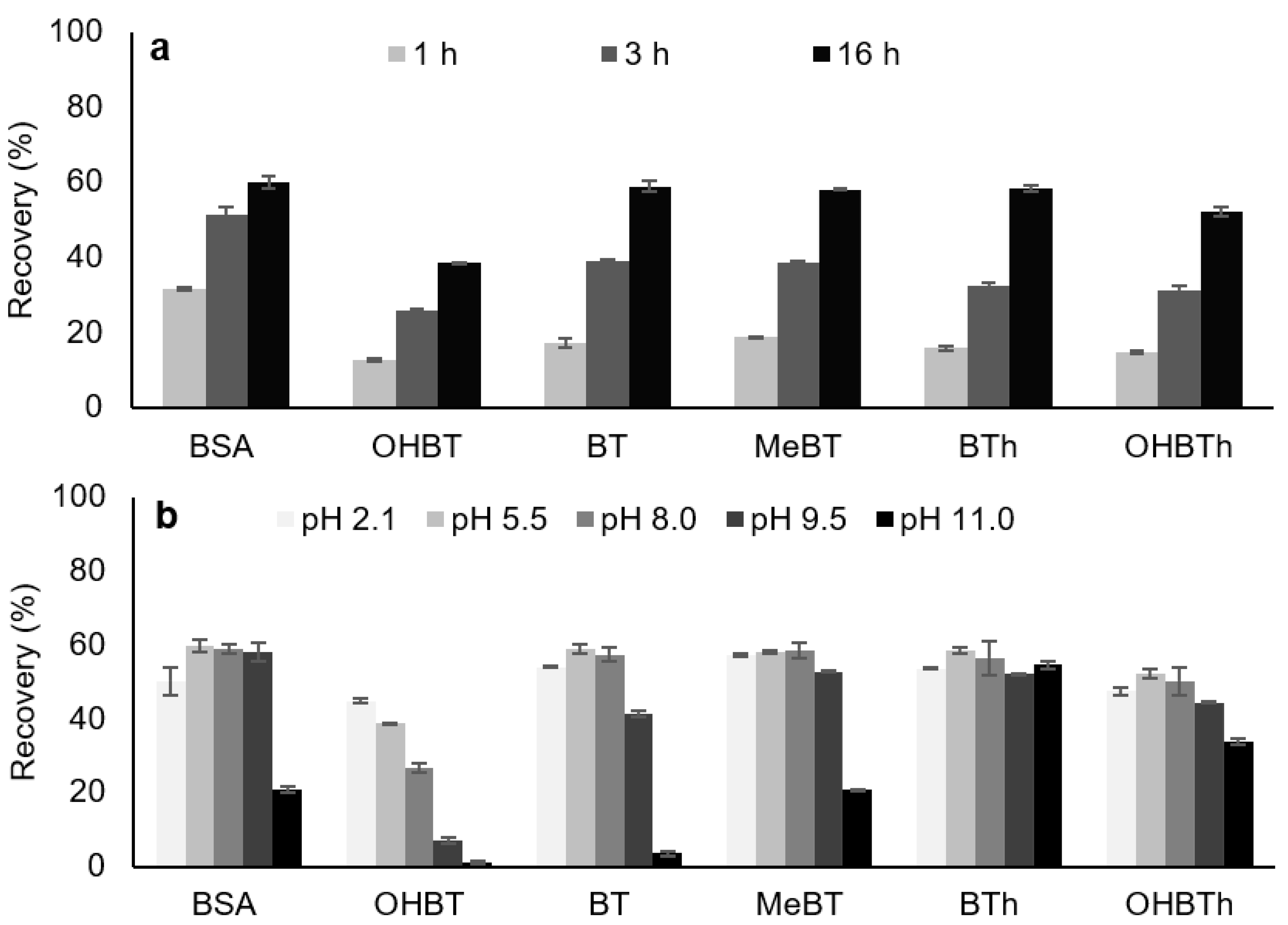
2.2.2. Back-Extraction Stage Conditions
2.2.3. Microextraction Stage Conditions
2.3. Validation Assessment
2.4. Comparison with Other Microextraction-Based Methodologies
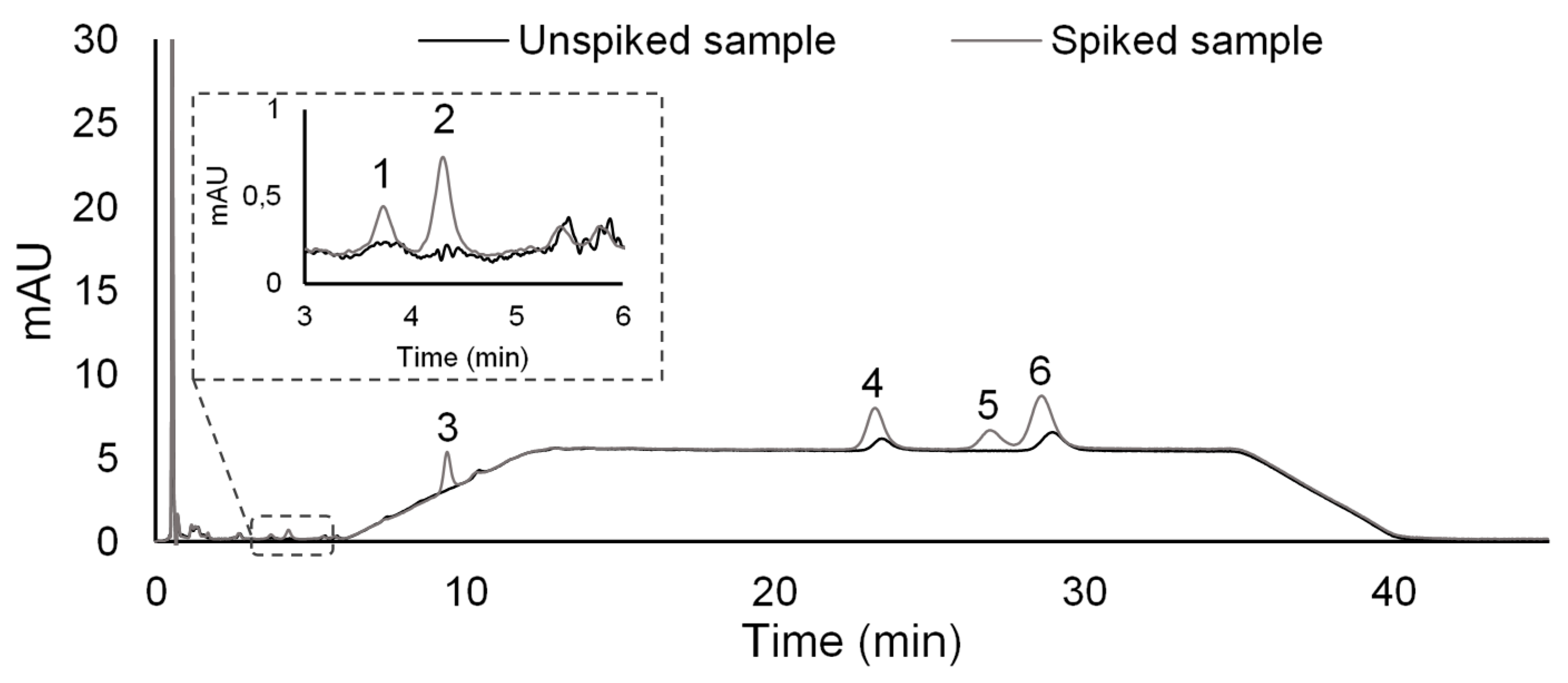
2.5. Application to Environmental Water Matrices
3. Experimental
3.1. Standards and Materials
3.2. BAµE-µLD Assays
3.3. Assays on Real Water Matrices
3.4. Instrumental Set-Up
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Herrero, P.; Borrull, F.; Pocurull, E.; Marcé, R.M. An overview of analytical methods and occurrence of benzotriazoles, benzothiazoles and benzenesulfonamides in the environment. TrAC - Trends Anal. Chem. 2014, 62, 46–55. [Google Scholar] [CrossRef]
- Shi, Z.Q.; Liu, Y.S.; Xiong, Q.; Cai, W.W.; Ying, G.G. Occurrence, toxicity and transformation of six typical benzotriazoles in the environment: A review. Sci. Total Environ. 2019, 661, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Kim, U.J.; Kannan, K. A Review of Environmental Occurrence, Fate, Exposure, and Toxicity of Benzothiazoles. Environ. Sci. Technol. 2018, 52, 5007–5026. [Google Scholar] [CrossRef]
- Richardson, S.D.; Ternes, T.A. Water Analysis: Emerging Contaminants and Current Issues. Anal. Chem. 2018, 90, 398–428. [Google Scholar] [CrossRef] [PubMed]
- Montesdeoca-Esponda, S.; Vega-Morales, T.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J. Extraction and determination methodologies for benzotriazole UV stabilizers in personal-care products in environmental and biological samples. TrAC - Trends Anal. Chem. 2013, 51, 23–32. [Google Scholar] [CrossRef]
- Chisvert, A.; Benedé, J.L.; Salvador, A. Current trends on the determination of organic UV filters in environmental water samples based on microextraction techniques–A review. Anal. Chim. Acta 2018, 1034, 22–38. [Google Scholar] [CrossRef]
- Kraševec, I.; Prosen, H. Solid-phase extraction of polar benzotriazoles as environmental pollutants: A review. Molecules 2018, 23, 2501. [Google Scholar] [CrossRef]
- Kraševec, I.; Prosen, H. Development of a dispersive liquid-liquid microextraction followed by LC-MS/MS for determination of benzotriazoles in environmental waters. Acta Chim. Slov. 2019, 66, 247–254. [Google Scholar] [CrossRef]
- Lu, J.; Wang, M.M.; Wang, Q.; LI, H.P.; Yang, Z.G. Determination of Benzotriazole and Its Derivatives in Aqueous Sample with Air-assisted Liquid-Liquid Microextraction Followed by High-performance Liquid Chromatography. Chinese J. Anal. Chem. 2018, 46, e1817–e1825. [Google Scholar] [CrossRef]
