
Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin

 ,
,  ,
,  ,
, 


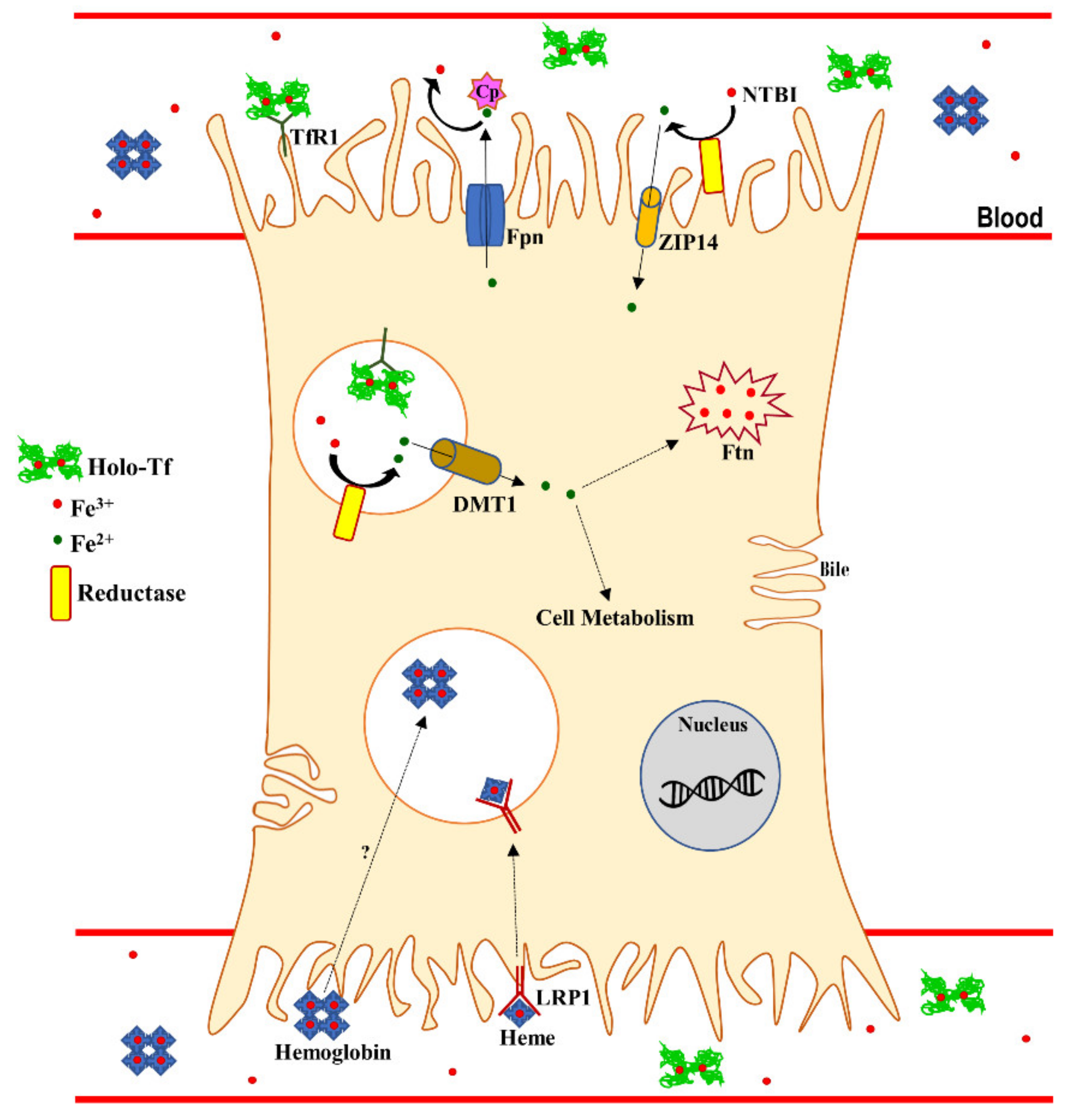{kind=link}
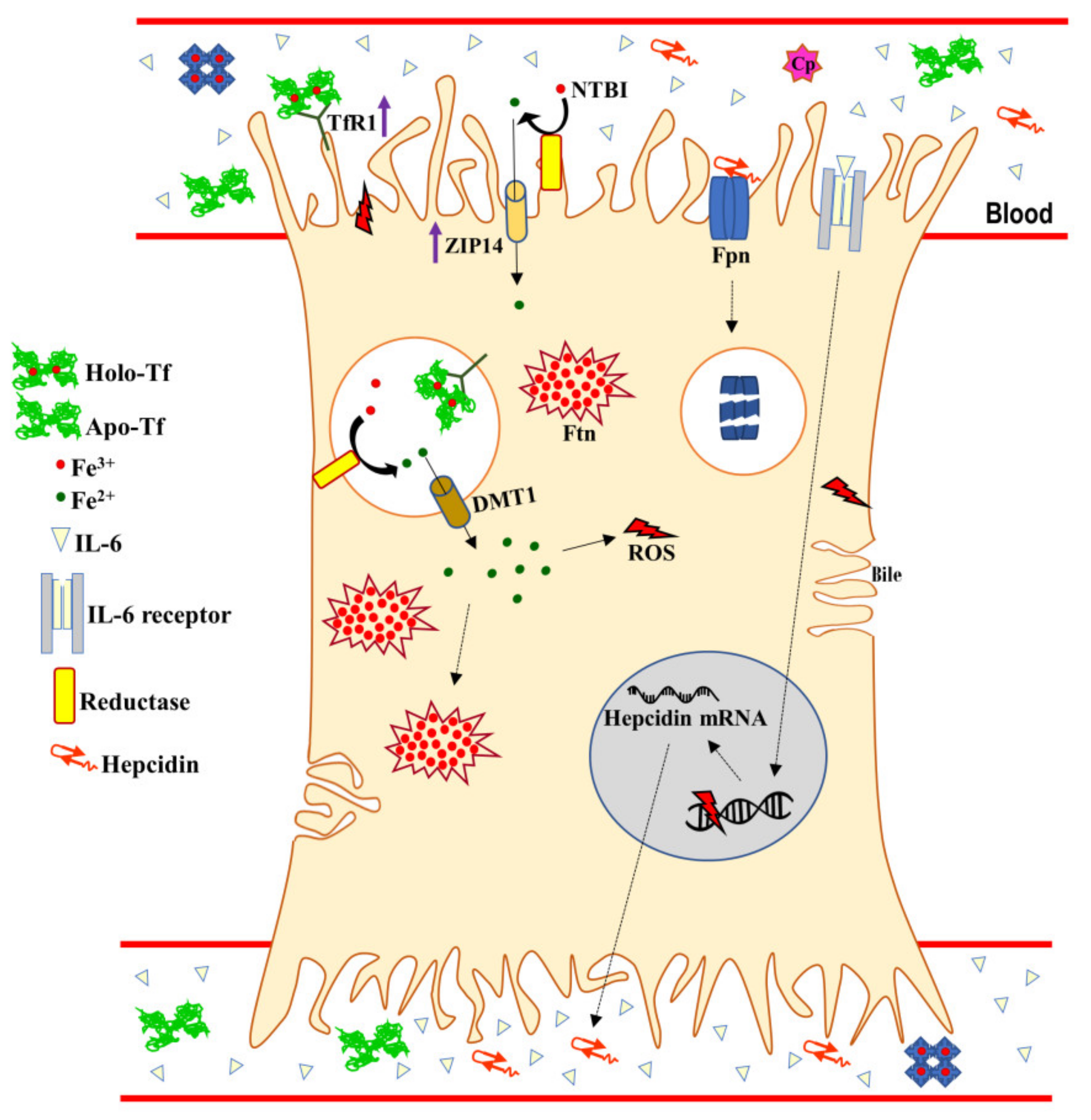{kind=link}
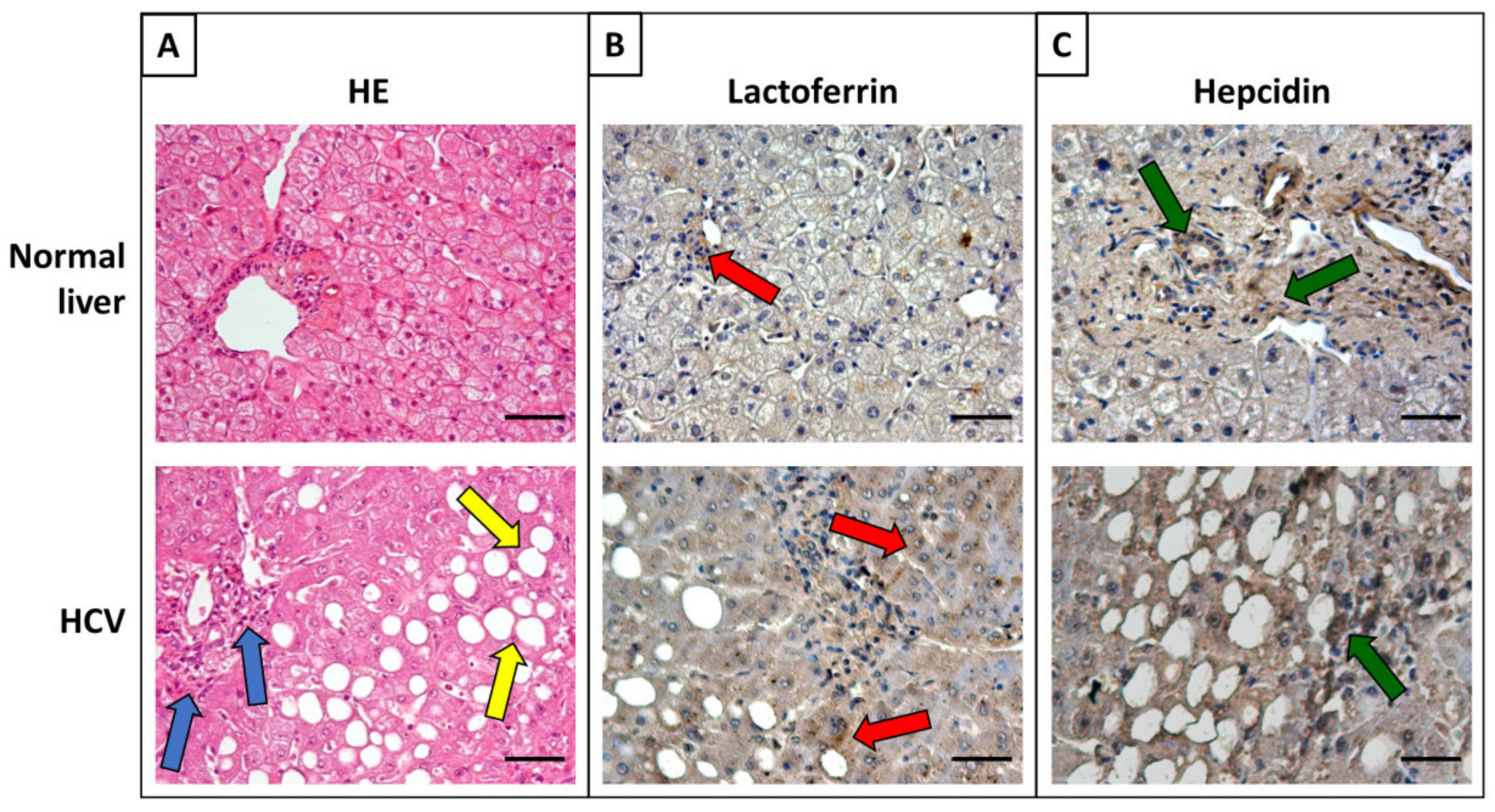{kind=link}
Abstract
1. The Liver as Central Immunological Organ
2. Viral Infections of the Liver: Hepatitis
3. Systemic and Hepatic Iron Homeostasis
4. Lactoferrin and its Receptors
5. Antiviral Activity of Lactoferrin in Apo- and Metal-Saturated Forms
6. Iron Proteins and Lactoferrin in Viral Hepatitis
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Wang, C.Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Son, G.; Kremer, M.; Hines, I.N. Contribution of gut bacteria to liver pathobiology. Gastroenterol. Res. Pr. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 2014, 6, 237ra66. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Thimme, R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014, 146, 1193–1207. [Google Scholar] [CrossRef]
- Sheth, K.; Bankey, P. The liver as an immune organ. Curr. Opin. Crit. Care 2001, 7, 99–104. [Google Scholar] [CrossRef]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Notas, G.; Kisseleva, T.; Brenner, D. NK and NKT cells in liver injury and fibrosis. Clin. Immunol. 2009, 130, 16–26. [Google Scholar] [CrossRef]
- Matsumura, T.; Ito, A.; Takii, T.; Hayashi, H.; Onozaki, K. Endotoxin and cytokine regulation of toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J. Interferon Cytokine Res. 2000, 20, 915–921. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef]
- Sato, T.; Yamamoto, H.; Sasaki, C.; Wake, K. Maturation of rat dendritic cells during intrahepatic translocation evaluated using monoclonal antibodies and electron microscopy. Cell Tissue Res. 1998, 294, 503–514. [Google Scholar] [CrossRef]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef]
- Li, G.; Kim, Y.J.; Broxmeyer, H.E. Macrophage colony-stimulating factor drives cord blood monocyte differentiation into IL-10(high)IL-12absent dendritic cells with tolerogenic potential. J. Immunol. 2005, 174, 4706–4717. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Jürgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef]
- Tu, Z.; Bozorgzadeh, A.; Pierce, R.H.; Kurtis, J.; Crispe, I.N.; Orloff, M.S. TLR-dependent cross talk between human Kupffer cells and NK cells. J. Exp. Med. 2008, 205, 233–244. [Google Scholar] [CrossRef]
- De Winter, B.; Hesselink, D.A.; Kamar, N. Dosing ribavirin in hepatitis E-infected solid organ transplant recipients. Pharm. Res. 2018, 130, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Abravanel, F.; Lhomme, S.; Rostaing, L.; Izopet, J. Hepatitis E virus: Chronic infection, extra-hepatic manifestations, and treatment. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.; Grytdal, S.; Wasley, A. Centers for Disease Control and Prevention (CDC). Surveillance for acute viral hepatitis - United States, 2007. MMWR Surveill. Summ. 2009, 58, 1–27. [Google Scholar] [PubMed]
- Zuckerman, A.J.; Baron, S. (Eds.) Source Medical Microbiology. In Hepatitis Viruses, 4th ed.; Galveston (TX): University of Texas Medical Branch: Texas, TX, USA, 1996. [Google Scholar]
- Mehta, P.; Reddivari, A.K.R. SourceStatPearls [Internet]. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554549/ (accessed on 28 January 2020).
- Ryder, S.D.; Beckingham, I.J. ABC of diseases of liver, pancreas, and biliary system: Acute hepatitis. B.M.J. 2001, 322, 51–53. [Google Scholar] [CrossRef]
- Sánchez, G.; Bosch, A.; Pintó, R.M. Hepatitis A virus detection in food: Current and future prospects. Lett. Appl. Microbiol. 2007, 45, 1–5. [Google Scholar] [CrossRef]
- Koenig, K.L.; Shastry, S.; Burns, M.J. Hepatitis A Virus: Essential Knowledge and a Novel Identify-Isolate-Inform Tool for Frontline Healthcare Providers. West. J. Emerg. Med. 2017, 18, 1000–1007. [Google Scholar] [CrossRef]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology. 2009, 49, S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, J.; Lu, Y.; Luo, S.; Zhang, J.; Zhu, P. Cryo-EM structure of native spherical subviral particles isolated from HBV carriers. Virus Res. 2019, 259, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yang, Z.; Liu, Y.; Zheng, M. The role of HBV-induced autophagy in HBV replication and HBV related-HCC. Life Sci. 2018, 205, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol 2018, 3, 383–403. [Google Scholar] [CrossRef]
