
Guanine Radicals Generated in Telomeric G-Quadruplexes by Direct Absorption of Low-Energy UV Photons: Effect of Potassium Ions



Abstract
1. Introduction
2. Results and Discussion
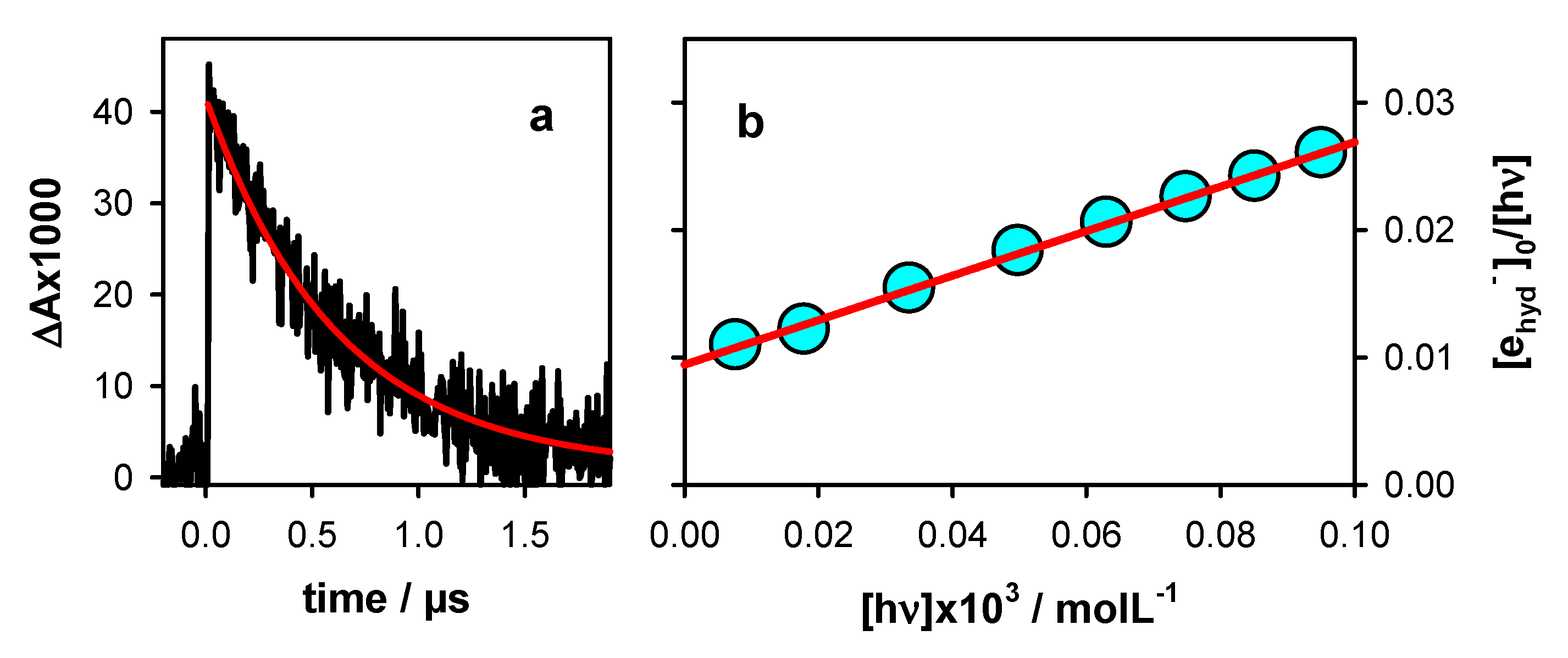
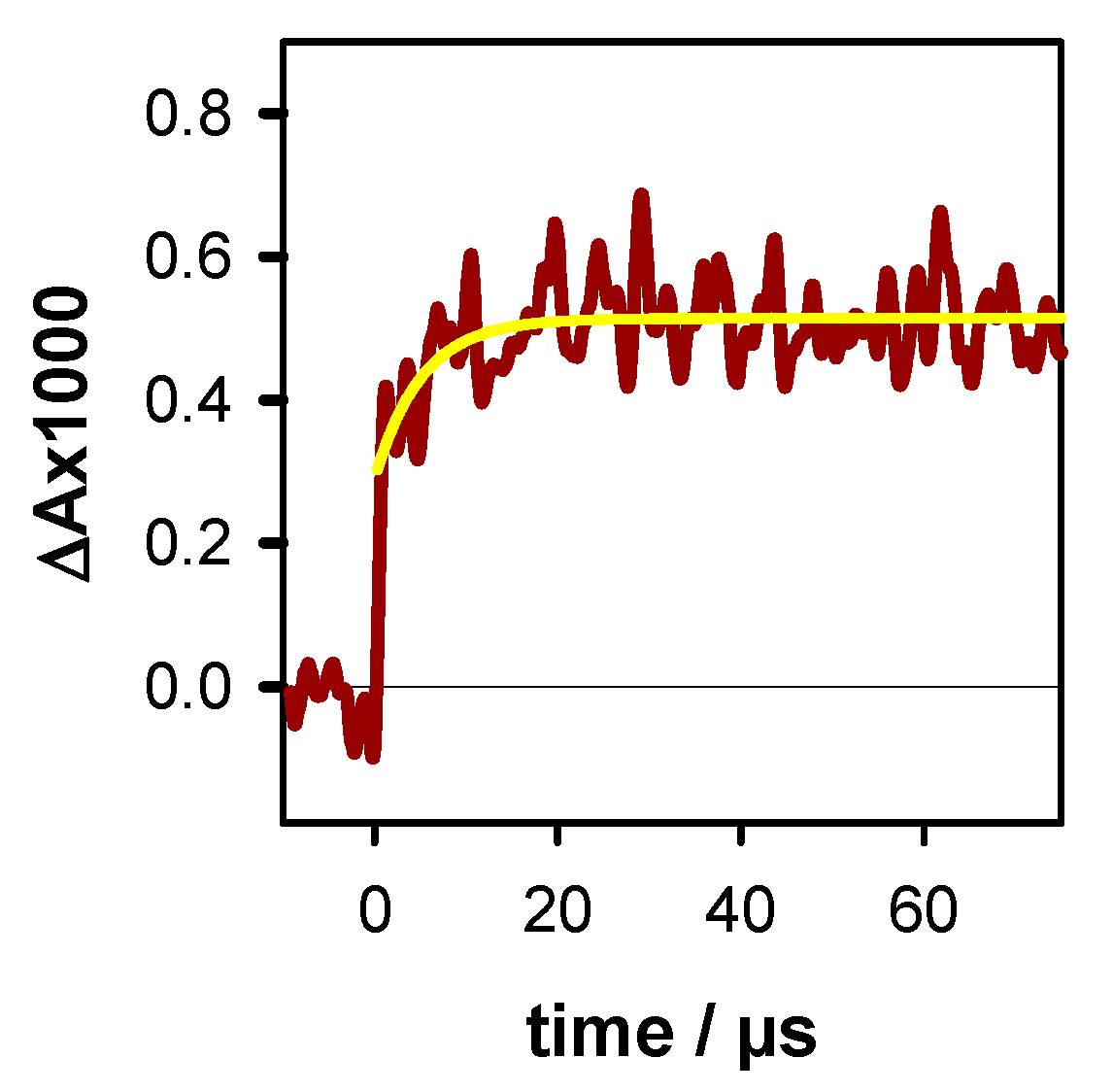
2.1. Photo-Ionization
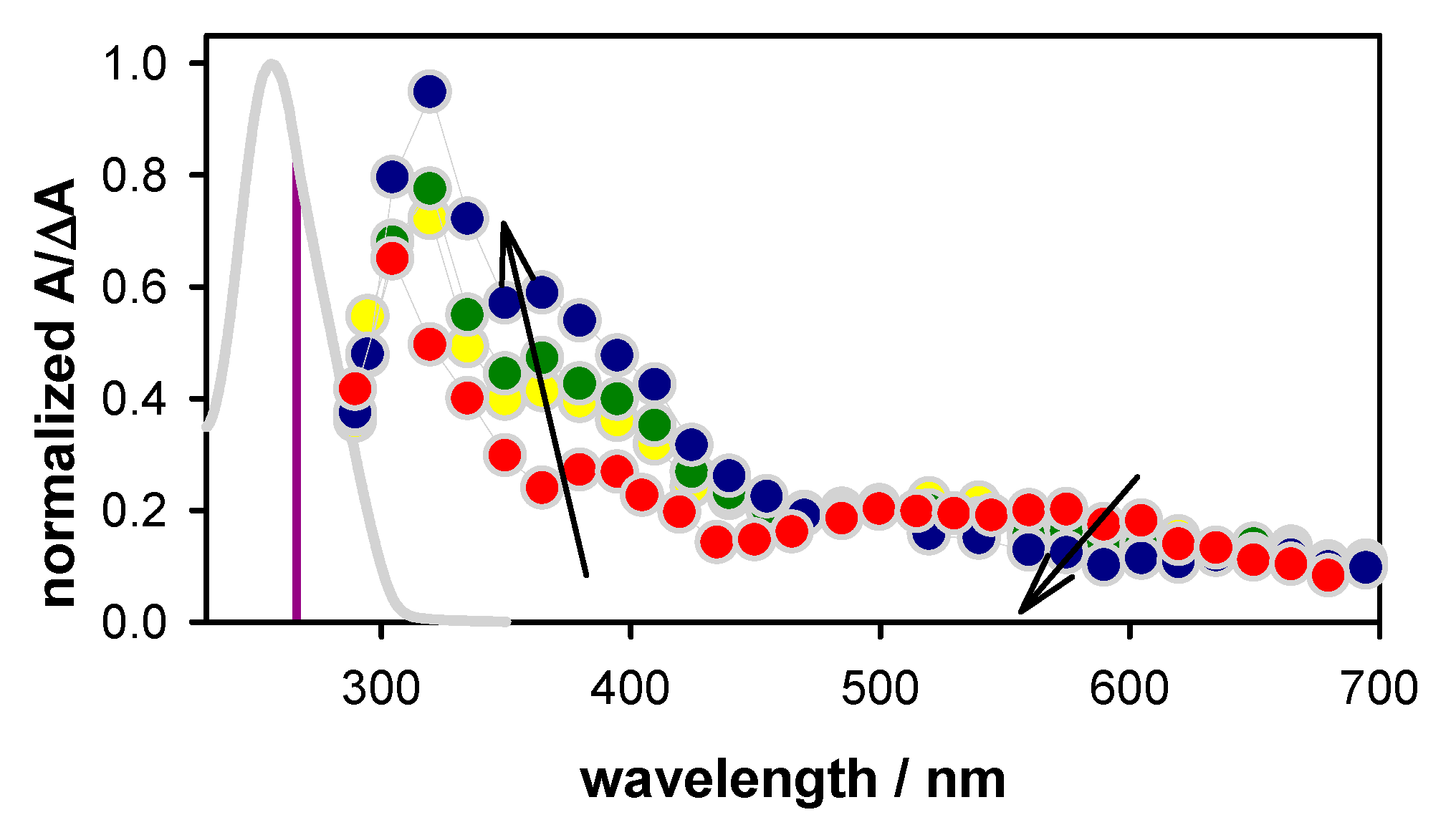
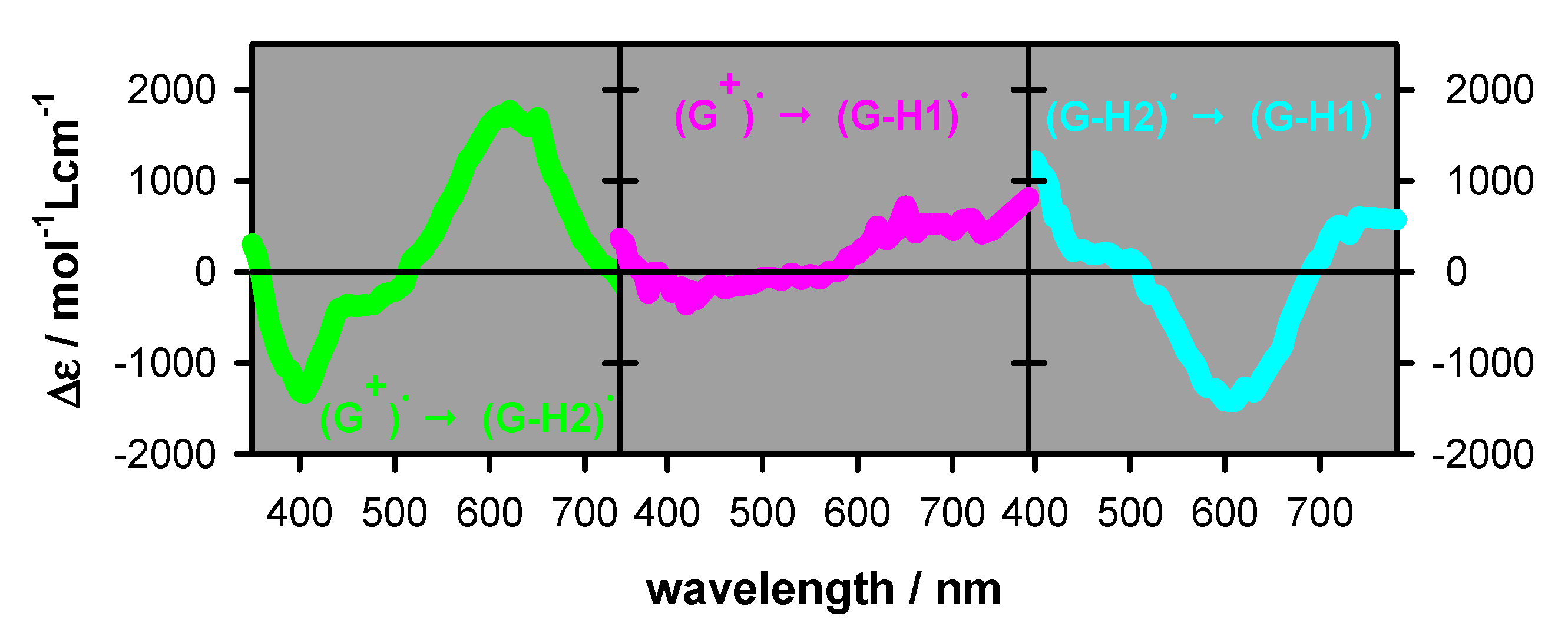
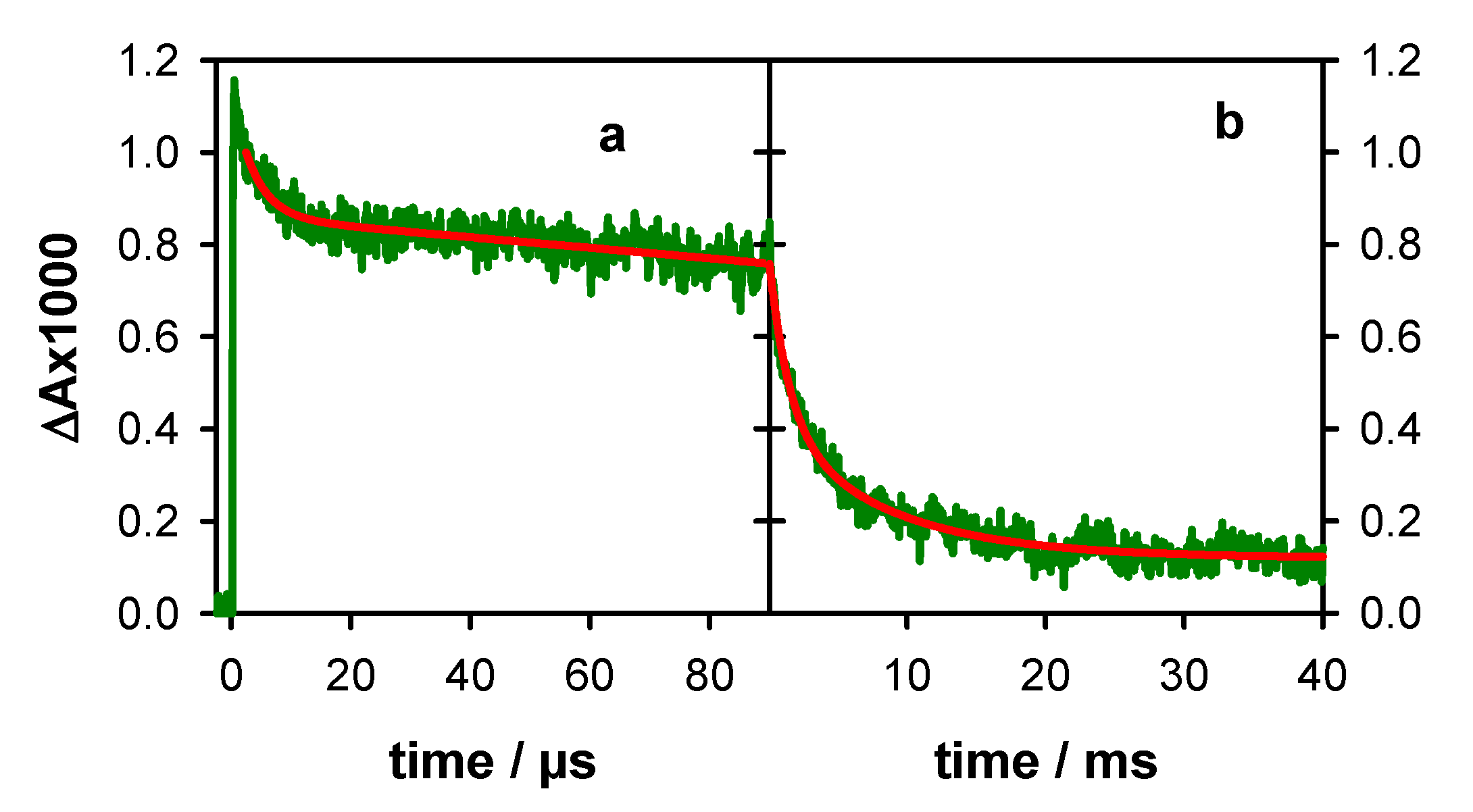
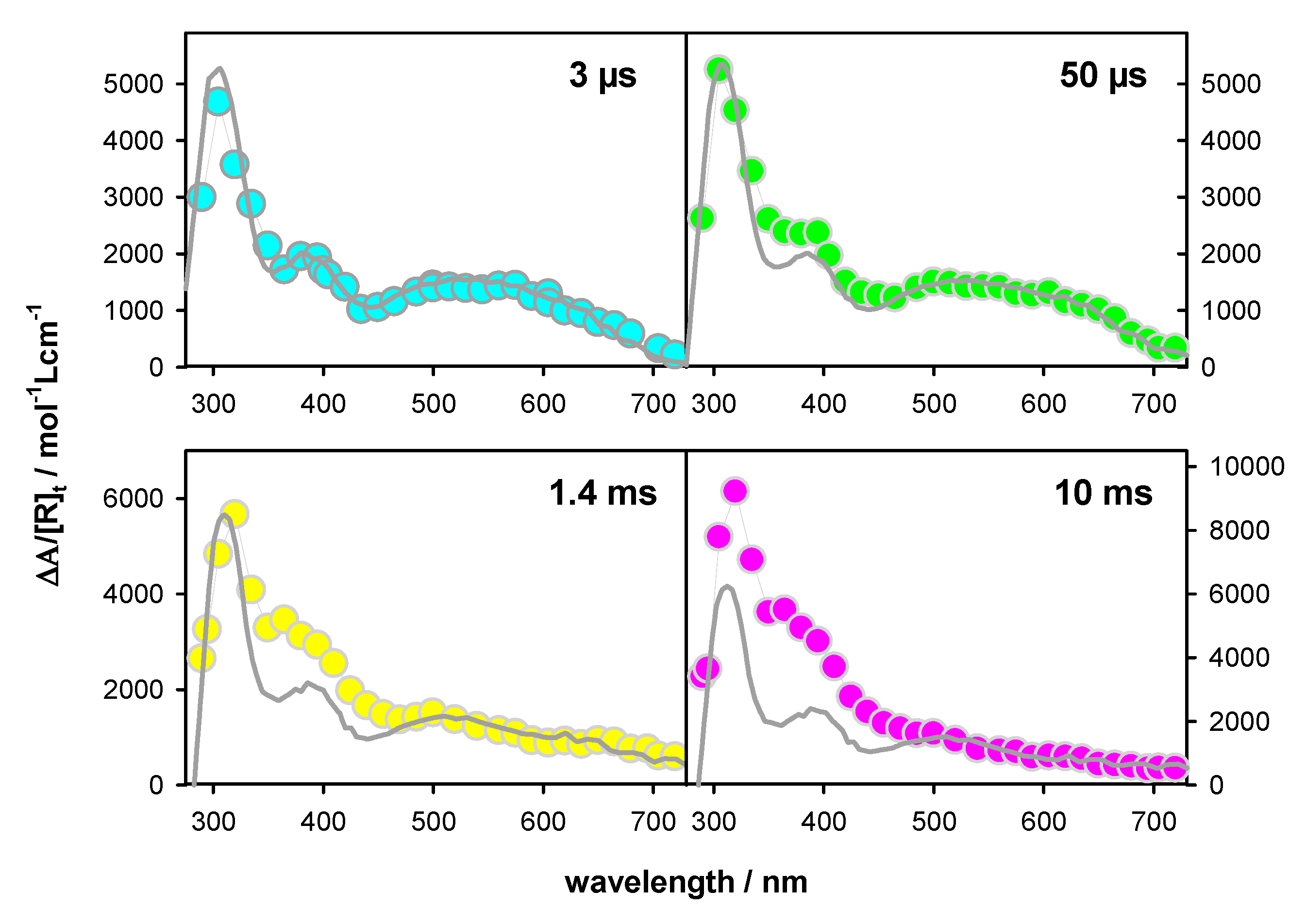
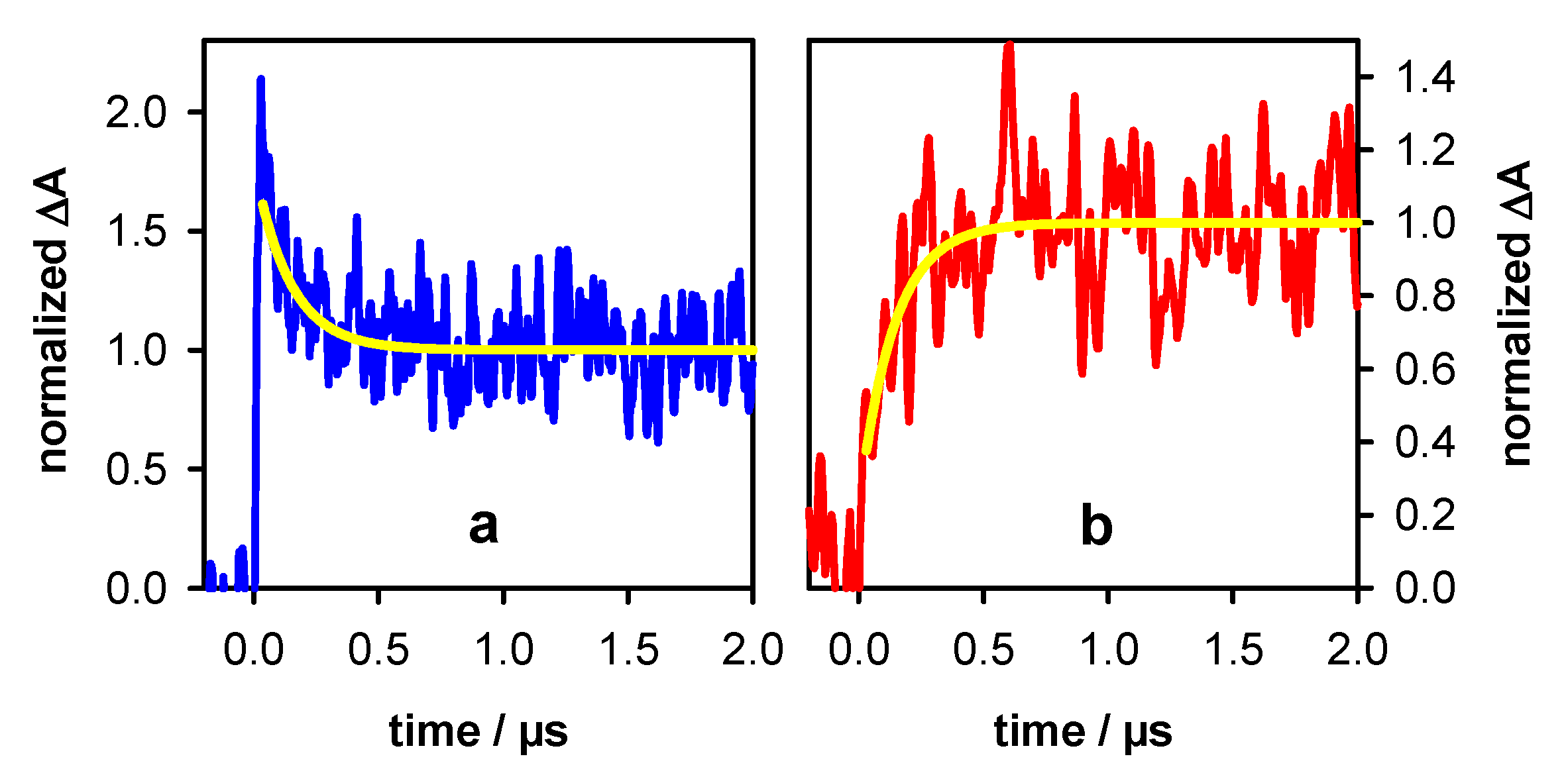
2.2. Transient Species
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kim, M.G.; Ko, S.B.; Kim, D.H.; Lee, B.J.; Macgregor, R.B.; Shim, G.; Oh, Y.K. Stemmed DNA nanostructure for the selective delivery of therapeutics. Nanoscale 2018, 10, 7511–7518. [Google Scholar] [CrossRef] [PubMed]
- Mergny, J.L.; Sen, D. DNA Quadruple Helices in Nanotechnology. Chem. Rev. 2019, 119, 6290–6325. [Google Scholar] [CrossRef]