- Fries, E. Determination of benzothiazole in untreated wastewater using polar-phase stir bar sorptive extraction and gas chromatography-mass spectrometry. Anal. Chim. Acta 2011, 689, 65–68. [Google Scholar] [CrossRef]
- Naccarato, A.; Gionfriddo, E.; Sindona, G.; Tagarelli, A. Simultaneous determination of benzothiazoles, benzotriazoles and benzosulfonamides by solid phase microextraction-gas chromatography-triple quadrupole mass spectrometry in environmental aqueous matrices and human urine. J. Chromatogr. A 2014, 1338, 164–173. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon Marvin 6.2.2 2014. Available online: https://chemaxon.com/products/marvin (accessed on 1 April 2020).
- Ahmad, S.M.; Nogueira, J.M.F. High throughput bar adsorptive microextraction: A simple and effective analytical approach for the determination of nicotine and cotinine in urine samples. J. Chromatogr. A 2020, 1615. [Google Scholar] [CrossRef] [PubMed]
- Neng, N.R.; Nogueira, J.M.F. Monitoring trace levels of hydroxy aromatic compounds in urine matrices by bar adsorptive microextraction (BAμE). Anal. Methods 2017, 9, 5260–5265. [Google Scholar] [CrossRef]
- Neng, N.R.; Mestre, A.S.; Carvalho, A.P.; Nogueira, J.M.F. Cork-based activated carbons as supported adsorbent materials for trace level analysis of ibuprofen and clofibric acid in environmental and biological matrices. J. Chromatogr. A 2011, 1218, 6263–6270. [Google Scholar] [CrossRef]
- Neng, N.R.; Mestre, A.S.; Carvalho, A.P.; Nogueira, J.M.F. Powdered activated carbons as effective phases for bar adsorptive micro-extraction (BAμE) to monitor levels of triazinic herbicides in environmental water matrices. Talanta 2011, 83, 1643–1649. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Almeida, C.; Neng, N.R.; Nogueira, J.M.F. Application of bar adsorptive microextraction (BAμE) for anti-doping control screening of anabolic steroids in urine matrices. J. Chromatogr. B Anal. 2014, 969, 35–41. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Mestre, A.S.; Neng, N.R.; Ania, C.O.; Carvalho, A.P.; Nogueira, J.M.F. Carbon-Based Sorbent Coatings for the Determination of Pharmaceutical Compounds by Bar Adsorptive Microextraction. ACS Appl. Bio Mater. 2020, 3, 2078–2091. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Almeida, C.; Neng, N.R.; Nogueira, J.M.F. Bar adsorptive microextraction (BAμE) coated with mixed sorbent phases-Enhanced selectivity for the determination of non-steroidal anti-inflammatory drugs in real matrices in combination with capillary electrophoresis. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2016, 1008, 115–124. [Google Scholar] [CrossRef]
