Discovery of Novel Pyridazine-Based Cyclooxygenase-2 Inhibitors with a Promising Gastric Safety Profile



Abstract

1. Introduction
2. Results and Discussion
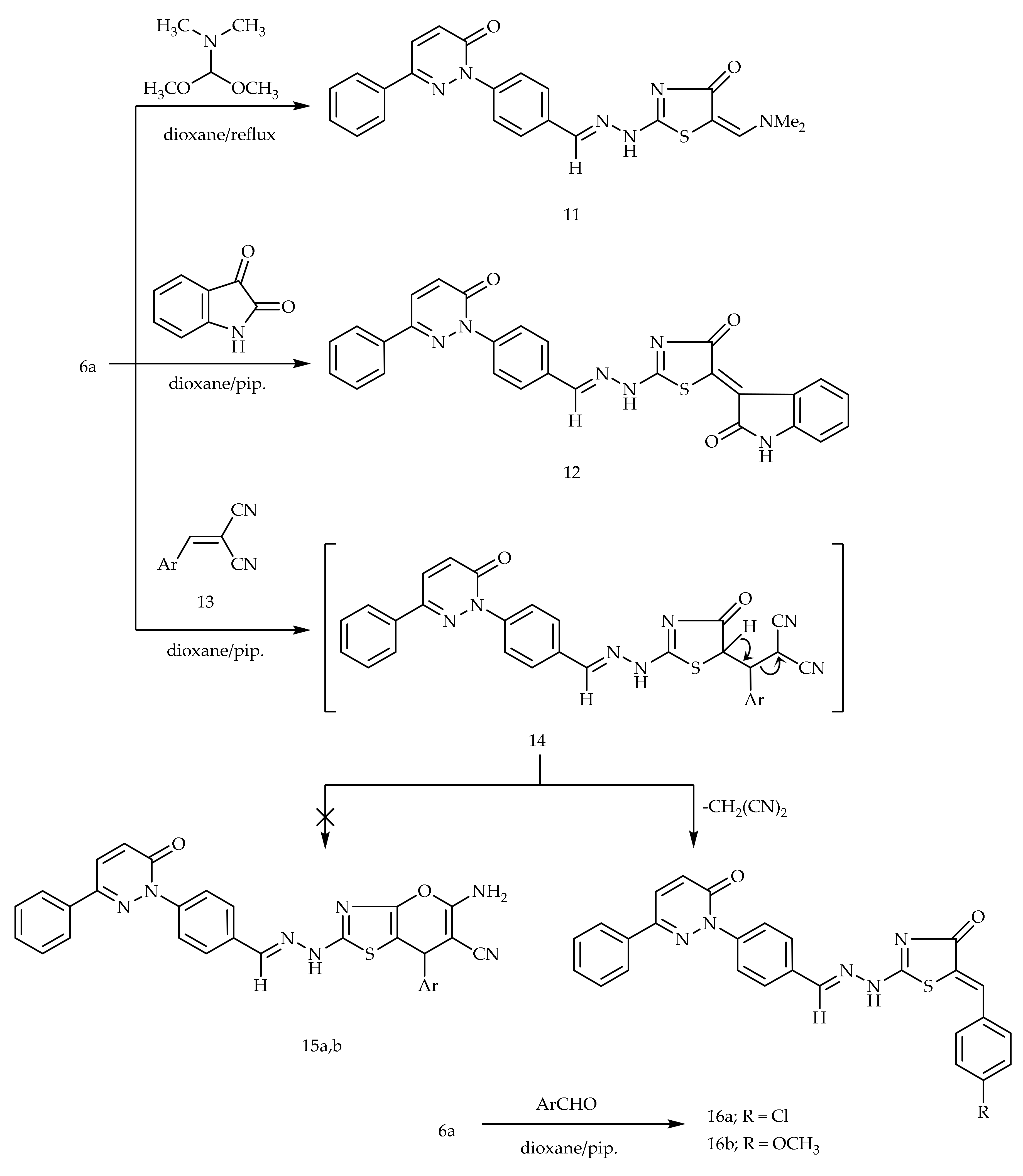
2.1. Chemistry
2.2. Biological Activity
2.2.1. In Vitro COX Inhibitory Action
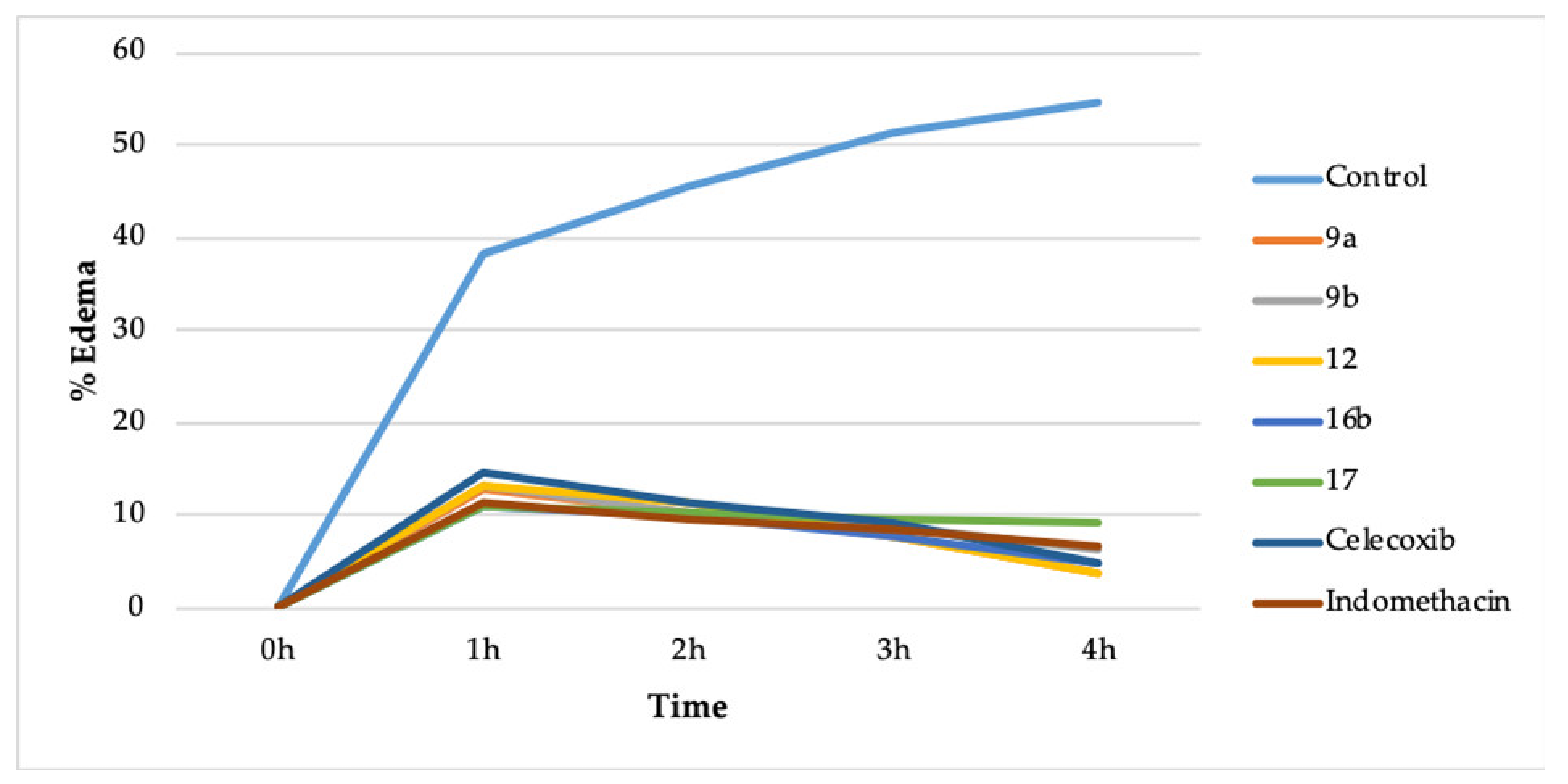
2.2.2. In Vivo Anti-Inflammatory Activity
2.2.3. Ulcerogenic Activity
2.2.4. Lipid Peroxidation Studies
2.3. Molecular Modelling
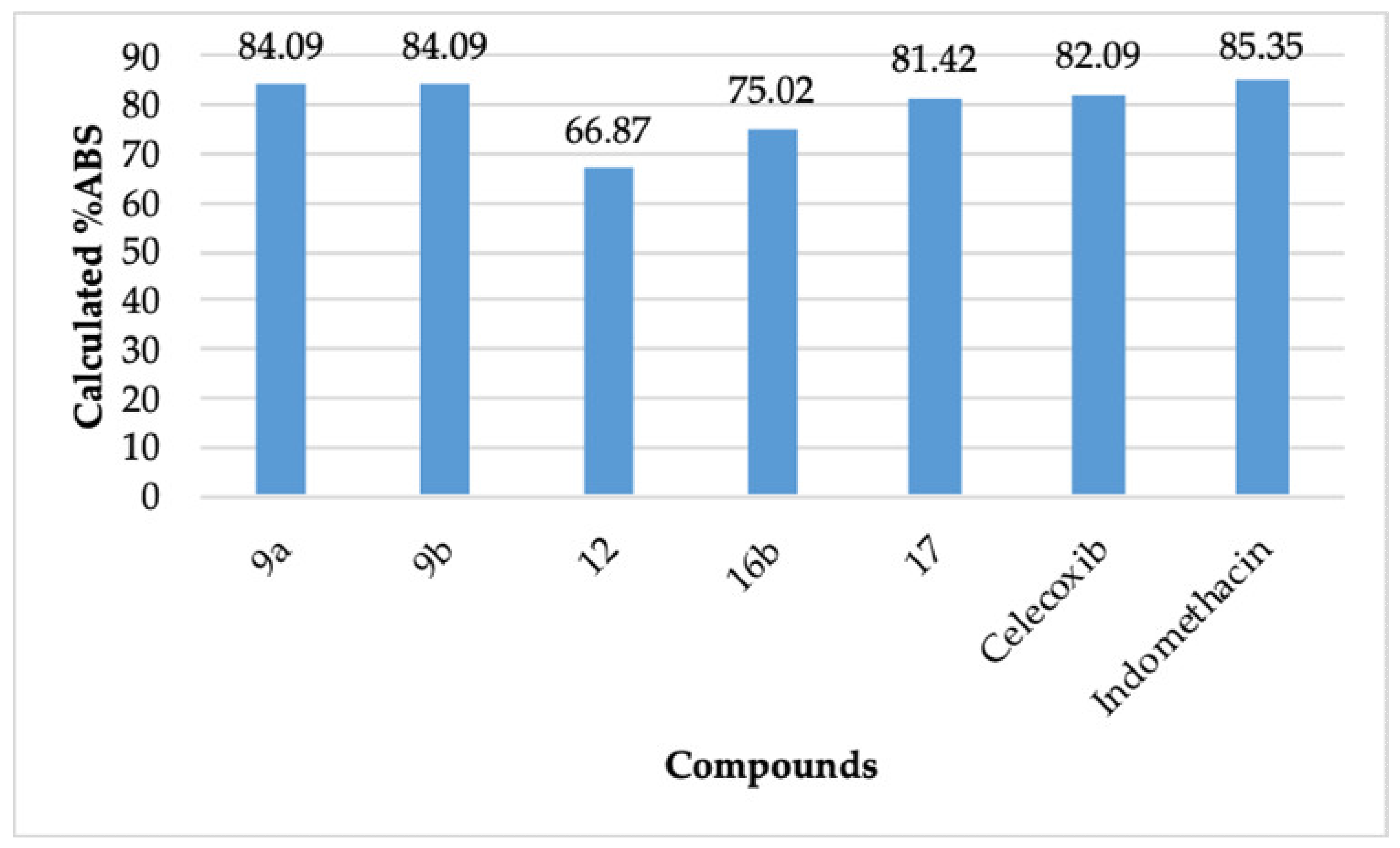
2.4. Physicochemical Parameters
3. Materials and Methods
3.1. Chemistry
3.2. Biological Activity
3.2.1. In Vitro COX-1 and COX-2 Inhibition Assay
3.2.2. In Vivo Anti-Inflammatory Activity
3.2.3. Gastric Ulcerogenic Activity
3.2.4. Lipid Peroxidation Inhibitory Activity
3.3. Molecular Docking
3.4. Physicochemical Parameters
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute Inflammation and Metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef]
- Nasef, N.A.; Mehta, S.; Ferguson, L.R. Susceptibility to chronic inflammation: An update. Arch. Toxicol. 2017, 91, 1131–1141. [Google Scholar] [CrossRef]
- Al Rashoud, A.S.; Abboud, R.J.; Wang, W.; Wigderowitz, C. Efficacy of low-level laser therapy applied at acupuncture points in knee osteoarthritis: A randomised double-blind comparative trial. Physiotherapy 2014, 100, 242–248. [Google Scholar] [CrossRef]
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; Van Wijnen, A.J.; Im, H.J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447. [Google Scholar] [CrossRef]
- Sehajpal, S.; Prasad, D.N.; Singh, R.K. Prodrugs of Non-steroidal Anti-inflammatory Drugs (NSAIDs): A Long March Towards Synthesis of Safer NSAIDs. Mini Rev. Med. Chem. 2018, 18, 1199–1219. [Google Scholar] [CrossRef]