- You, C.R.; Lee, S.W.; Jang, J.W.; Yoon, S.K. Update on hepatitis B virus infection. World J. Gastroenterol. 2014, 20, 13293–13305. [Google Scholar] [CrossRef]
- Li, H.C.; Lo, S.Y. Hepatitis C virus: Virology, diagnosis and treatment. World J. Hepatol. 2015, 7, 1377–1389. [Google Scholar] [CrossRef]
- Wang, X.; Yan, Y.; Gan, T.; Yang, X.; Li, D.; Zhou, D.; Sun, Q.; Huang, Z.; Zhong, J. A trivalent HCV vaccine elicits broad and synergistic polyclonal antibody response in mice and rhesus monkey. Gut 2019, 68, 140–149. [Google Scholar] [CrossRef]
- Rizzetto, M.; Hepatitis, D. Virus: Introduction and Epidemiology. Cold Spring. Harb. Perspect. Med. 2015, 5, a021576. [Google Scholar] [CrossRef]
- Cao, Y.; Bing, Z.; Guan, S.; Zhang, Z.; Wang, X. Development of new hepatitis E vaccines. Hum. Vaccin. Immunother. 2018, 14, 2254–2262. [Google Scholar] [CrossRef]
- Pérez-Gracia, M.T.; García, M.; Suay, B.; Mateos-Lindemann, M.L. Current Knowledge on Hepatitis E. J. Clin. Transl. Hepatol. 2015, 3, 117–126. [Google Scholar] [CrossRef]
- Liberal, R.; Grant, C.R.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2013, 41, 126–139. [Google Scholar] [CrossRef]
- Chayanupatkul, M.; Liangpunsakul, S. Alcoholic hepatitis: A comprehensive review of pathogenesis and treatment. World J. Gastroenterol. 2014, 20, 6279–6286. [Google Scholar] [CrossRef]
- Gao, Y.H.; Wang, J.Y.; Liu, P.Y.; Sun, J.; Wang, X.M.; Wu, R.H.; He, X.T.; Tu, Z.K.; Wang, C.G.; Xu, H.Q.; et al. Iron metabolism disorders in patients with hepatitis B-related liver diseases. World J. Clin. Cases 2018, 6, 600–610. [Google Scholar] [CrossRef]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 41, 1986–1995, Erratum in: 2000, 342, 364. [Google Scholar] [CrossRef]
- Bullen, J.J.; Rogers, H.J.; Griffiths, E. Role of iron in bacterial infection. Curr. Top. Microbiol. Immunol. 1978, 80, 1–35. [Google Scholar]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A Natural Glycoprotein Involved in Iron and Inflammatory Homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell. Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Marques., L.; Auriac, A.; Willemetz, A.; Banha, J.; Silva, B.; Canonne-Hergaux, F.; Costa, L. Immune cells and hepatocytes express glycosylphosphatidylinositol-anchored ceruloplasmin at their cell surface. Blood Cells Mol. Dis 2012, 48, 110–120. [Google Scholar] [CrossRef]
- Bonaccorsi di Patti, M.C.; Cutone, A.; Polticelli, F.; Rosa, L.; Lepanto, M.S.; Valenti, P.; Musci, G. The ferroportin-ceruloplasmin system and the mammalian iron homeostasis machine: Regulatory pathways and the role of lactoferrin. Biometals 2018, 31, 399–414. [Google Scholar] [CrossRef]
- Luck, A.N.; Mason, A.B. Transferrin-mediated cellular iron delivery. Curr. Top. Membr. 2012, 69, 3–35. [Google Scholar] [CrossRef]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The Intracellular Trafficking Pathway of Transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281. [Google Scholar] [CrossRef]
- Knutson, M.D. Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J. Biol. Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef]
- Brissot, P.; Wright, T.L.; Ma, W.L.; Weisiger, R.A. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Invest. 1985, 76, 1463–1470. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-i -induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Neitz, S.; Mägert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 1480, 147–150. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Lee, P.; Peng, H.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; VujicSpasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef]
- Cutone, A.; Frioni, A.; Berlutti, F.; Valenti, P.; Musci, G.; Bonaccorsi di Patti, M.C. Lactoferrin prevents LPS-induced decrease of the iron exporter ferroportin in human monocytes/macrophages. Biometals 2014, 27, 807–813. [Google Scholar] [CrossRef]
- Cutone, A.; Rosa, L.; Lepanto, M.S.; Scotti, M.J.; Berlutti, F.; Bonaccorsi di Patti, M.C.; Musci, G.; Valenti, P. Lactoferrin Efficiently Counteracts the Inflammation- Induced Changes of the Iron Homeostasis System in Macrophages. Front. Immunol. 2017, 8, 705. [Google Scholar] [CrossRef]
