
First-Principles Study of Nitrogen Adsorption and Dissociation on PuH2 (111) Surface

 and
and
Abstract
1. Introduction
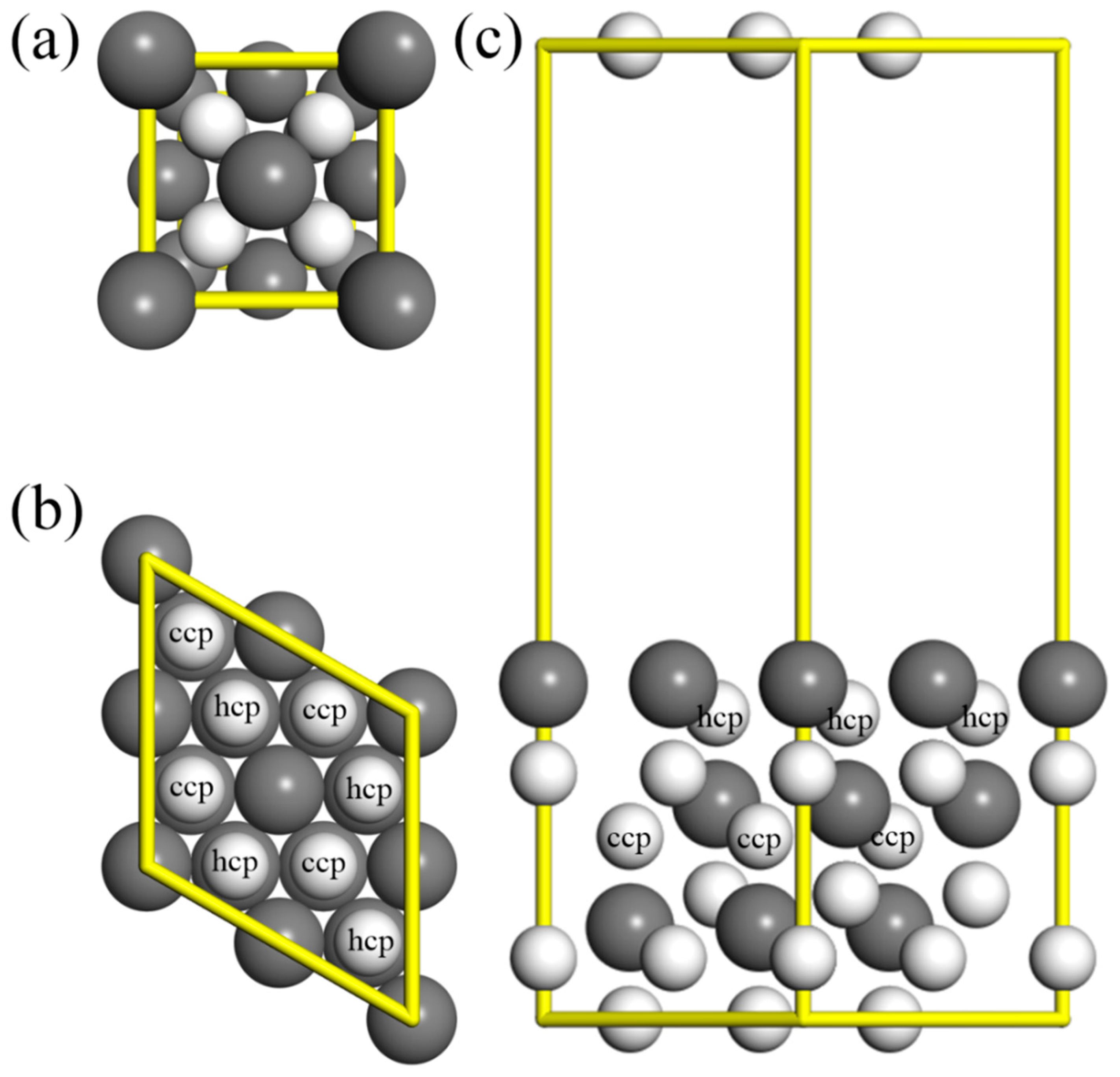
2. Theoretical Methods and Models
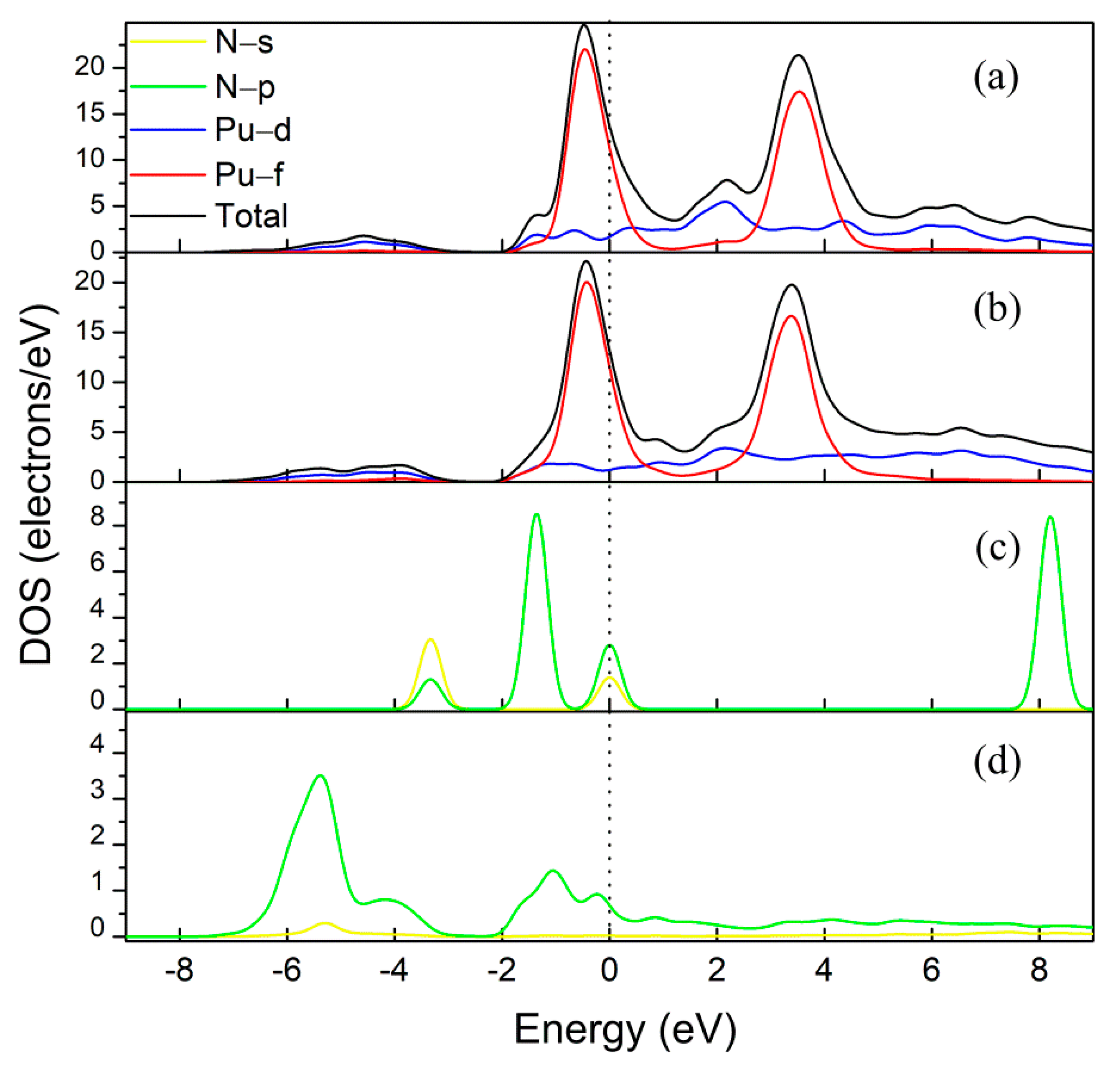
3. Results and Discussion
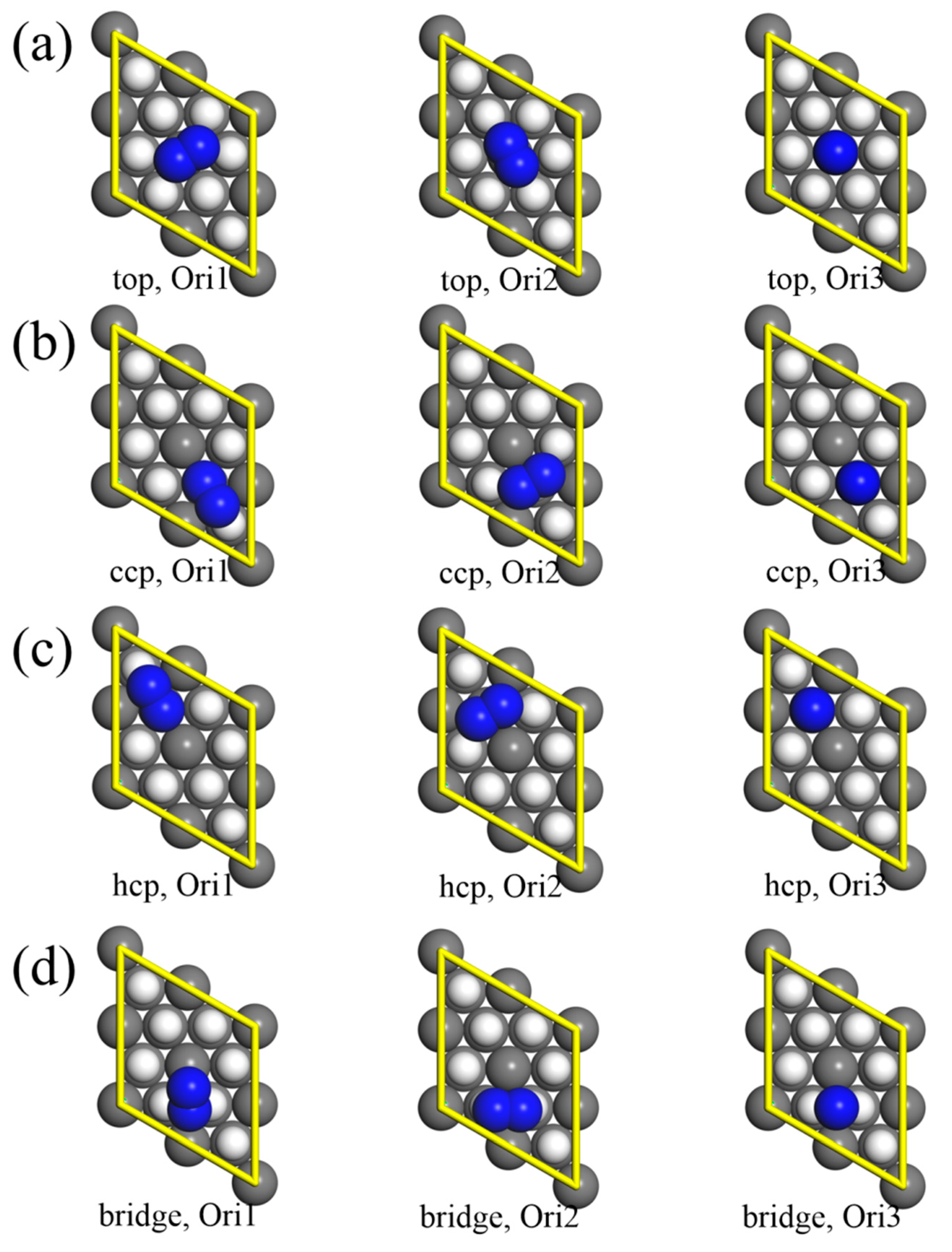
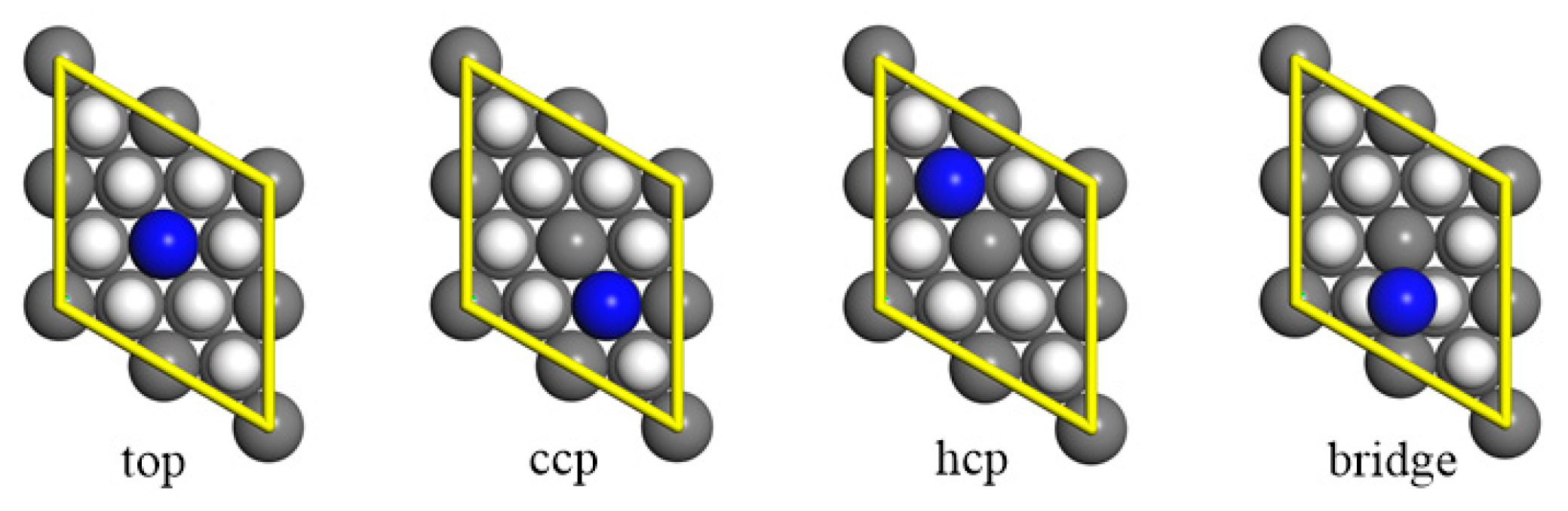
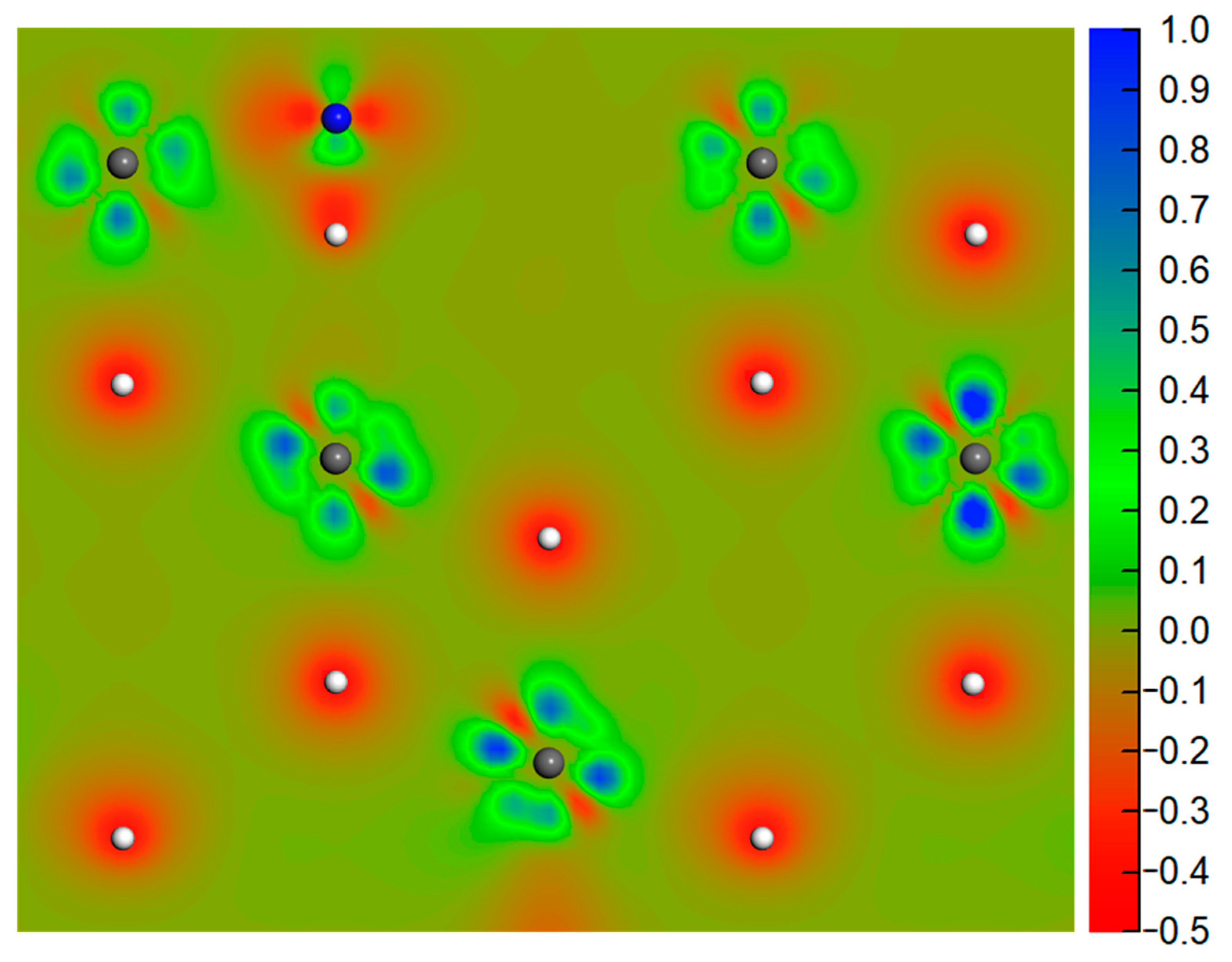
3.1. Molecular Adsorption
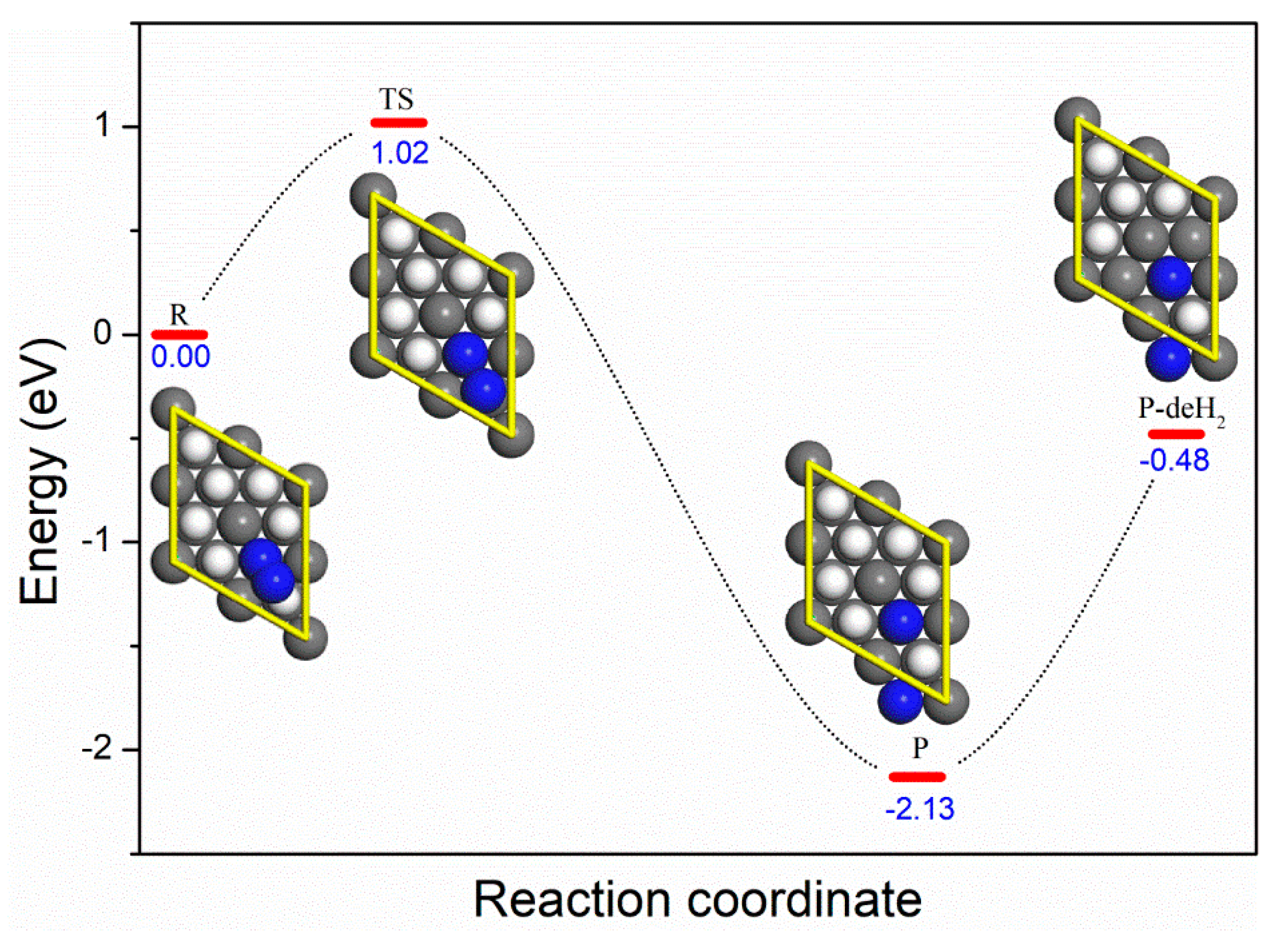
3.2. Dissociative Adsorption
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lashley, J.C.; Lawson, A.; McQueeney, R.J.; Lander, G.H. Absence of magnetic moments in plutonium. Phys. Rev. B 2005, 72, 054416. [Google Scholar] [CrossRef]