- Radi, Z.A.; Khan, K.N. Cardio-renal safety of non-steroidal anti-inflammatory drugs. J. Toxicol. Sci. 2019, 44, 373–391. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Cyclooxygenase pathways. Acta Biochim. Pol. 2014, 61, 639–649. [Google Scholar] [CrossRef]
- Tortorella, M.D.; Zhang, Y.; Talley, J. Desirable Properties for 3rd Generation Cyclooxygenase-2 Inhibitors. Mini Rev. Med. Chem. 2016, 16, 1284–1289. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Gupta, A.K.; Kumar, S. Developments in synthesis of the anti-inflammatory drug, celecoxib: A review. Recent Pat. Inflamm. Allergy Drug Discov. 2013, 7, 124–134. [Google Scholar] [CrossRef]
- Martina, S.D.; Vesta, K.S.; Ripley, T.L. Etoricoxib: A highly selective COX-2 inhibitor. Ann. Pharmacother. 2005, 39, 854–862. [Google Scholar] [CrossRef]
- Burnier, M. The safety of rofecoxib. Expert Opin. Drug Saf. 2005, 4, 491–499. [Google Scholar] [CrossRef]
- Fanelli, A.; Ghisi, D.; Aprile, P.L.; Lapi, F. Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: Latest evidence and clinical implications. Ther. Adv. Drug Saf. 2017, 8, 173–182. [Google Scholar] [CrossRef]
- Carullo, G.; Galligano, F.; Aiello, F. Structure-activity relationships for the synthesis of selective cyclooxygenase 2 inhibitors: An overview (2009–2016). MedChemComm 2016, 8, 492–500. [Google Scholar] [CrossRef]
- Khalil, N.A.; Ahmed, E.M.; Mohamed, K.O.; Nissan, Y.M.; Zaitone, S.A. Synthesis and biological evaluation of new pyrazolone-pyridazine conjugates as anti-inflammatory and analgesic agents. Bioorg. Med. Chem. 2014, 22, 2080–2089. [Google Scholar] [CrossRef]
- Saeed, M.M.; Khalil, N.A.; Ahmed, E.M.; Eissa, K.I. Synthesis and anti-inflammatory activity of novel pyridazine and pyridazinone derivatives as non-ulcerogenic agents. Arch. Pharm. Res. 2012, 35, 2077–2092. [Google Scholar] [CrossRef]