- Livshits, G.I.; Stern, A.; Rotem, D.; Borovok, N.; Eidelshtein, G.; Migliore, A.; Penzo, E.; Wind, S.J.; Di Felice, R.; Skourtis, S.S.; et al. Long-range charge transport in single G-quadruplex DNA molecules. Nat. Nanotech. 2014, 9, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Meng, Z.; Lu, Y.; Shao, F. Efficient Long-Range Hole Transport Through G-Quadruplexes. Chem. Eur. J. 2017, 23, 13980–13985. [Google Scholar] [CrossRef] [PubMed]
- Thazhathveetil, A.K.; Harris, M.A.; Young, R.M.; Wasielewski, M.R.; Lewis, F.D. Efficient Charge Transport via DNA G-Quadruplexes. J. Am. Chem. Soc. 2017, 139, 1730–1733. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of Low-Energy UV Radiation by Human Telomere G-Quadruplexes Generates Long-Lived Guanine Radical Cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef]
- Banyasz, A.; Balanikas, E.; Martinez-Fernandez, L.; Baldacchino, G.; Douki, T.; Improta, R.; Markovitsi, D. Radicals generated in tetramolecular guanine quadruplexes by photo-ionization: Spectral and dynamical features. J. Phys. Chem. B 2019, 123, 4950–4957. [Google Scholar] [CrossRef]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA strands: Direct versus Indirect Generation. Molecules 2019, 24, 2347. [Google Scholar] [CrossRef]
