Effects of the Combination of β-Hydroxy-β-Methyl Butyrate and R(+) Lipoic Acid in a Cellular Model of Sarcopenia

 ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Pharmacological Treatments
2.3. Cell Viability Assay
2.4. Caspase-3 Activity
2.5. Superoxide Dismutase-Inhibitable Superoxide Anion (O2−) Production Evaluated by Cytochrome C Assay
2.6. Protein Extraction and Quantification
2.7. Carbonylated Protein Evaluation
2.8. Western Immunoblot Analysis
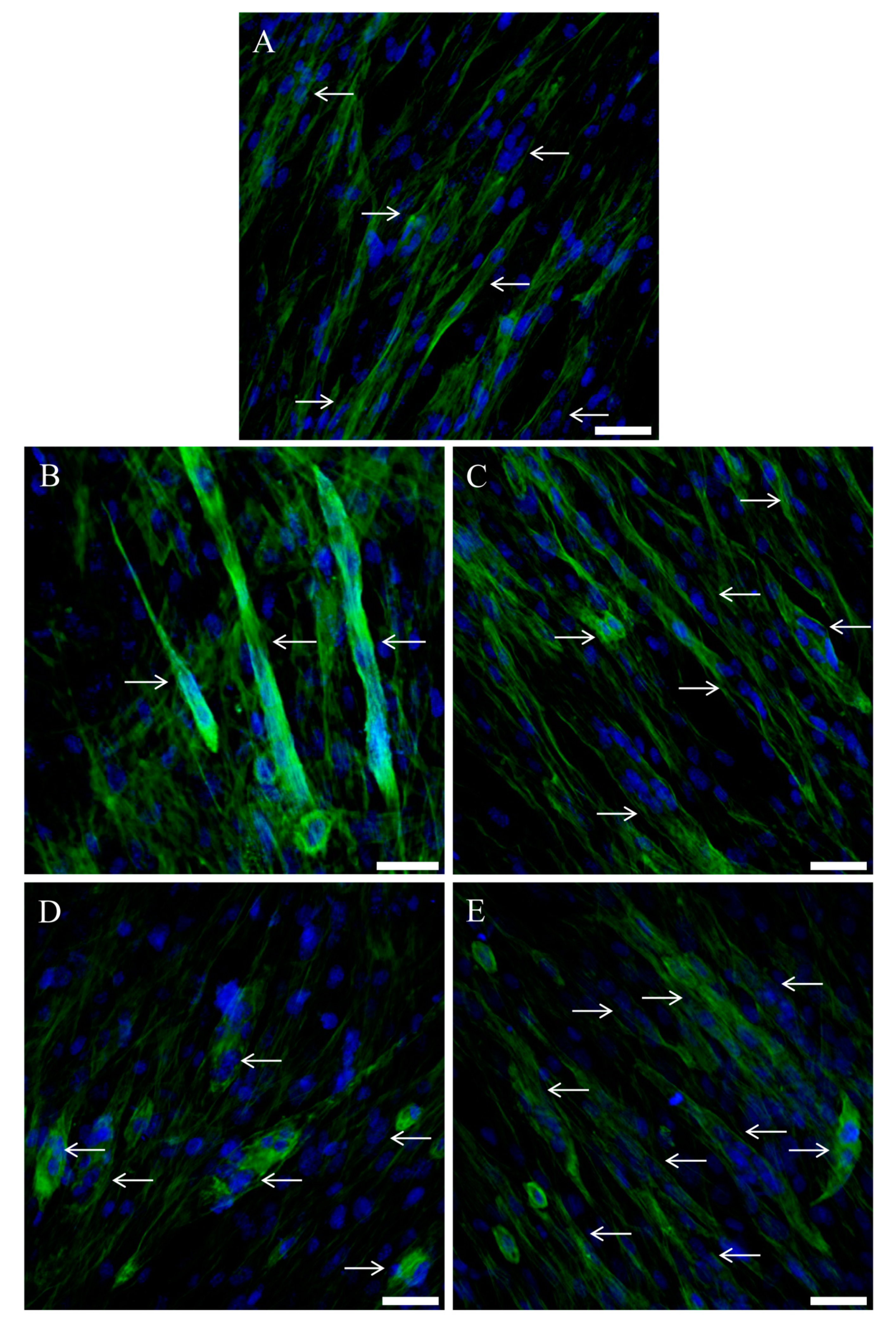
2.9. Immunofluorescence
2.10. Morphologic Evaluations
2.11. Statistical Analysis
3. Results
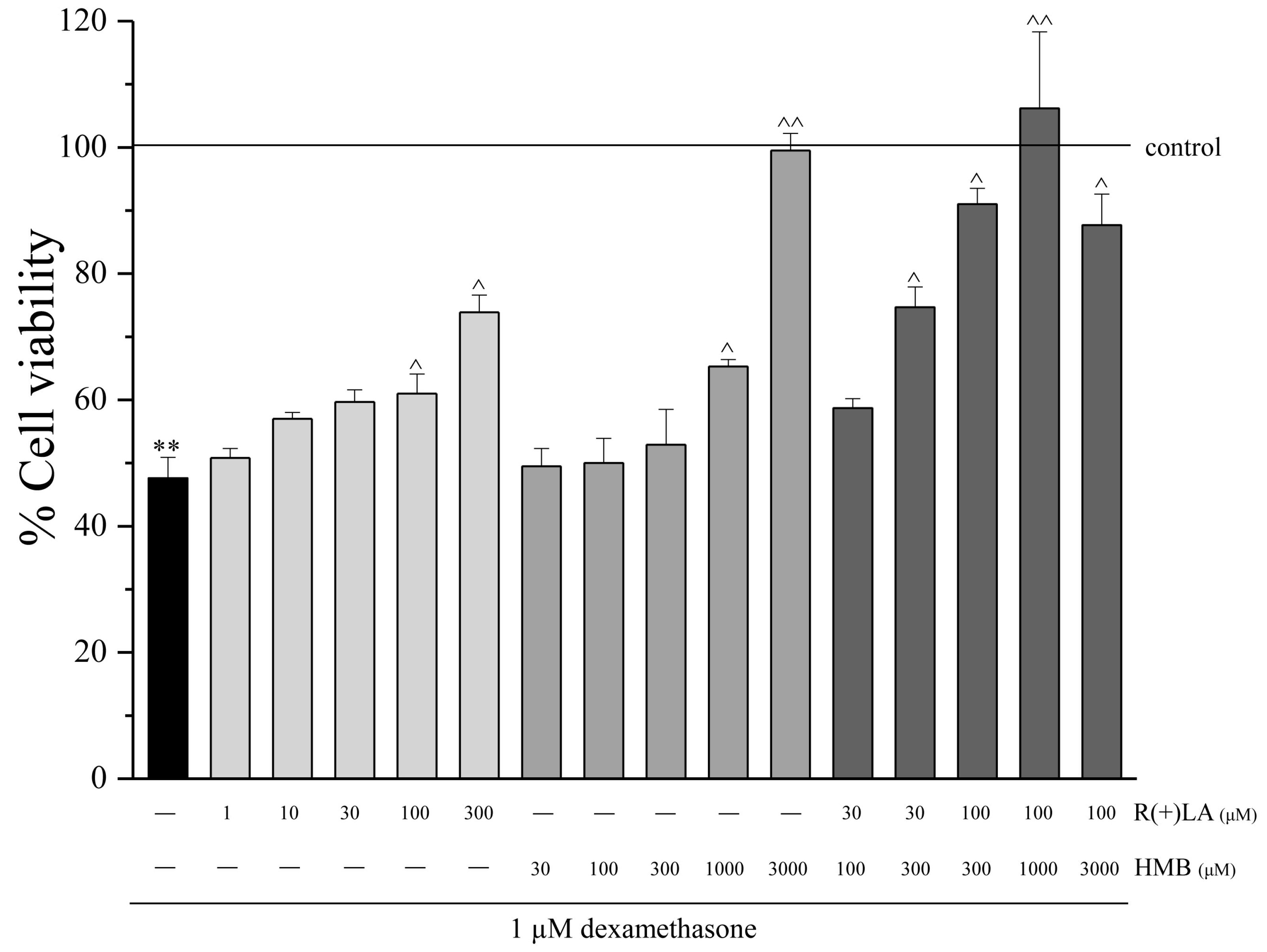
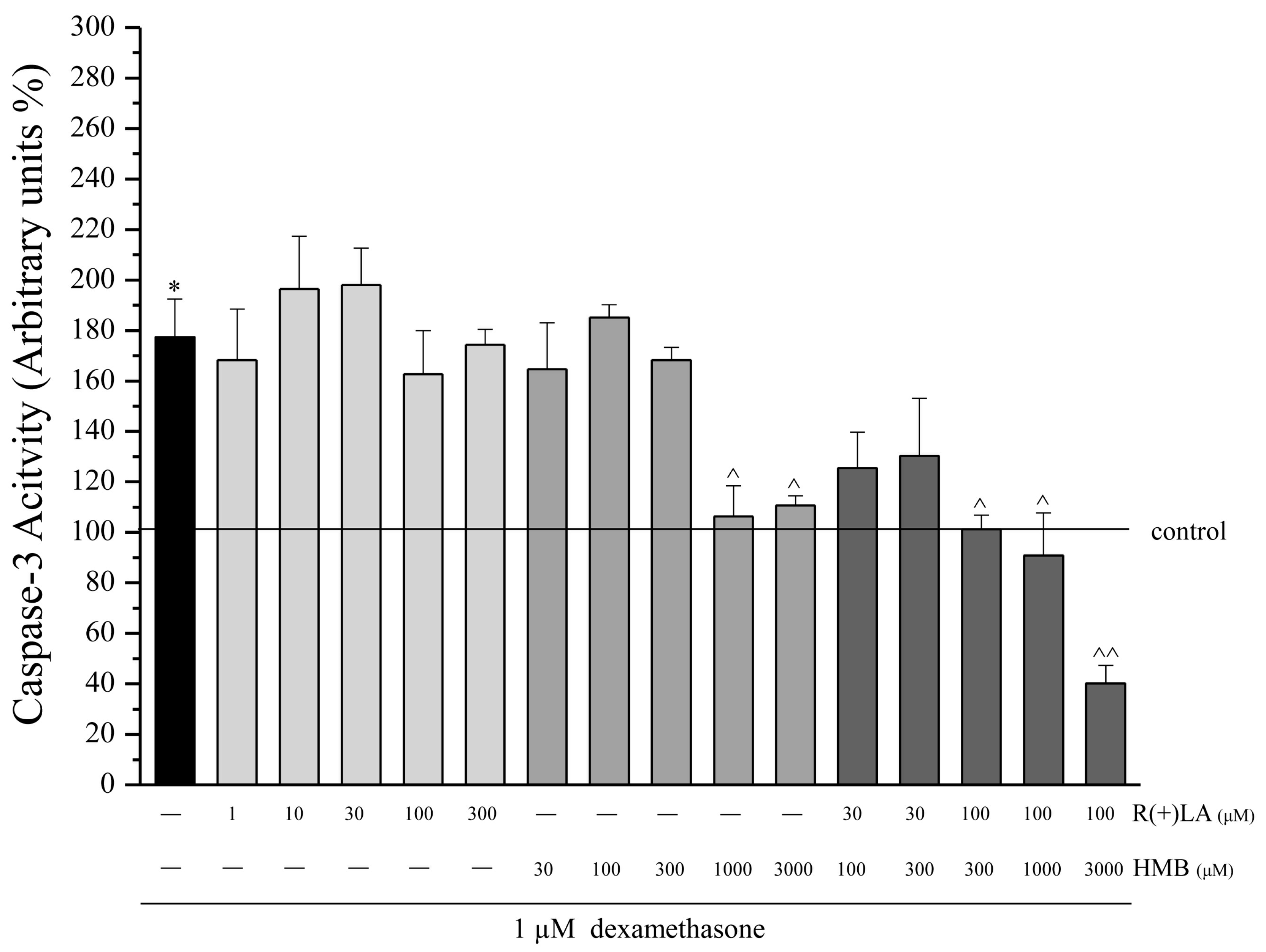
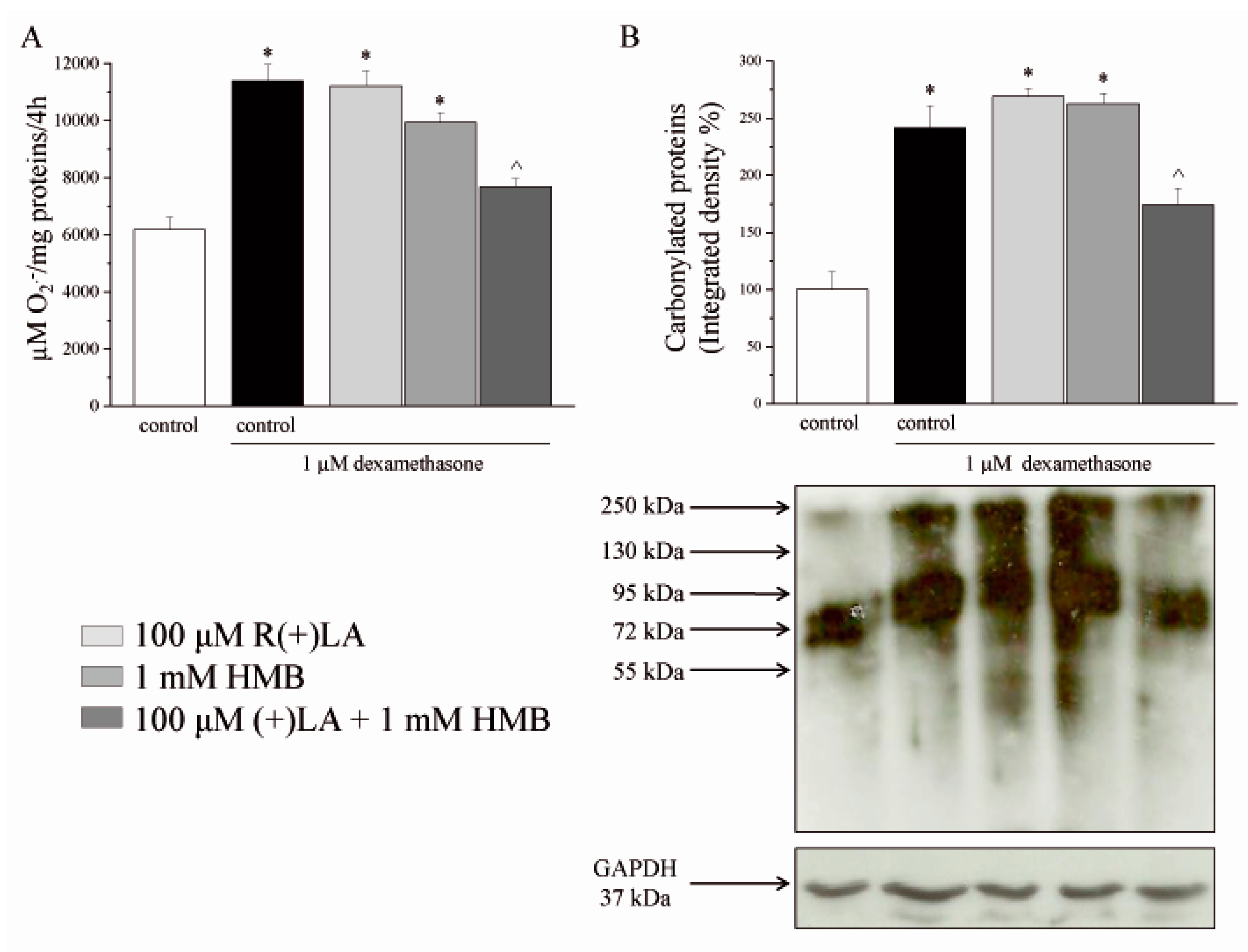
3.1. C2C12 Myoblasts
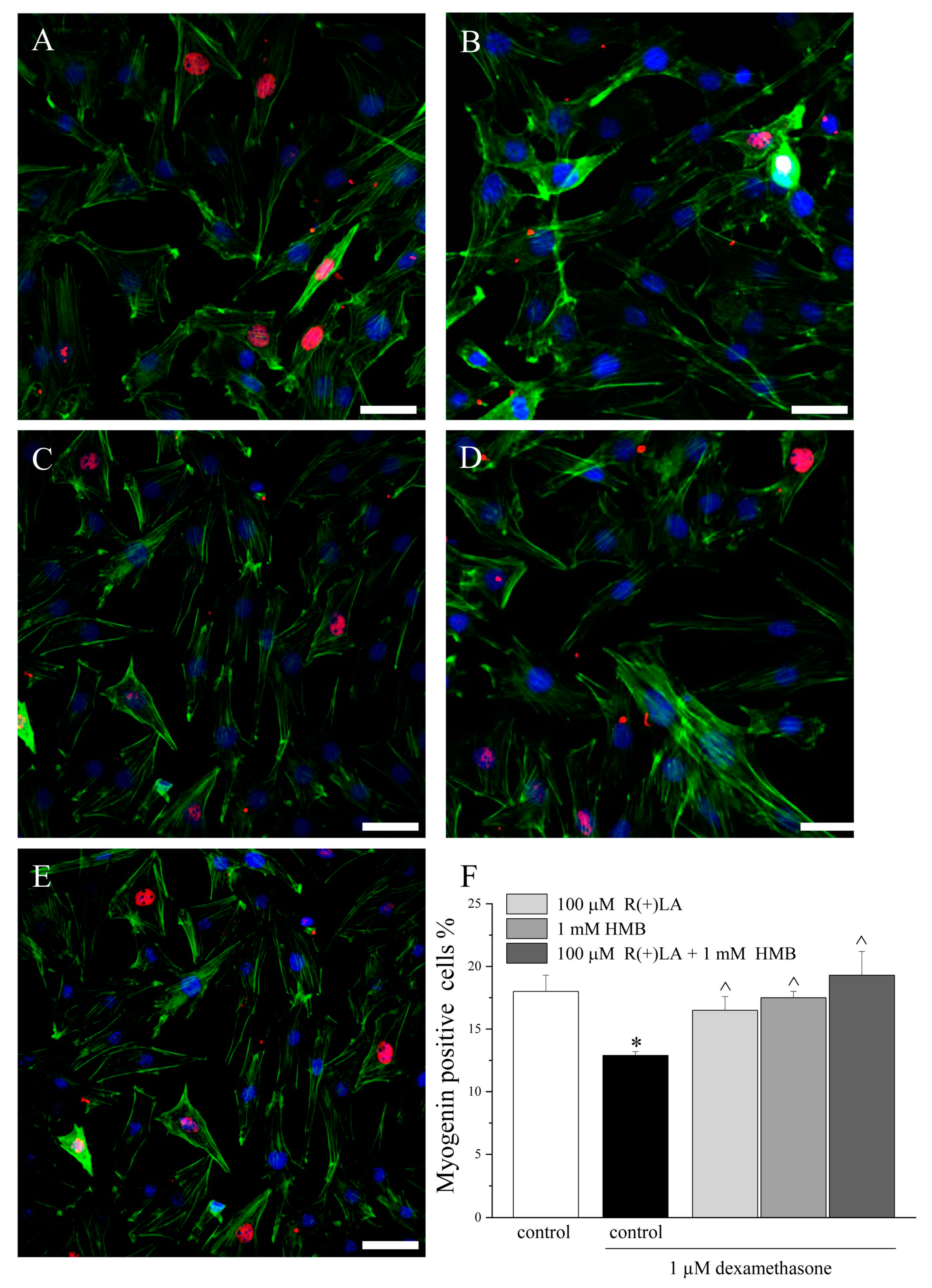
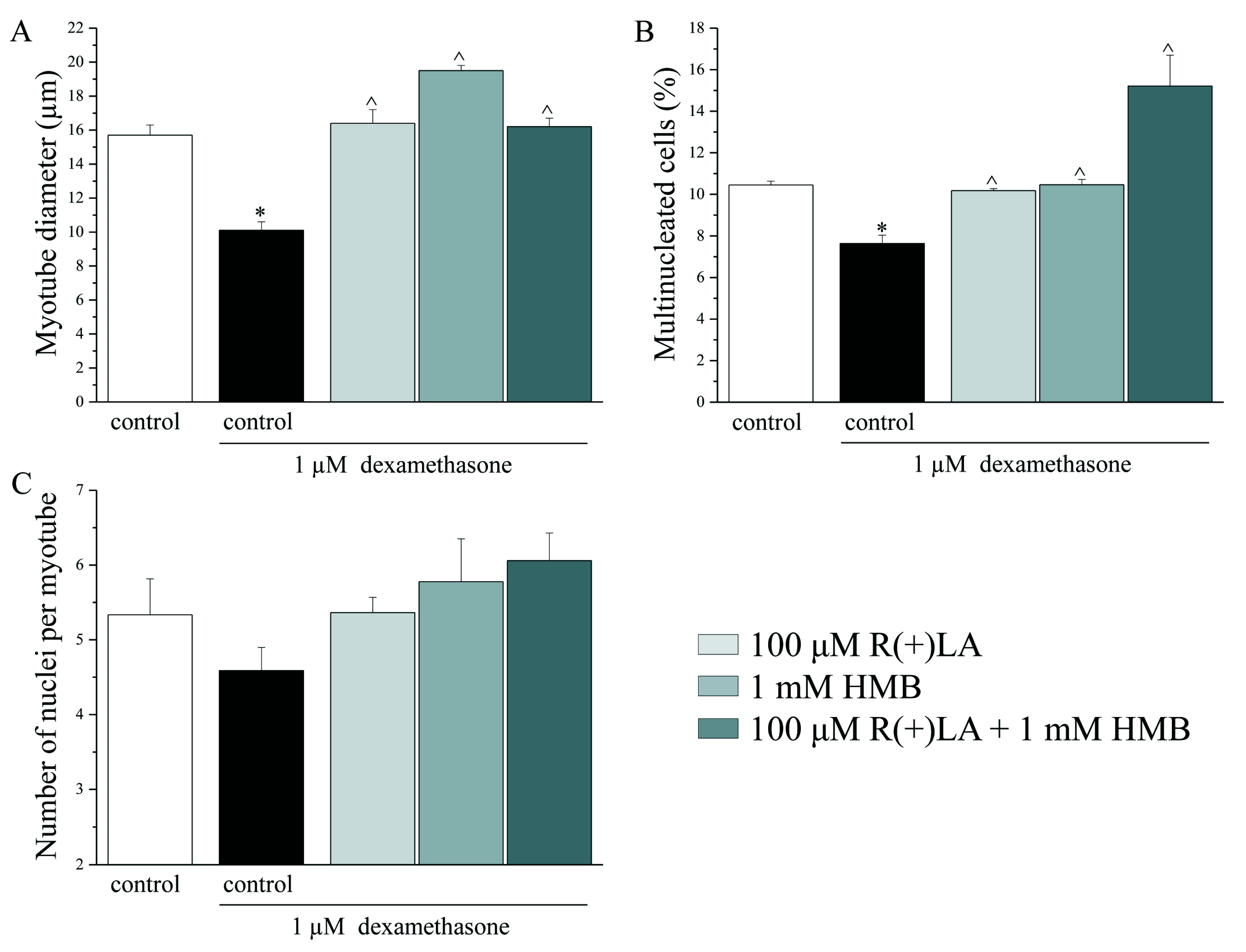
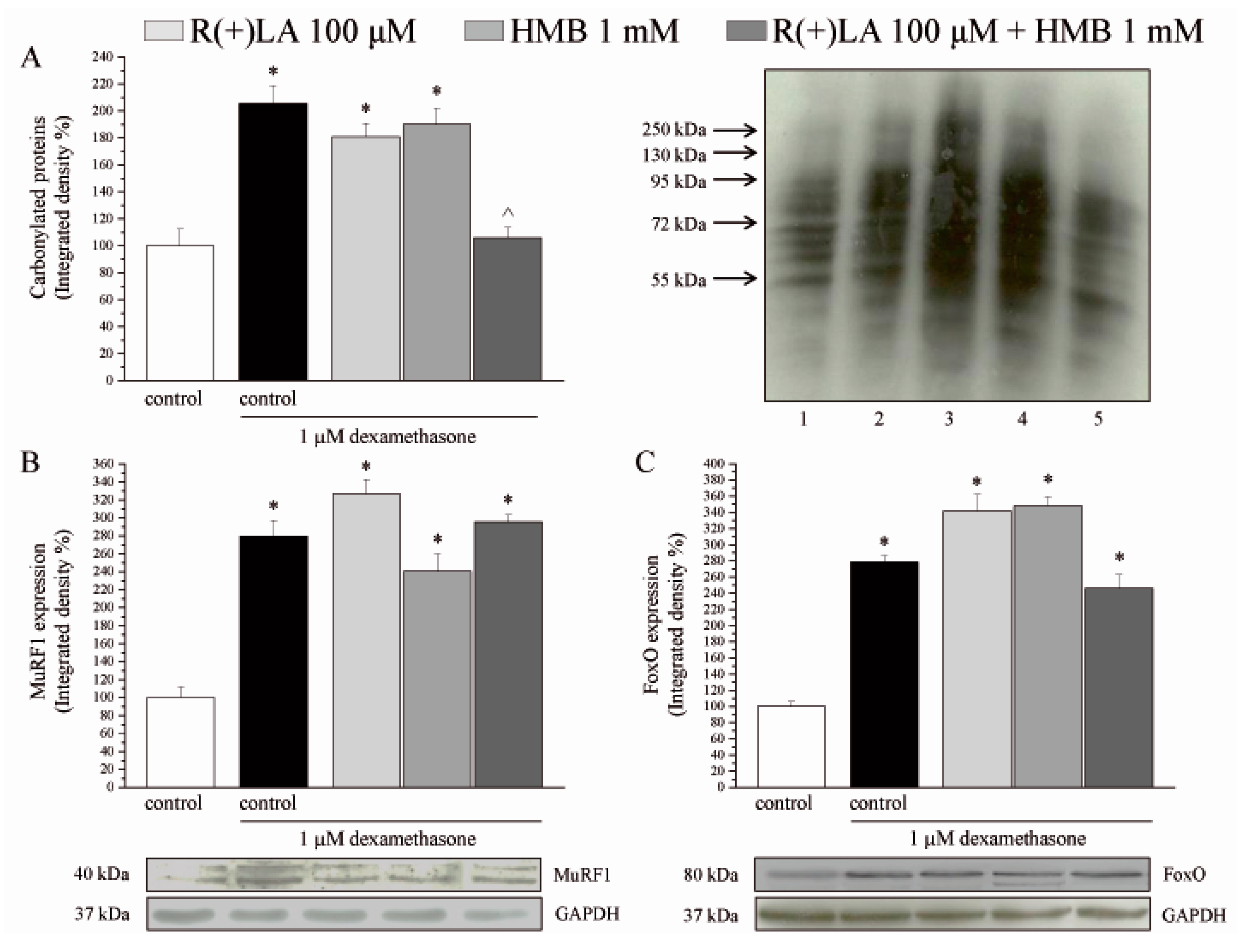
3.2. C2C12 Myotubes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HMB | β-Hydroxy-β–methyl butyrate |
| R(+)LA | R(+) stereoisomer of lipoic acid |
| DEX | dexamethasone |
| FoxO | forkhead box O |
| MuRF1 | muscle ring-finger protein 1 |
| ROS | reactive oxygen species |
| mTOR | mammalian target of rapamycin |
| LA | lipoic acid |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | fetal bovine serum |
| DM | differentiation medium |
| MTT | 3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide |
| HCl | hydrogen chloride |
| EDTA | ethylenediaminetetraacetic acid |
| SOD | Superoxide dismutase |
| PBS | phosphate-buffered saline |
| DNPH | 2,4-dinitrophenyl hydrazine |
| SDS-PAGE | sodium dodecyl sulphate-poly-acrylamide gel electrophoresis |
| BSA | bovine serum albumin |
| PBST | PBS with 0.1% Tween 20 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| FKHR | forkhead in rhabdomyosarcoma |
| ECL | Enhanced chemiluminescence |
| DAPI | 4′,6-Diamidine-2′-phenylindole dihydrochloride |
| AMPK | 5’AMP-activated protein kinase |
| SIRT1 | Sirtuin 1 |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-α |
| AKT | protein kinase B |
| p70S6K | p70S6 kinase |
| HMG–CoA | beta-hydroxy-beta-methylglutaryl coenzyme A |
| PI3K | phosphoinositide 3-kinase. |
References
- Baker, B.A. Efficacy of Age-Specific High-Intensity Stretch-Shortening Contractions in Reversing Dynapenia, Sarcopenia, and Loss of Skeletal Muscle Quality. J. Funct. Morphol. Kinesiol. 