- Bingham, S.; Beswick, P.J.; Bountra, C.; Brown, T.; Campbell, I.B.; Chessell, I.P.; Clayton, N.; Collins, S.D.; Davey, P.T.; Goodland, H.; et al. The cyclooxygenase-2 inhibitor GW406381X [2-(4-ethoxyphenyl)-3-[4-(methylsulfonyl)phenyl]-pyrazolo[1,5-b]pyridazine] is effective in animal models of neuropathic pain and central sensitization. J. Pharmacol. Exp. Ther. 2005, 312, 1161–1169. [Google Scholar] [CrossRef]
- Ahmed, E.M.; Kassab, A.E.; El-Malah, A.A.; Hassan, M.S.A. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019, 171, 25–37. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, D.; Bansal, R. Pyridazinone: An attractive lead for anti-inflammatory and analgesic drug discovery. Future Med. Chem. 2017, 9, 95–127. [Google Scholar] [CrossRef]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef]
- Fabiola, G.; Damodharan, L.; Pattabhi, V.; Nagarajan, K. Cyclooxygenase-2—An attractive target for fruitful drug design. Curr. Sci. 2001, 80, 26–34. [Google Scholar]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Abdelgawad, M.A.; Elshemy, H.A.; Alsayed, S.S. Design, synthesis and biological screening of new 4-thiazolidinone derivatives with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2016, 64, 1–12. [Google Scholar] [CrossRef]
- Kaminskyy, D.; Kryshchyshyn, A.; Lesyk, R. 5-Ene-4-thiazolidinones—An efficient tool in medicinal chemistry. Eur. J. Med. Chem. 2017, 140, 542–594. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.M.; Abdel-Moty, S.G. Synthesis, biological evaluation and docking study of 1,3,4-thiadiazole-thiazolidinone hybrids as anti-inflammatory agents with dual inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Gupta, S.J.; Fatima, G.N.; Sonar, P.K.; Verma, A.; Saraf, S.K. 4-Thiazolidinones: The advances continue. Eur. J. Med. Chem. 2014, 72, 52–77. [Google Scholar] [CrossRef]