- Malik, I.A.; Naz, N.; Sheikh, N.; Khan, S.; Moriconi, F.; Blaschke, M.; Ramadori, G. Comparison of changes in gene expression of transferrin receptor-1 and other iron-regulatory proteins in rat liver and brain during acute-phase response. Cell Tissue Res. 2011, 344, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Lichten, L.A.; Liuzzi, J.P.; Cousins, R.J. Interleukin-1beta contributes via nitric oxide to the upregulation and functional activity of the zinc transporter Zip14 (Slc39a14) in murine hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G860–G867. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Anderson, G.J. The orchestration of body iron intake: How and where do enterocytes receive their cues? Blood Cells Mol. Dis. 2003, 30, 288–297. [Google Scholar] [CrossRef]
- Lepanto, M.S.; Rosa, L.; Cutone, A.; Conte, M.P.; Paesano, R.; Valenti, P. Efficacy of Lactoferrin Oral Administration in the Treatment of Anemia and Anemia of Inflammation in Pregnant and Non-pregnant Women: An Interventional Study. Front. Immunol. 2018, 9, 2123. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.D. Iron withholding: A defense against viral infections. BioMetals 1996, 9, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef]
- Johnson, E.E.; Wessling-Resnick, M. Iron metabolism and the innate immune response to infection. Microbes Infect. 2012, 14, 207–216. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Crossing the Iron Gate: Why and How Transferrin Receptors Mediate Viral Entry. Annu. Rev. Nutr. 2018, 38, 431–458. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A.M. Hepcidin and the iron-infection axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef]
- Lepanto, M.S.; Rosa, L.; Cutone, A.; Scotti, M.J.; Conte, A.L.; Marazzato, M.; Zagaglia, C.; Longhi, C.; Berlutti, F.; Musci, G.; et al. Bovine Lactoferrin Pre-Treatment Induces Intracellular Killing of AIEC LF82 and Reduces Bacteria-Induced DNA Damage in Differentiated Human Enterocytes. Int. J. Mol. Sci. 2019, 20, 5666. [Google Scholar] [CrossRef]
- Berlutti, F.; Schippa, S.; Morea, C.; Sarli, S.; Perfetto, B.; Donnarumma, G.; Valenti, P. Lactoferrin downregulates pro-inflammatory cytokines up-expressed in intestinal epithelial cells infected with invasive or noninvasive Escherichia coli strains. Biochem. Cell Biol. 2006, 84, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Frioni, A.; Conte, M.P.; Cutone, A.; Longhi, C.; Musci, G.; di Patti, M.C.; Natalizi, T.; Marazzato, M.; Lepanto, M.S.; Puddu, P.; et al. Lactoferrin differently modulates the inflammatory response in epithelial models mimicking human inflammatory and infectious diseases. Biometals 2014, 27, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Valenti, P.; Frioni, A.; Rossi, A.; Ranucci, S.; De Fino, I.; Cutone, A.; Rosa, L.; Bragonzi, A.; Berlutti, F. Aerosolized bovine lactoferrin reduces neutrophils and pro-inflammatory cytokines in mouse models of Pseudomonas aeruginosa lung infections. Biochem. Cell Biol. 2017, 95, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Di Pietro, M.; Filardo, S.; Bressan, A.; Rosa, L.; Cutone, A.; Frioni, A.; Berlutti, F.; Paesano, R.; Valenti, P. Effect of bovine lactoferrin on Chlamydia trachomatis infection and inflammation. Biochem. Cell Biol. 2017, 95, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Di Pietro, M.; Filardo, S.; Bressan, A.; Mastromarino, P.; Biasucci, A.V.; Rosa, L.; Cutone, A.; Berlutti, F.; Paesano, R.; et al. Lactobacilli-lactoferrin interplay in Chlamydia trachomatis infection. Pathog. Dis. 2017, 75, 1–9. [Google Scholar] [CrossRef]
- Cutone, A.; Lepanto, M.S.; Rosa, L.; Scotti, M.J.; Rossi, A.; Ranucci, S.; De Fino, I.; Bragonzi, A.; Valenti, P.; Musci, G.; et al. Aerosolized Bovine Lactoferrin Counteracts Infection, Inflammation and Iron Dysbalance in A Cystic Fibrosis Mouse Model of Pseudomonas aeruginosa Chronic Lung Infection. Int. J. Mol. Sci. 2019, 20, 2128. [Google Scholar] [CrossRef]
- Armitage, A.E.; Stacey, A.R.; Giannoulatou, E.; Marshall, E.; Sturges, P.; Chatha, K.; Smith, N.M.; Huang, X.; Xu, X.; Pasricha, S.R. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. PNAS 2014, 111, 12187–12192. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef]