2018, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Anker, S.D.; Von Haehling, S. Prevalence, incidence, and clinical impact of sarcopenia: Facts, numbers, and epidemiology-update 2014. J. Cachex- Sarcopenia Muscle 2014, 5, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; A Mackenzie, T.; Barre, L.K.; Lopez-Jimenez, F.; Bartels, S.J. Sarcopenia, sarcopenic obesity and mortality in older adults: Results from the National Health and Nutrition Examination Survey III. Eur. J. Clin. Nutr. 2014, 68, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.; Kuh, D.; Hardy, R.; Mortality Review Group. FALCon and HALCyon Study Teams Objectively measured physical capability levels and mortality: Systematic review and meta-analysis. BMJ 2010, 341. [Google Scholar] [CrossRef]
- Toth, M.J.; Ades, P.A.; Tischler, M.D.; Tracy, R.P.; LeWinter, M.M. Immune activation is associated with reduced skeletal muscle mass and physical function in chronic heart failure. Int. J. Cardiol. 2006, 109, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Pahor, M.; Taaffe, D.R.; Goodpaster, B.H.; Simonsick, E.M.; Newman, A.B.; Nevitt, M.; Harris, T.B. Relationship of Interleukin-6 and Tumor Necrosis Factor- With Muscle Mass and Muscle Strength in Elderly Men and Women: The Health ABC Study. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2002, 57. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Weindruch, R. Interventions Based on the Possibility That Oxidative Stress Contributes to Sarcopenia. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 1995, 50, 157–161. [Google Scholar] [CrossRef]
- Delsite, R.L.; Rasmussen, L.J.; Rasmussen, A.K.; Kalen, A.; Goswami, P.; Singh, K.K. Mitochondrial impairment is accompanied by impaired oxidative DNA repair in the nucleus. Mutagenesis 2003, 18, 497–503. [Google Scholar] [CrossRef]
- Sullivan-Gunn, M.; Lewandowski, P. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 2013, 13, 104. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int. J. Biochem. Cell Boil. 2013, 45, 2288–2301. [Google Scholar] [CrossRef]
- Busquets, S.; Alvarez, B.; Llovera, M.; Agell, N.; López-Soriano, F.J.; Argilés, J.M. Branched-chain amino acids inhibit proteolysis in rat skeletal muscle: mechanisms involved. J. Cell. Physiol. 2000, 184, 380–384. [Google Scholar] [CrossRef]
- Glass, D.J. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nature 2003, 5, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Bodine-Fowler, S. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Invest. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Mechanism of attenuation of muscle protein degradation induced by tumor necrosis factor-alpha and angiotensin II by beta-hydroxy-beta-methylbutyrate. Am. J. Physiol. Endocrinol. Metab. 2008, 295. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Calvani, R.; Joseph, A.-M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Boil. Chem. 2013, 394, 393–414. [Google Scholar] [CrossRef]