- Unsal-Tan, O.; Ozadali, K.; Piskin, K.; Balkan, A. Molecular modeling, synthesis and screening of some new 4-thiazolidinone derivatives with promising selective COX-2 inhibitory activity. Eur. J. Med. Chem. 2012, 57, 59–64. [Google Scholar] [CrossRef]
- Verma, A.; Saraf, S.K. 4-thiazolidinone—A biologically activescaffold. Eur. J. Med. Chem. 2008, 43, 897–905. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Imran, M.; Nayeem, N. Design, Synthesis, and Anti-Inflammatory Activity of Novel Quinazolines. Orient. J. Chem. 2017, 33, 707–716. [Google Scholar] [CrossRef]
- Khan, A.; Diwan, A.; Thabet, H.K.; Imran, M. Synthesis of novel N-substitutedphenyl-6-oxo-3-phenyl pyridazine derivatives as cyclooxygenase-2 inhibitors. Drug Dev. Res. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Abd El-Samii, Z.K.; Osman, N.A.; Lashine, J.; Kamel, M.A.; Thabet, H.K. Synthesis, molecular docking and anti-inflammatory screening of novel quinoline incorporated pyrazole derivatives using the Pfitzinger reaction II. Bioorg. Chem. 2015, 58, 104–116. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Hamdy, K.T.; Mustafa, T.U. Synthesis, molecular modeling and anti-inflammatory screening of novel fluorinated quinoline incorporated benzimidazole derivatives using the Pfitzinger reaction. J. Fluor. Chem. 2014, 161, 87–94. [Google Scholar] [CrossRef]
- Matheus, M.E.; ViolanteFde, A.; Garden, S.J.; Pinto, A.C.; Fernandes, P.D. Isatins inhibit cyclooxygenase-2 and inducible nitric oxide synthase in a mouse macrophage cell line. Eur. J. Pharmacol. 