- Schmidt, S.M. The role of iron in viral infections. Front. Biosci. (Landmark Ed.) 2020, 25, 893–911. [Google Scholar] [CrossRef]
- Lawrence, C.M.; Ray, S.; Babyonyshev, M.; Galluser, R.; Borhani, D.W.; Harrison, S.C. Crystal structure of the ectodomain of human transferrin receptor. Science 1999, 286, 779–782. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Giannetti, A.M.; Snow, P.M.; Zak, O.; Bjorkman, P.J. Mechanism for multiple ligand recognition by the human transferrin receptor. PLOS Biol. 2003, 1, e51. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Murphy, W.J.; Wang, D.; O’Brien, S.J.; Parrish, C.R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 2001, 75, 3896–3902. [Google Scholar] [CrossRef] [PubMed]
- Park, G.S.; Best, S.M.; Bloom, M.E. Two mink parvoviruses use different cellular receptors for entry into CRFK cells. Virology 2005, 340, 1–9. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Sarute, N.; Ross, S.R. New World arenavirus biology. Annu. Rev. Virol. 2017, 4, 141–158. [Google Scholar] [CrossRef]
- Fillebeen, C.; Pantopoulos, K. Hepatitis C virus infection causes iron deficiency in Huh7.5.1 cells. Plos One 2013, 8, e83307. [Google Scholar] [CrossRef]
- Haberger, V.; Elgner, F.; Roos, J.; Bender, D.; Hildt, E. Regulation of the Transferrin Receptor Recycling in Hepatitis C Virus-Replicating Cells. Front. Cell. Dev. Biol. 2020, 8, 44. [Google Scholar] [CrossRef]
- Anderson, B.F.; Baker, H.M.; Dodson, E.J.; Norris, G.E.; Rumball, S.V.; Waters, J.M.; Baker, E.N. Structure of human lactoferrin at 3.2-A resolution. Proc. Natl. Acad. Sci. Usa 1987, 84, 1769–1773. [Google Scholar] [CrossRef]
- Anderson, B.F.; Baker, H.M.; Norris, G.E.; Rice, D.W.; Baker, E.N. Structure of human lactoferrin: Crystallographic structure analysis and refinement at 2.8 A resolution. J. Mol. Biol. 1989, 209, 711–734. [Google Scholar] [CrossRef]
- Baker, E.N.; Rumball, S.V.; Anderson, B.F. Transferrins: Insights into structure and function from studies on lactoferrin. Trends Biochem. Sci. 1987, 12, 350–353. [Google Scholar] [CrossRef]
- Bruns, C.M.; Nowalk, A.J.; Arvai, A.S.; McTigue, M.A.; Vaughan, K.G.; Mietzner, T.A.; McRee, D.E. Structure of Haemophilus influenza Fe(+3)-binding protein reveals convergent evolution within a superfamily. Nat. Struct. Biol. 1997, 4, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Scotti, M.J.; Conte, M.P.; Paesano, R.; Valenti, P. Physico-chemical properties influence the functions and efficacy of commercial bovine lactoferrins. Biometals 2018, 31, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.F.; Baker, H.M.; Norris, G.E.; Rumball, S.V.; Baker, E.N. Apolactoferrin structure demonstrates ligand-induced conformational change in transferrins. Nature 1990, 344, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Spik, G.; Brunet, B.; Mazurier-Dehaine, C.; Fontaine, G.; Montreuil, J. Characterization and properties of the human and bovine lactotransferrins extracted from the faeces of newborn infants. Acta Paediatr. Scand. 1982, 71, 979–985. [Google Scholar] [CrossRef]
- Van Berkel, P.H.; van Veen, H.A.; Geerts, M.E.; de Boer, H.A.; Nuijens, J.H. Heterogeneity in utilization of N-glycosylation sites Asn624 and Asn138 in human lactoferrin: A study with glycosylation-site mutants. Biochem. J. 1996, 319, 117–122. [Google Scholar] [CrossRef]
- Wang, C.S.; Chan, W.Y.; Kloer, U.H. Comparative studies on the chemical and immunochemical properties of human milk, human pancreatic juice and bovine milk lactoferrin. Comp. Biochem. Physiol. B. 1984, 78, 575–580. [Google Scholar] [CrossRef]
- Wei, Z.; Nishimura, T.; Yoshida, S. Presence of a glycan at a potential N-glycosylation site, Asn-281, of bovine lactoferrin. J. Dairy Sci. 2000, 83, 683–689. [Google Scholar] [CrossRef]
- Valenti, P.; Antonini, G. Lactoferrin: An important host defence against microbial and viral attack. Cell. Mol. Life Sci. 2005, 62, 2576–2587. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.D. Human immunodeficiency virus infection, anemia, and survival. Clin. Infect. Dis. 1999, 29, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Friis, H.; Gomo, E.; Nyazema, N.; Ndhlovu, P.; Krarup, H.; Madsen, P.H.; Michaelsen, K.F. Iron, haptoglobin phenotype, and HIV-1 viral load: A cross-sectional study among pregnant Zimbabwean women. J. Acquir. Immune Defic. Syndr. 2003, 33, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Onojobi, G.; Schneider, M.F.; Dawkins, F.W.; Delapenha, R.; Voloshin, Y.; von Wyl, V.; Bacon, M.; Minkoff, H.; Levine, A.; et al. The association of serum ferritin and transferrin receptor concentrations with mortality in women with human immunodeficiency virus infection. Haematologica 2006, 91, 739–743. [Google Scholar]