- Behmand, B.; Balanikas, E.; Martinez-Fernandez, L.; Improta, R.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Potassium Ions Enhance Guanine Radical Generation upon Absorption of Low-Energy Photons by G-quadruplexes and Modify Their Reactivity. J. Phys. Chem. Lett. 2020, 11, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Behmand, B.; Balanikas, E.; Martinez-Fernandez, L.; Improta, R.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Correction to “Potassium Ions Enhance Guanine Radical Generation upon Absorption of Low-Energy Photons by G-quadruplexes and Modify Their Reactivity”. J. Phys. Chem. Lett. 2020, 11, 2742. [Google Scholar] [CrossRef] [PubMed]
- Candeias, L.P.; Steenken, S. Ionization of purine nucleosides and nucleotides and their components by 193-nm laser photolysis in aqueous solution: Model studies for oxidative damage of DNA. J. Am. Chem. Soc. 1992, 114, 699–704. [Google Scholar] [CrossRef]
- Rokhlenko, Y.; Cadet, J.; Geacintov, N.E.; Shafirovich, V. Mechanistic Aspects of Hydration of Guanine Radical Cations in DNA. J. Am. Chem. Soc. 2014, 136, 5956–5962. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Improta, R.; Ketola, T.M.; Balty, C.; Markovitsi, D. Radicals generated in alternating guanine-cytosine duplexes by direct absorption of low-energy UV radiation. Phys. Chem. Chem. Phys. 2018, 20, 21381–21389. [Google Scholar] [CrossRef]
- Wu, L.D.; Liu, K.H.; Jie, J.L.; Song, D.; Su, H.M. Direct Observation of Guanine Radical Cation Deprotonation in G-Quadruplex DNA. J. Am. Chem. Soc. 2015, 137, 259–266. [Google Scholar] [CrossRef]
- Phan, A.T.; Kuryavyi, V.; Luu, K.N.; Patel, D.J. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucl. Ac. Res. 2007, 35, 6517–6525. [Google Scholar] [CrossRef]
- Dai, J.X.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D.Z. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: Insights into structure polymorphism of the human telomeric sequence. Nucl. Ac. Res. 2007, 35, 4927–4940. [Google Scholar] [CrossRef]