- Tomassoni, D.; Amenta, F.; Amantini, C.; Farfariello, V.; Mannelli, L.D.C.; Nwankwo, E.; Marini, C.; Tayebati, S.K. Brain Activity of Thioctic Acid Enantiomers: In Vitro and in Vivo Studies in an Animal Model of Cerebrovascular Injury. Int. J. Mol. Sci. 2013, 14, 4580–4595. [Google Scholar] [CrossRef] [PubMed]
- Bear, D.E.; Langan, A.; Dimidi, E.; Wandrag, L.; Harridge, S.D.R.; Hart, N.; Connolly, B.; Whelan, K. β-Hydroxy-β-methylbutyrate and its impact on skeletal muscle mass and physical function in clinical practice: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2019, 109, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Molfino, A.; Gioia, G.; Fanelli, F.R.; Molfino, A. Beta-hydroxy-beta-methylbutyrate supplementation in health and disease: a systematic review of randomized trials. Amino Acids. 2013, 45, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.H.; Feleke, G.; Din, M.; Yasmin, T.; Singh, G.; Khan, F.A.; Rathmacher, J.A. Nutritional treatment for acquired immunodeficiency virus-associated wasting using beta-hydroxy beta-methylbutyrate, glutamine, and arginine: A randomized, double-blind, placebo-controlled study. JPEN J. Parenter Enteral Nutr. 2000, 24, 133–139. [Google Scholar] [CrossRef]
- Vukovich, M.D.; Stubbs, N.B.; Bohlken, R.M. Body composition in 70-year-old adults responds to dietary beta-hydroxy-beta-methylbutyrate similarly to that of young adults. J. Nutr. 2001, 131, 2049–2052. [Google Scholar] [CrossRef]
- Eley, H.L.; Russell, S.T.; Baxter, J.H.; Mukerji, P.; Tisdale, M.J. Signaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuli. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E923–E931. [Google Scholar] [CrossRef]
- Smith, H.J. Mechanism of the Attenuation of Proteolysis-Inducing Factor Stimulated Protein Degradation in Muscle by -Hydroxy- -Methylbutyrate. Cancer Res. 2004, 64, 8731–8735. [Google Scholar] [CrossRef]
- Wilson, G.J.; Wilson, J.M.; Manninen, A.H. Effects of beta-hydroxy-beta-methylbutyrate (HMB) on exercise performance and body composition across varying levels of age, sex, and training experience: A review. Nutr. Metab. 2008, 5, 1. [Google Scholar] [CrossRef]
- Zanchi, N.; Gerlinger-Romero, F.; Guimarães-Ferreira, L.; Filho, M.A.D.S.; Felitti, V.; Lira, F.S.; Seelaender, M.C.L.; Lancha, A.H. HMB supplementation: Clinical and athletic performance-related effects and mechanisms of action. Amino Acids. 2010, 40, 1015–1025. [Google Scholar] [CrossRef]