- Puddu, P.; Borghi, P.; Gessani, S.; Valenti, P.; Belardelli, F.; Seganti, L. Antiviral effect of bovine lactoferrin saturated with metal ions on early steps of human immunodeficiency virus type 1 infection. Int. J. Biochem. Cell Biol. 1998, 30, 1055–1062. [Google Scholar] [CrossRef]
- Berkhout, B.; van Wamel, J.L.; Beljaars, L.; Meijer, D.K.; Visser, S.; Floris, R. Characterization of the anti-HIV effects of native lactoferrin and other milk proteins and protein-derived peptides. Antivir. Res. 2002, 55, 341–355. [Google Scholar] [CrossRef]
- Berlutti, F.; Pantanella, F.; Natalizi, T.; Frioni, A.; Paesano, R.; Polimeni, A.; Valenti, P. Antiviral properties of lactoferrin—A natural immunity molecule. Molecules 2011, 16, 6992–7018. [Google Scholar] [CrossRef]
- Valenti, P.; Rosa, L.; Capobianco, D.; Lepanto, M.S.; Schiavi, E.; Cutone, A.; Paesano, R.; Mastromarino, P. Role of Lactobacilli and Lactoferrin in the Mucosal Cervicovaginal Defense. Front. Immunol. 2018, 9, 376. [Google Scholar] [CrossRef]
- Legrand, D. Lactoferrin, a key molecule in immune and inflammatory processes. Biochem. Cell Biol. 2012, 90, 252–268. [Google Scholar] [CrossRef]
- Lepanto, M.S.; Rosa, L.; Paesano, P.; Valenti, P.; Cutone, A. Lactoferrin in Aseptic and Septic Inflammation. Molecules 2019, 24, 1323. [Google Scholar] [CrossRef]
- Cutone, A.; Rosa, L.; Ianiro, G.; Lepanto, M.S.; Bonaccorsi di Patti, M.C.; Valenti, P.; Musci, G. Lactoferrin’s Anti-Cancer Properties: Safety, Selectivity, and Wide Range of Action. Biomolecules 2020, 10, 456. [Google Scholar] [CrossRef]
- Ashida, K.; Sasaki, H.; Suzuki, Y.A.; Lönnerdal, B. Cellular internalization of lactoferrin in intestinal epithelial cells. Biometals 2004, 17, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.A.; Wong, H.; Ashida, K.Y.; Schryvers, A.B.; Lönnerdal, B. The N1 domain of human lactoferrin is required for internalization by Caco-2 cells and targeting to the nucleus. Biochemistry 2008, 47, 10915–10920. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Jiang, R.; Lönnerdal, B. Biochemical and molecular impacts of lactoferrin on small intestinal growth and development during early life. Biochem. Cell. Biol. 2012, 90, 476–484. [Google Scholar] [CrossRef]
- Jiang, R.; Lopez, V.; Kelleher, S.L.; Lönnerdal, B. Apo- and holo-lactoferrin are both internalized by lactoferrin receptor via clathrin-mediated endocytosis but differentially affect ERK-signaling and cell proliferation in Caco-2 cells. J. Cell Physiol. 2011, 226, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Legrand, D. Overview of Lactoferrin as a Natural Immune Modulator. J. Pediatr. 2016, 173, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.A.; Lopez, V.; Lönnerdal, B. Mammalian lactoferrin receptors: Structure and function. Cell. Mol. Life Sci. 2005, 62, 2560–2575. [Google Scholar] [CrossRef]
- Kawakami, H.; Lönnerdal, B. Isolation and function of a receptor for human lactoferrin in human fetal intestinal brush-border membranes. Am. J. Physiol. 1991, 261, G841–G846. [Google Scholar] [CrossRef]
- Suzuki, Y.A.; Shin, K.; Lönnerdal, B. Molecular cloning and functional expression of a human intestinal lactoferrin receptor. Biochemistry 2001, 40, 15771–15779. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Oshima, K.; Kuhara, T.; Shin, K.; Abe, F.; Iwatsuki, K.; Nadano, D.; Matsuda, T. A lactoferrin-receptor, intelectin 1, affects uptake, sub-cellular localization and release of immunochemically detectable lactoferrin by intestinal epithelial Caco-2 cells. J. Biochem. 2013, 154, 437–448. [Google Scholar] [CrossRef]
- Wrackmeyer, U.; Hansen, G.H.; Seya, T.; Danielsen, E.M. Intelectin: A novel lipid raft-associated protein in the enterocyte brush border. Biochemistry 2006, 45, 9188–91897. [Google Scholar] [CrossRef]