2007, 556, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animaltissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Banerjee, A.G.; Das, N.; Shengule, S.A.; Sharma, P.A.; Srivastava, R.S.; Shrivastava, S.K. Design, synthesis, evaluation and molecular modelling studies of some novel 5,6-diphenyl-1,2,4-triazin-3(2H)-ones bearing five-member heterocyclic moieties as potential COX-2 inhibitors: A hybrid pharmacophore approach. Bioorg. Chem. 2016, 69, 102–120. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef]
- Pérez-Villanueva, J.; Yépez-Mulia, L.; González-Sánchez, I.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; Sainz-Espuñes, T.D.R.; Cerbón, M.A.; Rodríguez-Villar, K.; Rodríguez-Vicente, A.K.; Cortés-Gines, M.; et al. Synthesis and Biological Evaluation of 2H-Indazole Derivatives: Towards Antimicrobial and Anti-Inflammatory Dual Agents. Molecules 2017, 22, 1864. [Google Scholar] [CrossRef]
- Jiao, J.; Yang, Y.; Wu, Z.; Li, B.; Zheng, Q.; Wei, S.; Wang, Y.; Yang, M. Screening cyclooxygenase-2 inhibitors from Andrographis paniculata to treat inflammation based on bio-affinity ultrafiltration coupled with UPLC-Q-TOF-MS. Fitoterapia 2019, 137, 104259. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: www.molinspiration.com/services/properties.html (accessed on 25 February 2020).
- Lipinski, C.A.; Lombardo, L.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estime solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, F.; Imran, M.; Haq, N.; Alshehri, S.; Anwer, M.K. Synthesis, Characterization and Solubility Determination of 6-Phenyl-pyridazin-3(2H)-one in Different Pharmaceutical Solvents. Molecules 2019, 24, 3404. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.H.; Mohammed, A.A.; Abdellatif, K.R. Novel4-methylsulfonylphenylderivatives as NSAIDS with preferentialCOX-2inhibition. Future Med. Chem. 2018, 10, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Abraham, M.H.; Lee, J. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COX-1 (IC50, nM *) | %COX-1 Inhibition | COX-2 (IC50, nM *) | %COX-2 Inhibition | SI | %SI |