- Liu, J.; Head, E.; Gharib, A.M.; Yuan, W.; Ingersoll, R.T.; Hagen, T.M.; Cotman, C.W.; Ames, B.N. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: Partial reversal by feeding acetyl-L-carnitine and/or R- -lipoic acid. Proc. Natl. Acad. Sci. USA 2002, 99, 2356–2361. [Google Scholar] [CrossRef]
- Palaniappan, A.R.; Dai, A. Mitochondrial Ageing and the Beneficial Role of α-Lipoic Acid. Neurochem. Res. 2007, 32, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The Effects and Mechanisms of Mitochondrial Nutrient α-Lipoic Acid on Improving Age-Associated Mitochondrial and Cognitive Dysfunction: An Overview. Neurochem. Res. 2007, 33, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.-S.; Sibille, B.; Murdaca, J.; Mothe-Satney, I.; Grimaldi, P.A.; Neels, J. α-Lipoic acid up-regulates expression of peroxisome proliferator-activated receptor b in skeletal muscle: Involvement of the JNK signaling pathway. Faseb J. 2015, 30, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Hong, O.; Son, J.W.; Kwon, H.-S.; Lee, S.-S.; Kim, S.R.; Yoo, S.J. Alpha-lipoic acid preserves skeletal muscle mass in type 2 diabetic OLETF rats. Nutr. Metab. 2018, 15, 66. [Google Scholar] [CrossRef]
- Tomassoni, D. Morpho-functional Evaluation of the Activity of Stereoisomers of the Antioxidant Alpha-lipoic (Tioctic) Acid in a Model of Compressive Neuropathy. 2010, p. 166. Available online: https://www.torrossa.com/it/resources/an/2492010 (accessed on 30 April 2020). [CrossRef]
- Kaasik, P.; Umnova, M.; Pehme, A.; Alev, K.; Aru, M.; Selart, A.; Seene, T. Ageing and dexamethasone associated sarcopenia: Peculiarities of regeneration. J. Steroid Biochem. Mol. Boil. 2007, 105, 85–90. [Google Scholar] [CrossRef]
- Chiu, C.-S.; Weber, H.; Adamski, S.; Rauch, A.; A Gentile, M.; E Alves, S.; Kath, G.; Flores, O.; A Wilkinson, H. Non-invasive muscle contraction assay to study rodent models of sarcopenia. BMC Musculoskelet. Disord. 2011, 12, 246. [Google Scholar] [CrossRef]
- Rieu, I.; Sornet, C.; Grizard, J.; Dardevet, M. Glucocorticoid excess induces a prolonged leucine resistance on muscle protein synthesis in old rats. Exp. Gerontol. 2004, 39, 1315–1321. [Google Scholar] [CrossRef]
- Shen, S.; Liao, Q.; Liu, J.; Pan, R.; Lee, S.M.-Y.; Lin, L.-G. Myricanol rescues dexamethasone-induced muscle dysfunction via a sirtuin 1-dependent mechanism. J. Cachex- Sarcopenia Muscle 2019, 10, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Lofberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergstrom, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar] [CrossRef]