- Mancinelli, R.; Olivero, F.; Carpino, G.; Overi, D.; Rosa, L.; Lepanto, M.S.; Cutone, A.; Franchitto, A.; Alpini, G.; Onori, P.; et al. Role of lactoferrin and its receptors on biliary epithelium. Biometals 2018, 31, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.M. The Role of Lactoferrin in Gastrointestinal and Immune Development and Function: A Preclinical Perspective. J. Pediatr. 2016, 173, S16–S28. [Google Scholar] [CrossRef] [PubMed]
- Demmelmair, H.; Prell, C.; Timby, N.; Lönnerdal, B. Benefits of Lactoferrin, Osteopontin and Milk Fat Globule Membranes for Infants. Nutrients 2017, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Iigo, M.; Kuhara, T.; Ushida, Y.; Sekine, K.; Moore, M.A.; Tsuda, H. Inhibitory effects of bovine lactoferrin on colon carcinoma 26 lung metastasis in mice. Clin. Exp. Metastasis 1999, 17, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kuhara, T.; Iigo, M.; Itoh, T.; Ushida, Y.; Sekine, K.; Terada, N.; Okamura, H.; Tsuda, H. Orally administered lactoferrin exerts an antimetastatic effect and enhances production of IL-18 in the intestinal epithelium. Nutr. Cancer 2000, 38, 192–199. [Google Scholar] [CrossRef]
- Herz, J.; Strickland, D.K. LRP: A multifunctional scavenger and signaling receptor. J. Clin. Invest. 2001, 108, 779–784. [Google Scholar] [CrossRef]
- Jeon, H.; Blacklow, S.C. Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar] [CrossRef]
- Lin, L.; Hu, K. LRP-1: Functions, Signalling and Implications in Kidney and Other Diseases. Int. J. Mol. Sci. 2014, 15, 22887–22901. [Google Scholar] [CrossRef]
- Hofer, F.; Gruenberger, M.; Kowalski, H.; Machat, H.; Huettinger, M.; Kuechler, E.; Blaas, D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1839–1842. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Gern, J.E. Rhinoviruses and Their Receptors: Implications for Allergic Disease. Curr. Allergy Asthma Rep. 2016, 16, 30. [Google Scholar] [CrossRef]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A.; He., J.J. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med 2000, 6, 1380–1387. [Google Scholar] [CrossRef]
- Caruz, A.; Neukam, K.; Rivero-Juárez, A.; Herrero, R.; Real, L.M.; Camacho, A.; Barreiro, P.; Labarga, P.; Rivero, A.; Pineda, J.A. Association of low-density lipoprotein receptor genotypes with hepatitis C viral load. Genes Immun. 2014, 15, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef]
- Herker, E.; Ott, M. Emerging role of lipid droplets in host/pathogen interactions. J. Biol. Chem. 2012, 287, 2280–2287. [Google Scholar] [CrossRef]
- Vieyres, G.; Pietschmann, T. HCV Pit Stop at the Lipid Droplet: Refuel Lipids and Put on a Lipoprotein Coat before Exit. Cells. 2019, 8, 233. [Google Scholar] [CrossRef]
- Syed, G.H.; Khan, M.; Yang, S.; Siddiqui, A. Hepatitis C Virus Lipoviroparticles Assemble in the Endoplasmic Reticulum (ER) and Bud off from the ER to the Golgi Compartment in COPII Vesicles. J. Virol. 2017, 91, e00499-17. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Legrand, D.; Vigié, K.; Said, E.A.; Elass, E.; Masson, M.; Slomianny, M.C.; Carpentier, M.; Briand, J.P.; Mazurier, J.; Hovanessian, A.G. Surface nucleolin participates in both the binding and endocytosis of lactoferrin in target cells. Eur. J. Biochem. 2004, 271, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Losfeld, M.E.; Khoury, D.E.; Mariot, P.; Carpentier, M.; Krust, B.; Briand, J.P.; Mazurier, J.; Hovanessian, A.G.; Legrand, D. The cell surface expressed nucleolin is a glycoprotein that triggers calcium entry into mammalian cells. Exp. Cell. Res. 2009, 315, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Wakabayashi, H.; Yamauchi, K.; Yaeshima, T.; Iwatsuki, K. Recombinant human intelectin binds bovine lactoferrin and its peptides. Biol. Pharm. Bull. 2008, 31, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Paesano, R.; Natalizi, T.; Berlutti, F.; Valenti, P. Body iron delocalization: The serious drawback in iron disorders in both developing and developed countries. Pathog. Glob. Health 2012, 106, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hangoc, G.; Oliff, A.; Chen, L.T.; Shen, R.N.; Broxmeyer, H.E. Protective influence of lactoferrin on mice infected with the polycythemia-inducing strain of Friend virus complex. Cancer Res. 1987, 47, 4184–4188. [Google Scholar]