|---|---|---|---|---|---|---|
| 4a | 235 ± 0.11 | 93.61 | 26.19 ± 0.50 | 67.92 | 8.97 | 49.88 |
| 6a | 260 ± 0.22 | 84.61 | 25.10 ± 0.15 | 70.87 | 10.35 | 57.56 |
| 6b | 280 ± 0.50 | 78.57 | 22.75 ± 0.10 | 78.19 | 12.30 | 68.40 |
| 8 | 250 ± 0.12 | 88.0 | 24.18 ± 0.14 | 73.57 | 10.33 | 57.45 |
| 9a | 330 ± 0.28 | 66.66 | 15.50 ± 0.55 | 114.77 | 21.29 | 118.40 |
| 9b | 275 ± 0.42 | 80.0 | 17.50 ± 0.60 | 101.65 | 15.71 | 87.37 |
| 10 | 285 ± 0.15 | 77.19 | 22.11 ± 0.55 | 80.46 | 12.89 | 71.69 |
| 11 | 270 ± 0.24 | 81.48 | 25.11 ± 0.11 | 70.84 | 10.75 | 59.78 |
| 12 | 295 ± 0.28 | 74.57 | 17.10 ± 0.19 | 104.03 | 17.25 | 95.93 |
| 16a | 275 ± 0.10 | 80.0 | 22.16 ± 0.44 | 80.27 | 12.40 | 68.96 |
| 16b | 315 ± 0.33 | 69.84 | 16.90 ± 0.16 | 105.26 | 18.63 | 103.61 |
| 17 | 285 ± 0.16 | 77.19 | 17.70 ± 0.18 | 100.5 | 16.10 | 89.54 |
| 18 | 255 ± 0.20 | 86.27 | 23.15 ± 0.50 | 76.84 | 11.01 | 61.23 |
| Celecoxib | 320 ± 0.01 | 68.75 | 17.79 ± 0.69 | 100.0 | 17.98 | 100.0 |
| Indomethacin | 220 ± 0.01 | 100.0 | 67.72 ± 0.62 | 26.26 | 3.24 | 18.02 |
| Compound | 0 h | 1 h | 2 h | 3 h | 4 h | ||||
|---|---|---|---|---|---|---|---|---|---|
| PD * | PD | % Edema | PD | % Edema | PD | % Edema | PD | % Edema | |
| Control | 3.40 ± 0.20 | 4.70 ± 0.42 | 38.23 | 4.95 ± 0.21 | 45.58 | 5.15 ± 0.10 | 51.47 | 5.25 ± 0.32 | 54.41 |
| 9a | 3.45 ± 0.20 | 3.89 ± 0.14 | 12.75 | 3.81 ± 0.12 | 10.43 | 3.72 ± 0.14 | 7.82 | 3.58 ± 0.36 | 3.76 |
| 9b | 3.44 ± 0.18 | 3.89 ± 0.10 | 13.08 | 3.80 ± 0.28 | 10.46 | 3.74 ± 0.16 | 8.72 | 3.66 ± 0.40 | 6.39 |