- Moghadam-Kia, S.; Werth, V.P. Prevention and treatment of systemic glucocorticoid side effects. Int. J. Dermatol. 2010, 49, 239–248. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Rando, T.A. Dystrophin mutations predict cellular susceptibility to oxidative stress. Muscle Nerve. 2000, 23, 784–792. [Google Scholar] [CrossRef]
- Lee, C.S.; Yi, J.-S.; Jung, S.-Y.; Kim, B.-W.; Lee, N.-R.; Choo, H.-J.; Jang, S.-Y.; Han, J.; Chi, S.-G.; Park, M.; et al. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 2010, 17, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Auclair, D.; Garrel, D.R.; Zerouala, A.C.; Ferland, L.H. Activation of the ubiquitin pathway in rat skeletal muscle by catabolic doses of glucocorticoids. Am. J. Physiol. Physiol. 1997, 272. [Google Scholar] [CrossRef] [PubMed]
- Czerwinski, S.M.; Wegrzyn, L.E.; Hickson, R.C. Glutamine Prevents Down-Regulation of Myosin Heavy Chain Synthesis and Muscle Atrophy from Glucocorticoids. Med. Sci. Sports Exerc. 1995, 27. [Google Scholar] [CrossRef]
- Kayali, A.G.; Young, V.R.; Goodman, M.N. Sensitivity of myofibrillar proteins to glucocorticoid-induced muscle proteolysis. Am. J. Physiol. Metab. 1987, 252. [Google Scholar] [CrossRef]
- Santidrian, S.; Moreyra, M.; Munro, H.N.; Young, V.R. Effect of corticosterone and its route of administration on muscle protein breakdown, measured in vivo by urinary excretion of N tau-methylhistidine in rats: response to different levels of dietary protein and energy. Metabolism 1981, 30, 798–804. [Google Scholar] [CrossRef]
- Thompson, M.G.; Thom, A.; Partridge, K.; Garden, K.; Campbell, G.P.; Calder, G.; Palmer, R.M. Stimulation of myofibrillar protein degradation and expression of mRNA encoding the ubiquitin-proteasome system in C(2)C(12) myotubes by dexamethasone: effect of the proteasome inhibitor MG-132. J. Cell. Physiol. 1999, 181, 455–461. [Google Scholar] [CrossRef]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell. 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Menconi, M.; Gonnella, P.; Petkova, V.; Lecker, S.; Hasselgren, P.-O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 2008, 105. [Google Scholar] [CrossRef]
- Gueugneau, M.; D’Hose, D.; Barbé, C.; De Barsy, M.; Lause, P.; Maiter, D.; Bindels, L.B.; Delzenne, N.M.; Schaeffer, L.; Gangloff, Y.-G.; et al. Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy. J. Cachex- Sarcopenia Muscle 2018, 9, 929–946. [Google Scholar] [CrossRef]