- Almeida, C.; Strzelczyk, R.; Nogueira, J.M.F. Improvements on bar adsorptive microextraction (BAμE) technique–application for the determination of insecticide repellents in environmental water matrices. Talanta 2014, 120, 126–134. [Google Scholar] [CrossRef]
- Mafra, G.; Oenning, A.L.; Dias, A.N.; Merib, J.; Budziak, D.; da Silveira, C.B.; Carasek, E. Low-cost approach to increase the analysis throughput of bar adsorptive microextraction (BAµE) combined with environmentally-friendly renewable sorbent phase of recycled diatomaceous earth. Talanta 2018, 178, 886–893. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Ide, A.H.; Neng, N.R.; Nogueira, J.M.F. Application of bar adsorptive microextraction to determine trace organic micro-pollutants in environmental water matrices. Int. J. Environ. Anal. Chem. 2017, 97, 484–498. [Google Scholar] [CrossRef]
- Almeida, C.; Stepkowska, A.; Alegre, A.; Nogueira, J.M.F. Determination of trace levels of benzophenone-type ultra-violet filters in real matrices by bar adsorptive micro-extraction using selective sorbent phases. J. Chromatogr. A 2013, 1311, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.M.F. Novel sorption-based methodologies for static microextraction analysis: A review on SBSE and related techniques. Anal. Chim. Acta 2012, 757, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.; Nogueira, J.M.F. Comparison of the selectivity of different sorbent phases for bar adsorptive microextraction--application to trace level analysis of fungicides in real matrices. J. Chromatogr. A 2012, 1265, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Neng, N.R.; Nogueira, J.M.F. Development of a bar adsorptive micro-extraction-large-volume injection-gas chromatography-mass spectrometric method for pharmaceuticals and personal care products in environmental water matrices. Anal. Bioanal. Chem. 2012, 402, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.; Rodríguez, I.; Ramil, M.; Cela, R. Polyethersulfone solid-phase microextraction followed by liquid chromatography quadrupole time-of-flight mass spectrometry for benzotriazoles determination in water samples. J. Chromatogr. A 2013, 1299, 40–47. [Google Scholar] [CrossRef]