- Wakabayashi, H.; Oda, H.; Yamauchi, K.; Abe, F. Lactoferrin for prevention of common viral infections. J. Infect. Chemother. 2014, 20, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Longhi, C.; Conte, M.P.; Pisani, S.; Valenti, P.; Seganti, L. Lactoferrin inhibits herpes simplex virus type 1 adsorption to Vero cells. Antiviral Res. 1996, 29, 221–231. [Google Scholar] [CrossRef]
- Marchetti, M.; Pisani, S.; Antonini, G.; Valenti, P.; Seganti, L.; Orsi, N. Metal complexes of bovine lactoferrin inhibit in vitro replication of herpes simplex virus type 1 and 2. Biometals 1998, 11, 89–94. [Google Scholar] [CrossRef]
- Marchetti, M.; Superti, F.; Ammendolia, M.G.; Rossi, P.; Valenti, P.; Seganti, L. Inhibition of poliovirus type 1 infection by iron-, manganese- and zinc-saturated lactoferrin. Med. Microbiol. Immunol. 1999, 187, 199–204. [Google Scholar] [CrossRef]
- Siciliano, R.; Rega, B.; Marchetti, M.; Seganti, L.; Antonini, G.; Valenti, P. Bovine lactoferrin peptidic fragments involved in inhibition of herpes simplex virus type 1 infection. Biochem. Biophys. Res. Commun. 1999, 264, 19–23. [Google Scholar] [CrossRef]
- Superti, F.; Siciliano, R.; Rega, B.; Giansanti, F.; Valenti, P.; Antonini, G. Involvement of bovine lactoferrin metal saturation, sialic acid and protein fragments in the inhibition of rotavirus infection. Biochim. Biophys. Acta. 2001, 1528, 107–115. [Google Scholar] [CrossRef]
- El Yazidi-Belkoura, I.; Legrand, D.; Nuijens, J.; Slomianny, M.C.; van Berkel, P.; Spik, G. The binding of lactoferrin to glycosaminoglycans on enterocyte-like HT29–18-C1 cells is mediated through basic residues located in the N-terminus. Biochim. Biophys. Acta 2001, 1568, 197–204. [Google Scholar] [CrossRef]
- Groot, F.; Geijtenbeek, T.B.; Sanders, R.W.; Baldwin, C.E.; Sanchez-Hernandez, M.; Floris, R.; van Kooyk, Y.; de Jong, E.C.; Berkhout, B. Lactoferrin prevents dendritic cell-mediated human immunodeficiency virus type 1 transmission by blocking the Dc-SIGN—gp120 interaction. J. Virol. 2005, 79, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.J.; Chen, W.J.; Hsu, W.L.; Chiou, S.S. Bovine lactoferrin inhibits japanese encephalitis virus by binding to heparan sulfate and receptor for low density lipoprotein. Virology 2008, 379, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Ikeda, M.; Saito, S.; Matsumoto, S.; Numata, K.; Kato, N.; Tanaka, K.; Sekihara, H. Lactoferrin inhibits hepatitis B virus infection in cultured human hepatocytes. Hepatol. Res. 2002, 24, 228. [Google Scholar] [CrossRef]
- Wang, J.; Dong, A.; Liu, G.; Anderson, G.J.; Hu, T.Y.; Shi, J.; Hu, Y.; Nie, G. Correlation of serum hepcidin levels with disease progression in hepatitis B virus-related disease assessed by nanopore film based assay. Sci. Rep. 2016, 6, 34252. [Google Scholar] [CrossRef]
- Bartolomei, G.; Cevik, R.E.; Marcello, A. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J. Gen. Virol. 2011, 92, 2072–2081. [Google Scholar] [CrossRef]
- Douam, F.; Lavillette, D.; Cosset, F.L. The mechanism of HCV entry into host cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–107. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Yoshimoto, T.; Zhang, H.; Murao, K. Roles of lipoprotein receptors in the entry of hepatitis C virus. World J. Hepatol. 2015, 7, 2535–2542. [Google Scholar] [CrossRef]
- Miyanishi, K.; Tanaka, S.; Sakamoto, H.; Kato, J. The role of iron in hepatic inflammation and hepatocellular carcinoma. Free Radic. Biol. Med. 2019, 133, 200–205. [Google Scholar] [CrossRef]
- Kato, M.; Kobune, T.; Nakamura, G.; Kuroiwa, K.; Takada, R.; Takimoto, R.; Sato, Y.; Fujikawa, K.; Takahashi, M.; Takayama, T.; et al. Normalization of elevated hepatic 8-hydroxy-2′-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001, 61, 8697–8702. [Google Scholar] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. PNAS 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.W.; Chiu, L.T.; Chen, C.H.; Shih, W.L.; Lu, T.P. Tumor-Infiltrating Leukocyte Composition and Prognostic Power in Hepatitis B- and Hepatitis C-Related Hepatocellular Carcinomas. Genes 2019, 10, 630. [Google Scholar] [CrossRef]
- Florian, P.E.; Macovei, A.; Lazar, C.; Milac, A.L.; Sokolowska, I.; Darie, C.C.; Evans, R.W.; Roseanu, A.; Branza-Nichita, N. Characterization of the anti-HBV activity of HLP1-23, a human lactoferrin-derived peptide. J. Med. Virol. 2013, 85, 780–788. [Google Scholar] [CrossRef]