- Marinovic, A.C.; Zheng, B.; Mitch, W.E.; Price, S.R. Tissue-specific regulation of ubiquitin (UbC) transcription by glucocorticoids: in vivo and in vitro analyses. Am. J. Physiol. Physiol. 2007, 292. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Reano, S.; Ferrara, M.; Angelino, E.; Gnocchi, V.F.; Prodam, F.; Ronchi, G.; Fagoonee, S.; Fornaro, M.; et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.-I.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between Glucocorticoid Receptor and Nutritional Sensor mTOR in Skeletal Muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.L. Molecular mechanisms regulating myogenic determination and differentiation. Front. Biosci. 2000, 5. [Google Scholar] [CrossRef]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin Inhibits Myoblast Differentiation by Down-regulating MyoD Expression. J. Boil. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef]
- Pas, M.F.T.; De Jong, P.R.; Verburg, F.J. Glucocorticoid inhibition of C2C12 proliferation rate and differentiation capacity in relation to mRNA levels of the MRF gene family. Mol. Boil. Rep. 2000, 27, 87–98. [Google Scholar] [CrossRef]
- Oshima, Y.; Kuroda, Y.; Kunishige, M.; Matsumoto, T.; Mitsui, T. Oxidative stress-associated mitochondrial dysfunction in corticosteroid-treated muscle cells. Muscle Nerve 2004, 30, 49–54. [Google Scholar] [CrossRef]
- Mitsui, T.; Azuma, H.; Nagasawa, M.; Iuchi, T.; Akaike, M.; Odomi, M.; Matsumoto, T. Chronic corticosteroid administration causes mitochondrial dysfunction in skeletal muscle. J. Neurol. 2002, 249, 1004–1009. [Google Scholar] [CrossRef]
- Winiarska, K.; Malińska, D.; Szymanski, K.; Dudziak, M.; Bryla, J. Lipoic acid ameliorates oxidative stress and renal injury in alloxan diabetic rabbits. Biochimie 2008, 90, 450–459. [Google Scholar] [CrossRef]
- Muller, F.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.-T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free. Radic. Boil. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Ow, J.R.; Yang, N.-D.; Taneja, R. Oxidative Stress-Mediated Skeletal Muscle Degeneration: Molecules, Mechanisms, and Therapies. Oxidative Med. Cell. Longev. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bilska, A.; Włodek, L. Lipoic acid-the drug of the future? Pharmacol. Rep. 2005, 57, 570–577. [Google Scholar]