- Casado, J.; Nescatelli, R.; Rodríguez, I.; Ramil, M.; Marini, F.; Cela, R. Determination of benzotriazoles in water samples by concurrent derivatization-dispersive liquid-liquid microextraction followed by gas chromatography-mass spectrometry. J. Chromatogr. A 2014, 1336, 1–9. [Google Scholar] [CrossRef]
- Pena, M.T.; Vecino-Bello, X.; Casais, M.C.; Mejuto, M.C.; Cela, R. Optimization of a dispersive liquid-liquid microextraction method for the analysis of benzotriazoles and benzothiazoles in water samples. Anal. Bioanal. Chem. 2012, 402, 1679–1695. [Google Scholar] [CrossRef]
- Neng, N.R.; Silva, A.R.; Nogueira, J.M.F. Adsorptive micro-extraction techniques--novel analytical tools for trace levels of polar solutes in aqueous media. J. Chromatogr. A 2010, 1217, 7303–7310. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Compound | LODs(μg L−1) | LOQs(μg L−1) | Recovery (%) ± RSD (%) | |
|---|---|---|---|---|
| Intraday Assays (n = 5) | Interday Assays (n = 9) | |||
| BSA | 1.0 | 3.3 | 59 ± 3 | 58 ± 6 |
| OHBT | 1.4 | 4.0 | 38 ± 9 | 38 ± 9 |
| BT | 1.0 | 3.3 | 56 ± 3 | 56 ± 9 |
| MeBT | 1.2 | 4.0 | 59 ± 2 | 59 ± 2 |
| BTh | 1.2 | 4.0 | 57 ± 6 | 56 ± 9 |
| OHBTh | 1.0 | 3.3 | 53 ± 4 | 53 ± 3 |
| Microextraction Technique | SPME | DLLME | SBSE | DLLME | AALLME | DLLME | SPME | BAμE |
|---|---|---|---|---|---|---|---|---|
| Instrumental System | GC-MS/MS | LC-MS/MS | GC-MS | HPLC-FLD-UV | HPLC-UV | GC-MS | LC-qTOF/MS | HPLC-DAD |
| Sample Type | Tap, river, and effluent waste water | Ground, river, influent, and effluent waste water | Influent waste water | Tap, river, industrial waters, as well as effluent and influent wastewaters | Tap, lake, river, as well as effluent and influent wastewaters | Tap and effluent wastewaters | Tap, as well as influent and effluent wastewaters | Tap, estuarine, rain, and effluent wastewaters |
| Sorbent or Solvent Phase Used for Microextraction | Polyacrylate | Chloroform/Carbon tetrachloride/ACN | Polyacrylate | Tri-n-butylphosphate/methanol | 1-Hexanol | Toluene/ACN | Polyethersulfone | Mixed ACs |
| LODs (μg L−1) | 0.0001–7.5 | 0.04–0.75 | 0.256 | 0.04–2.2 | 0.8–1.4 | 0.007–0.080 a | 0.005–0.1 a | 1.0–1.4 |
| Linear Range(μg L−1) | 1.0–100.0 | 0.01–50.0 | 0–10.0 | 2.4–536.1 | 5.0–10000.0 | 0.05–20.0 | 0.1–50.0 | 5.0–120.0 |
| Precision (%) | ≤ 24.6 | ≤ 44.0 | ≤ 9.8 | ≤ 8.4 | ≤ 7.8 | ≤ 8.0 | ≤ 8.0 | ≤ 9.3 |
| Absolute Recovery (%) | n.a. | 5.0–42.0 | n.a. | 67.4–97.1 | n.a. | 24.0–46.0 | n.a. | 37.9–59.2 |