- Yi, M.; Kaneko, S.; Yu, D.Y.; Murakami, S. Hepatitis C virus envelope proteins bind lactoferrin. J. Virol. 1997, 71, 5997–6002. [Google Scholar] [CrossRef]
- El-Fakharany, E.M.; Sánchez, L.; Al-Mehdar, H.A.; Redwan, E.M. Effectiveness of human, camel, bovine and sheep lactoferrin on the hepatitis C virus cellular infectivity: Comparison study. Virol. J. 2013, 10, 199. [Google Scholar] [CrossRef]
- Picard-Jean, F.; Bouchard, S.; Larivée, G.; Bisaillon, M. The intracellular inhibition of HCV replication represents a novel mechanism of action by the innate immune Lactoferrin protein. Antiviral Res. 2014, 111, 13–22. [Google Scholar] [CrossRef]
- Tanaka, K.; Ikeda, M.; Nozaki, A.; Kato, N.; Tsuda, H.; Saito, S.; Sekihara, H. Lactoferrin inhibits hepatitis C virus viremia in patients with chronic hepatitis C: A pilot study. Jpn. J. Cancer Res. 1999, 90, 367–371. [Google Scholar] [CrossRef]
- Okada, S.; Tanaka, K.; Sato, T.; Ueno, H.; Saito, S.; Okusaka, T.; Sato, K.; Yamamoto, S.; Kakizoe, T. Dose-response trial of lactoferrin in patients with chronic hepatitis C. Jpn. J. Cancer Res. 2002, 93, 1063–1069. [Google Scholar] [CrossRef]
- Ueno, H.; Sato, T.; Yamamoto, S.; Tanaka, K.; Ohkawa, S.; Takagi, H. Randomized, double-blind, placebo-controlled trial of bovine lactoferrin in patients with chronic hepatitis C. Cancer Sci. 2006, 97, 1105–1110. [Google Scholar] [CrossRef]
- Hirashima, N.; Orito, E.; Ohba, K.; Kondo, H.; Sakamoto, T.; Matsunaga, S.; Kato, A.; Nukaya, H.; Sakakibara, K.; Ohno, T.; et al. A randomized controlled trial of consensus interferon with or without lactoferrin for chronic hepatitis C patients with genotype 1b and high viral load. Hepatol. Res. 2004, 29, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Takeda, K.; Tsukidate, N.; Miyazaki, H.; Ohira, K.; Dosaka-Akita, H.; Nishimura, M. Randomized placebo-controlled trial of interferon alpha-2b plus ribavirin with and without lactoferrin for chronic hepatitis C. Hepatol. Res. 2005, 32, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kaito, M.; Iwasa, M.; Fujita, N.; Kobayashi, Y.; Kojima, Y.; Ikoma, J.; Imoto, I.; Adachi, Y.; Hamano, H.; Koji, Y. Effect of lactoferrin in patients with chronic hepatitis C: Combination therapy with interferon and ribavirin. J. Gastroenterol. Hepatol. 2007, 22, 1894–1897. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Onori, P.; Gaudio, E.; Franchitto, A.; Carpino, G.; Ueno, Y.; Alvaro, D.; Annarale, L.P.; Demorrow, S.; Francis, H. Taurocholate feeding to bile duct ligated rats prevents caffeic acid-induced bile duct damage by changes in cholangiocyte VEGF expression. Exp. Biol. Med. (Maywood). 2009, 234, 462–474. [Google Scholar] [CrossRef]
- Mancinelli, R.; Glaser, S.; Francis, H.; Carpino, G.; Franchitto, A.; Vetuschi, A.; Sferra, R.; Pannarale, L.; Venter, J.; Meng, F.; et al. Ischemia reperfusion of the hepatic artery induces the functional damage of large bile ducts by changes in the expression of angiogenic factors. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G865–G873. [Google Scholar] [CrossRef]
- Kruzel, M.L.; Zimecki, M.; Actor, J.K. Lactoferrin in a Context of Inflammation-Induced Pathology. Front. Immunol. 2017, 8, 1438. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancinelli, R.; Rosa, L.; Cutone, A.; Lepanto, M.S.; Franchitto, A.; Onori, P.; Gaudio, E.; Valenti, P. Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin. Molecules 2020, 25, 1997. https://doi.org/10.3390/molecules25081997
Mancinelli R, Rosa L, Cutone A, Lepanto MS, Franchitto A, Onori P, Gaudio E, Valenti P. Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin. Molecules. 2020; 25(8):1997. https://doi.org/10.3390/molecules25081997
Chicago/Turabian StyleMancinelli, Romina, Luigi Rosa, Antimo Cutone, Maria Stefania Lepanto, Antonio Franchitto, Paolo Onori, Eugenio Gaudio, and Piera Valenti. 2020. "Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin" Molecules 25, no. 8: 1997. https://doi.org/10.3390/molecules25081997
APA StyleMancinelli, R., Rosa, L., Cutone, A., Lepanto, M. S., Franchitto, A., Onori, P., Gaudio, E., & Valenti, P. (2020). Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin. Molecules, 25(8), 1997. https://doi.org/10.3390/molecules25081997