- Arsenlis, A.; Wolfer, W.G.; Schwartz, A.J. Change in flow stress and ductility of δ-phase Pu-Ga alloys due to self-irradiation damage. J. Nucl. Mater. 2005, 336, 31–39. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Sadigh, B. Density-functional theory for plutonium. Adv. Phys. 2019, 68, 1–47. [Google Scholar] [CrossRef]
- Hernandez, S.; Freibert, F.; Wills, J. Density functional theory study of defects in unalloyed δ-Pu. Scr. Mater. 2017, 134, 57–60. [Google Scholar] [CrossRef]
- Ray, A.K.; Boettger, J.C. First-principles electronic structure study of the quantum size effects in (111) films of δ-plutonium. Phys. Rev. B 2004, 70, 085418. [Google Scholar] [CrossRef]
- Stakebake, J.L. The storage behavior of plutonium metal, alloys, and oxide: A review. J. Nucl. Mater. 1971, 38, 241–259. [Google Scholar] [CrossRef]
- Haschke, J.M.; Allen, T.H.; Morales, L.A. Reaction of plutonium dioxide with water: Formation and properties of PuO2+x. Science 2000, 287, 285–287. [Google Scholar] [CrossRef]
- Muromura, T.; Ouchi, K. Kinetic study of the nitrogenation of plutonium hydride with ammonia. J. Inorg. Nucl. Chem. 1976, 38, 1855–1859. [Google Scholar] [CrossRef]
- Stakebake, J.L. Kinetics for the reaction of hydrogen with plutonium powder. J. Alloy Compd. 1992, 187, 271–283. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Zhang, P. Dissociation mechanism of water molecules on the PuO2 (110) surface: An ab initio molecular dynamics study. J. Phys. Chem. C 2018, 122, 371–376. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Li, R.-S.; Lu, X.; Wang, J.-T.; Xin, D.-Q.; Yao, X.-G. A many-body perspective on dual 5f states in two plutonium hydrides. Chem. Phys. Lett. 2020, 740, 137079. [Google Scholar] [CrossRef]
- Söderlind, P.; Sadigh, B. Density-Functional calculations of α, β, γ, δ, δ′, and ϵ Plutonium. Phys. Rev. Lett. 2004, 92, 185702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y. First-principles thermodynamic calculations for δ-Pu and ϵ-Pu. J. Phys. Condens. Matter 2000, 12, L311. [Google Scholar] [CrossRef]
- Wallace, D.C. Electronic and phonon properties of six crystalline phases of Pu metal. Phys. Rev. B 1998, 58, 15433. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. A density functional study of atomic hydrogen adsorption on plutonium layers. Physica B 2004, 352, 5–17. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. An ab initio study of H2 interaction with the Pu (100) surface. Physica B 2005, 366, 95–109. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. Molecular hydrogen adsorption and dissociation on the plutonium (111) surface. Phys. Rev. B 2005, 72, 085101. [Google Scholar] [CrossRef]