| Reference | [11] | [8] | [10] | [29] | [9] | [28] | [27] | This work |
| Compounds | Slope | Intercept | r2 | Slope | Intercept | r2 | |
|---|---|---|---|---|---|---|---|
| Tap water | Estuarine water | ||||||
| BSA | 0.3000 | 0.0788 | 0.9938 | 0.2600 | 0.0676 | 0.9913 | |
| OHBT | 0.3000 | 0.0672 | 0.9920 | 0.2600 | 0.0608 | 0.9907 | |
| BT | 1.5600 | 1.1600 | 0.9989 | 5.1200 | 1.3560 | 0.9927 | |
| MeBT | 1.8400 | 1.6944 | 0.9987 | 0.4200 | 3.1112 | 0.9992 | |
| BTh | 0.5000 | 2.0740 | 0.9937 | 0.2000 | 2.1520 | 0.9962 | |
| OHBTh | 1.9800 | 5.5472 | 0.9957 | 0.8800 | 5.1488 | 0.9993 | |
| Rainwater | Waste water | ||||||
| BSA | 0.2800 | 0.0692 | 0.9926 | 0.2400 | 0.0680 | 0.9934 | |
| OHBT | 0.4400 | 0.0992 | 0.9907 | 0.0800 | 0.0314 | 0.9923 | |
| BT | 2.2000 | 1.4620 | 0.9973 | 4.2000 | 1.1480 | 0.9932 | |
| MeBT | 0.6400 | 3.0044 | 0.9988 | 31.0000 | 2.6044 | 0.9991 | |
| BTh | 0.2000 | 2.0480 | 0.9970 | 0.8000 | 2.0320 | 0.9995 | |
| OHBTh | 0.5000 | 4.6128 | 0.9989 | 47.1200 | 4.3584 | 0.9984 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.M.; Calado, B.B.C.; Oliveira, M.N.; Neng, N.R.; Nogueira, J.M.F. Bar Adsorptive Microextraction Coated with Carbon-Based Phase Mixtures for Performance-Enhancement to Monitor Selected Benzotriazoles, Benzothiazoles, and Benzenesulfonamides in Environmental Water Matrices. Molecules 2020, 25, 2133. https://doi.org/10.3390/molecules25092133
Ahmad SM, Calado BBC, Oliveira MN, Neng NR, Nogueira JMF. Bar Adsorptive Microextraction Coated with Carbon-Based Phase Mixtures for Performance-Enhancement to Monitor Selected Benzotriazoles, Benzothiazoles, and Benzenesulfonamides in Environmental Water Matrices. Molecules. 2020; 25(9):2133. https://doi.org/10.3390/molecules25092133
Chicago/Turabian StyleAhmad, Samir M., Bruno B.C. Calado, Mariana N. Oliveira, Nuno R. Neng, and J.M.F. Nogueira. 2020. "Bar Adsorptive Microextraction Coated with Carbon-Based Phase Mixtures for Performance-Enhancement to Monitor Selected Benzotriazoles, Benzothiazoles, and Benzenesulfonamides in Environmental Water Matrices" Molecules 25, no. 9: 2133. https://doi.org/10.3390/molecules25092133
APA StyleAhmad, S. M., Calado, B. B. C., Oliveira, M. N., Neng, N. R., & Nogueira, J. M. F. (2020). Bar Adsorptive Microextraction Coated with Carbon-Based Phase Mixtures for Performance-Enhancement to Monitor Selected Benzotriazoles, Benzothiazoles, and Benzenesulfonamides in Environmental Water Matrices. Molecules, 25(9), 2133. https://doi.org/10.3390/molecules25092133