- Matsugo, S.; Yan, L.-J.; Han, D.; Trischler, H.; Packer, L. Elucidation of Antioxidant Activity of α-Lipoic Acid Toward Hydroxyl Radical. Biochem. Biophys. Res. Commun. 1995, 208, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Borcea, V.; Nourooz-Zadeh, J.; Wolff, S.P.; Klevesath, M.; Hofmann, M.; Urich, H.; Wahl, P.; Ziegler, R.; Tritschler, H.; Halliwell, B.; et al. α-lipoic acid decreases oxidative stress even in diabetic patients with poor glycemic control and albuminuria. Free. Radic. Boil. Med. 1999, 26, 1495–1500. [Google Scholar] [CrossRef]
- Tibullo, D.; Volti, G.L.L.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Syndr. 2014, 6, 80. [Google Scholar] [CrossRef]
- Jurisic-Erzen, D.; Starcevic-Klasan, G.; Ivanac, D.; Peharec, S.; Girotto, D.; Jerkovic, R. The effects of alpha-lipoic acid on diabetic myopathy. J. Endocrinol. Investig. 2017, 41, 203–209. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Mahmoud, M.O.; Awaad, A.S.; Gamal, G.M.; Abdelfatah, D. Alpha lipoic acid protects against dexamethasone-induced metabolic abnormalities via APPL1 and PGC-1 α up regulation. Steroids 2019, 144, 1–7. [Google Scholar] [CrossRef]
- Kaczka, P.; Michalczyk, M.M.; Jastrząb, R.; Gawelczyk, M.; Kubicka, K. Mechanism of Action and the Effect of Beta-Hydroxy-Beta-Methylbutyrate (HMB) Supplementation on Different Types of Physical Performance-Systematic Review. J. Hum. Kinet. 2019, 68, 211–222. [Google Scholar] [CrossRef]
- Russell, S.T.; Tisdale, M.J. Mechanism of attenuation by beta-hydroxy-beta-methylbutyrate of muscle protein degradation induced by lipopolysaccharide. Mol. Cell. Biochem. 2009, 330, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Standley, R.A.; Distefano, G.; Pereira, S.L.; Tian, M.; Kelly, O.; Coen, P.M.; Deutz, N.E.P.; Wolfe, R.R.; Goodpaster, B.H. Effects of β-hydroxy-β-methylbutyrate on skeletal muscle mitochondrial content and dynamics, and lipids after 10 days of bed rest in older adults. J. Appl. Physiol. 2017, 123, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Lowery, R.P.; Joy, J.M.; Walters, J.A.; Baier, S.M.; Fuller, J.C.; Stout, J.R.; Norton, L.E.; Sikorski, E.M.; Wilson, S.M.C.; et al. β-Hydroxy-β-methylbutyrate free acid reduces markers of exercise-induced muscle damage and improves recovery in resistance-trained men. Br. J. Nutr. 2013, 110, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J. Beta-Hydroxy-Beta-Methyl Butyrate (HMB): From Experimental Data to Clinical Evidence in Sarcopenia. Curr. Protein Pept. Sci. 2018, 19, 668–672. [Google Scholar] [CrossRef]
- Kornasio, R.; Riederer, I.; Butler-Browne, G.; Mouly, V.; Uni, Z.; Halevy, O. Beta-hydroxy-beta-methylbutyrate (HMB) stimulates myogenic cell proliferation, differentiation and survival via the MAPK/ERK and PI3K/Akt pathways. Biochim. Biophys. Acta 2009, 1793, 755–763. [Google Scholar] [CrossRef]