| 12 | 3.51 ± 0.16 | 3.98 ± 0.62 | 13.39 | 3.91 ± 0.10 | 11.39 | 3.78 ± 0.22 | 7.69 | 3.64 ± 0.15 | 3.70 |
| 16b | 3.50 ± 0.32 | 3.89 ± 0.21 | 11.14 | 3.85 ± 0.30 | 10.0 | 3.77 ± 0.50 | 7.71 | 3.67 ± 0.42 | 4.85 |
| 17 | 3.49 ± 0.13 | 3.88 ± 0.10 | 11.17 | 3.85 ± 0.13 | 10.31 | 3.82 ± 0.12 | 9.45 | 3.81 ± 0.14 | 9.16 |
| Celecoxib | 3.45 ± 0.22 | 3.95 ± 0.22 | 14.49 | 3.84 ± 0.22 | 11.30 | 3.77 ± 0.33 | 9.27 | 3.62 ± 0.14 | 4.92 |
| Indomethacin | 3.48 ± 0.44 | 3.88 ± 0.26 | 11.49 | 3.81 ± 0.40 | 9.48 | 3.77 ± 0.14 | 8.33 | 3.71 ± 0.33 | 6.60 |
| Group | Score | MDA Content in nmol (100 mg Tissue) | |
|---|---|---|---|
| No of Gastric Ulcers * | Severity Lesions * | ||
| Control | 0 | 0 | 3.24 ± 0.32 |
| 9a | 0 | 0 | 3.05 ± 0.12 |
| 9b | 0.33 ± 0.38 | 0.26 ± 0.16 | 3.89 ± 0.50 |
| 12 | 0 | 0 | 2.95 ± 0.44 |
| 16b | 0.12 ± 0.30 | 0.18 ± 0.22 | 3.45 ± 0.28 |
| 17 | 0.42 ± 0.10 | 0.20 ± 0.50 | 4.11 ± 0.18 |
| Celecoxib | 2.51 ± 0.32 | 5.82 ± 0.44 | 6.11 ± 0.50 |
| Indomethacin | 8.40 ± 0.60 | 12.15 ± 0.10 | 8.90 ± 0.18 |
| Compound | Binding Affinity (kcal/mol) | Hydrogen Bonding Interacting Residues * | Other Interacting Residues |
|---|---|---|---|
| Celecoxib | −9.4 | MET 522 (-NH of sulphonamide group), ARG 120 (Fluorine of -CF3 group) | TRP 387, LEU 352, ALA 527, PHE 518, VAL 349, VAL 523, LEU 359 |
| 9a | −10.8 | CYS 47 (imine group of -CH=N-), TYR 30 (C=O of the pyridazinone) | TYR 136, GLY 135, PRO 156, PRO 154, PRO 153, LYS 468 |
| 9b | −10.2 | CYS 47 (imine group of -CH=N-), TYR 30 and PRO 153 (C=O of the pyridazinone) | PRO 514, PRO 156, SER 353, LYS 468 |
| 12 | −10.5 | ARG 44 (2o-amino group of =N-NH- moiety), HIS 122 (2o-amino group of isatin), TYR 130 (phenyl ring) | ASP 125, LYS 137, VAL 46, PRO 153, CYS 47, CYS 36 |