- Torche, F.; Marignier, J.L. Direct Evaluation of the Molar Absorption Coefficient of Hydrated Electron by the Isosbestic Point Method. J. Phys. Chem. B 2016, 120, 7201–7206. [Google Scholar] [CrossRef]
- Changenet-Barret, P.; Hua, Y.; Gustavsson, T.; Markovitsi, D. Electronic excitations in G-quadruplexes formed by the human telomeric sequence: A time-resolved fluorescence study. Photochem. Photobiol. 2015, 91, 759–765. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Changenet, P.; Banyasz, A.; Gustavsson, T.; Markovitsi, D.; Improta, I. A Comprehensive Study of Guanine Excited State Relaxation and Photoreactivity in G-Quadruplexes. J. Phys. Chem. Lett. 2019, 10, 6873–6877. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chan, R.C.T.; Chan, C.T.L.; Wong, A.K.W.; Kwok, W.M. Real-time Monitoring Excitation Dynamics of Human Telomeric Guanine Quadruplexes: Effect of Folding Topology, Metal Cation, and Confinement by Nanocavity Water Pool. J. Phys. Chem. Lett. 2019, 10, 7577–7585. [Google Scholar] [CrossRef] [PubMed]
- Candeias, L.P.; Steenken, S. Stucture and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Caminal, C.; Altieri, A.; Vougioukalakis, G.C.; Mulazzani, Q.G.; Gimisis, T.; Guerra, M. Tautomerism in the guanyl radical. J. Am. Chem. Soc. 2006, 128, 13796–13805. [Google Scholar] [CrossRef] [PubMed]
- Marguet, S.; Markovitsi, D. Time-resolved study of thymine dimer formation. J. Am. Chem. Soc. 2005, 127, 5780–5781. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Ketola, T.; Martinez-Fernandez, L.; Improta, R.; Markovitsi, D. Adenine radicals generated in alternating AT duplexes by direct absorption of low-energy UV radiation. Faraday Disc. 2018, 207, 181–197. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Ketola, T.; Muñoz-Losa, A.; Esposito, L.; Markovitsi, D.; Improta, R. Excited State Pathways Leading to Formation of Adenine Dimers. J. Phys. Chem. Lett. 2016, 7, 2020–2023. [Google Scholar] [CrossRef]