- Baptista, I.L.; Silva, W.J.; Artioli, G.G.; Guilherme, J.P.L.F.; Leal, M.L.; Aoki, M.S.; Miyabara, E.H.; Moriscot, A.S. Leucine and HMB differentially modulate proteasome system in skeletal muscle under different sarcopenic conditions. PLoS ONE 2013, 8, 76752. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Viability % | |||
|---|---|---|---|
| Dexamethasone Concentration (μM) | Incubation | ||
| 24 h | 48 h | ||
| 0 | 100.0 ± 0.9 | 100 ± 2.4 | |
| 0.01 | 87.32 ± 4.5 | 69.1 ± 1.8** | |
| 0.03 | 85.34 ± 6.7 | 62.4 ± 4.6** | |
| 0.1 | 68.9 ± 1.8** | 58.9 ± 4.3** | |
| 1 | 65.4 ± 1.7** | 47.6 ± 3.3** | |
| 3 | 62.2 ± 1.9** | 56.1 ± 5.0** | |
| 10 | 63.3 ± 1.8** | 52.3 ± 2.3** | |
| 30 | 62.5 ± 1.8** | 53.5 ± 6.7** | |
| 100 | 61.3 ± 0.4** | 52.4 ± 3.8** | |
| 300 | 59.5 ± 0.6** | 54.2 ± 8.1** | |
| Caspase-3 Activity % | |
|---|---|
| Dexamethasone Concentration (μM) | 48 h Incubation |
| 0 | 100 ± 3.4 |
| 0.01 | 160.6 ± 20.1* |
| 0.1 | 171.7 ± 14.9* |
| 0.3 | 170.3 ± 16.8* |
| 1 | 177.3 ± 15.2* |
| 10 | 106.2 ± 4.4 |
| 100 | 94.5 ± 5.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Cesare Mannelli, L.; Micheli, L.; Lucarini, E.; Parisio, C.; Toti, A.; Tenci, B.; Zanardelli, M.; Branca, J.J.V.; Pacini, A.; Ghelardini, C. Effects of the Combination of β-Hydroxy-β-Methyl Butyrate and R(+) Lipoic Acid in a Cellular Model of Sarcopenia. Molecules 2020, 25, 2117. https://doi.org/10.3390/molecules25092117
Di Cesare Mannelli L, Micheli L, Lucarini E, Parisio C, Toti A, Tenci B, Zanardelli M, Branca JJV, Pacini A, Ghelardini C. Effects of the Combination of β-Hydroxy-β-Methyl Butyrate and R(+) Lipoic Acid in a Cellular Model of Sarcopenia. Molecules. 2020; 25(9):2117. https://doi.org/10.3390/molecules25092117
Chicago/Turabian StyleDi Cesare Mannelli, Lorenzo, Laura Micheli, Elena Lucarini, Carmen Parisio, Alessandra Toti, Barbara Tenci, Matteo Zanardelli, Jacopo Junio Valerio Branca, Alessandra Pacini, and Carla Ghelardini. 2020. "Effects of the Combination of β-Hydroxy-β-Methyl Butyrate and R(+) Lipoic Acid in a Cellular Model of Sarcopenia" Molecules 25, no. 9: 2117. https://doi.org/10.3390/molecules25092117
APA StyleDi Cesare Mannelli, L., Micheli, L., Lucarini, E., Parisio, C., Toti, A., Tenci, B., Zanardelli, M., Branca, J. J. V., Pacini, A., & Ghelardini, C. (2020). Effects of the Combination of β-Hydroxy-β-Methyl Butyrate and R(+) Lipoic Acid in a Cellular Model of Sarcopenia. Molecules, 25(9), 2117. https://doi.org/10.3390/molecules25092117