| 16b | −10.6 | TYR 130 (C=O of the pyridazinone), ASN 34 (C=O of the thiazolidinoe) | TYR 136, ARG 469, LEU 152, PRO 153, CYS 47, CYS 36 |
| 17 | −9.5 | LYS 473 (phenyl ring) | VAL 89, PRO 84, TYR 115, GLU 524, PRO 86 |
| Compound | %ABS | tPSA | nrotb ≤ 10 | nON ≤ 10 | nOHNH ≤ 5 | miLogP ≤ 5 | MW ≤ 500 | n Violations ≤ 1 |
|---|---|---|---|---|---|---|---|---|
| 9a | 84.09 | 72.18 | 6 | 6 | 1 | 5.10 | 449.54 | 1 |
| 9b | 84.09 | 72.18 | 6 | 6 | 1 | 5.91 | 528.43 | 2 |
| 12 | 66.87 | 122.11 | 5 | 9 | 2 | 4.37 | 518.56 | 1 |
| 16b | 75.02 | 98.48 | 7 | 8 | 1 | 4.66 | 507.57 | 1 |
| 17 | 81.42 | 79.93 | 5 | 7 | 0 | 4.24 | 465.54 | 0 |
| Celecoxib | 82.09 | 77.99 | 4 | 5 | 2 | 3.61 | 381.38 | 0 |
| Indomethacin | 85.35 | 68.54 | 4 | 5 | 1 | 3.99 | 357.79 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Diwan, A.; Thabet, H.K.; Imran, M.; Bakht, M.A. Discovery of Novel Pyridazine-Based Cyclooxygenase-2 Inhibitors with a Promising Gastric Safety Profile. Molecules 2020, 25, 2002. https://doi.org/10.3390/molecules25092002
Khan A, Diwan A, Thabet HK, Imran M, Bakht MA. Discovery of Novel Pyridazine-Based Cyclooxygenase-2 Inhibitors with a Promising Gastric Safety Profile. Molecules. 2020; 25(9):2002. https://doi.org/10.3390/molecules25092002
Chicago/Turabian StyleKhan, Abida, Anupama Diwan, Hamdy Kh. Thabet, Mohd Imran, and Md. Afroz Bakht. 2020. "Discovery of Novel Pyridazine-Based Cyclooxygenase-2 Inhibitors with a Promising Gastric Safety Profile" Molecules 25, no. 9: 2002. https://doi.org/10.3390/molecules25092002
APA StyleKhan, A., Diwan, A., Thabet, H. K., Imran, M., & Bakht, M. A. (2020). Discovery of Novel Pyridazine-Based Cyclooxygenase-2 Inhibitors with a Promising Gastric Safety Profile. Molecules, 25(9), 2002. https://doi.org/10.3390/molecules25092002