- Banyasz, A.; Ketola, T.; Muñoz-Losa, A.; Rishi, S.; Adhikary, A.; Sevilla, M.D.; Martinez-Fernandez, L.; Improrta, R.; Markovitsi, D. UV-induced Adenine Radicals Induced in DNA A-tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett. 2016, 7, 3949–3953. [Google Scholar] [CrossRef]
- Abu-Ghazalah, R.M.; Rutledge, S.; Lau, L.W.Y.; Dubins, D.N.; Macgregor, R.B., Jr.; Helmy, A.S. Concentration-Dependent Structural Transitions of Human Telomeric DNA Sequences. Biochemistry 2012, 51, 7357–7366. [Google Scholar] [CrossRef]
- Amand, B.; Bensasson, R. Determination of triplet quantum yields by laser flash absorption spectroscopy. Chem. Phys. Lett. 1975, 34, 44–48. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TEL21/Na+ [8] | TEL25/Na+ [10] | (TG4T)4/Na+ [9] | (TG4T)4/K+ [11,12] | TEL21/K+ |
|---|---|---|---|---|
| 4.5 ± 0.6 | 5.2 ± 0.3 | 3.5 ± 0.5 [9] | 8.5 ± 0.5 | 9.4 ± 0.1 |
| Total 1 | (G)+● 2 | (G-H2)● 2 | (G-H1)● 2 | Error | |
|---|---|---|---|---|---|
| 3 µs | 1.00 | 0.60 | 0.40 | 0 | ±0.05 |
| 50 µs | 0.80 | 0.40 | 0.35 | 0.05 3 | ±0.1 |
| 1.4 ms | 0.50 | 0.0 | 0.0 | 0.5 | ±0.1 |
| 10 ms | 0.20 | 0.0 | 0.0 | 0.2 | ±0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Guanine Radicals Generated in Telomeric G-Quadruplexes by Direct Absorption of Low-Energy UV Photons: Effect of Potassium Ions. Molecules 2020, 25, 2094. https://doi.org/10.3390/molecules25092094
Balanikas E, Banyasz A, Baldacchino G, Markovitsi D. Guanine Radicals Generated in Telomeric G-Quadruplexes by Direct Absorption of Low-Energy UV Photons: Effect of Potassium Ions. Molecules. 2020; 25(9):2094. https://doi.org/10.3390/molecules25092094
Chicago/Turabian StyleBalanikas, Evangelos, Akos Banyasz, Gérard Baldacchino, and Dimitra Markovitsi. 2020. "Guanine Radicals Generated in Telomeric G-Quadruplexes by Direct Absorption of Low-Energy UV Photons: Effect of Potassium Ions" Molecules 25, no. 9: 2094. https://doi.org/10.3390/molecules25092094
APA StyleBalanikas, E., Banyasz, A., Baldacchino, G., & Markovitsi, D. (2020). Guanine Radicals Generated in Telomeric G-Quadruplexes by Direct Absorption of Low-Energy UV Photons: Effect of Potassium Ions. Molecules, 25(9), 2094. https://doi.org/10.3390/molecules25092094