- Goldman, N.; Aradi, B.; Lindsey, R.K.; Fried, L.E. Development of a Multicenter Density Functional Tight Binding Model for Plutonium Surface Hydriding. J. Chem. Theory Comput. 2018, 14, 2652–2660. [Google Scholar] [CrossRef]
- Goldman, N.; Morales, M.A. A First-principles study of hydrogen diffusivity and dissociation on δ-Pu (100) and (111) surfaces. J. Phys. Chem. C 2017, 121, 17950–17957. [Google Scholar] [CrossRef]
- Yu, H.L.; Tang, T.; Zheng, S.T.; Shi, Y.; Qiu, R.Z.; Luo, W.H.; Meng, D.Q. A theoretical study of hydrogen atoms adsorption and diffusion on PuO2 (110) surface. J. Alloy Compd. 2016, 666, 287–291. [Google Scholar] [CrossRef]
- Yu, H.L.; Li, G.; Li, H.B.; Qiu, R.Z.; Huang, H.; Meng, D.Q. Adsorption and dissociation of H2 on PuO2 (110) surface: A density functional theory study. J. Alloys Compd. 2016, 654, 567–573. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Zhang, P. Thermodynamics study of dehydriding reaction on the PuO2 (110) surface from first principles. J. Phys. Chem. C 2018, 122, 7790–7794. [Google Scholar] [CrossRef]
- Sun, B.; Liu, H.; Song, H.; Zhang, G.; Zheng, H.; Zhao, Z.G.; Zhang, P. The different roles of Pu-oxide overlayers in the hydrogenation of Pu-metal: An ab initio molecular dynamics study based on van der Waals density functional (vdW-DF)+U. J. Chem. Phys. 2014, 140, 164709. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, P. Hydriding and dehydriding energies of PuHx from ab initio calculations. Phys. Lett. A 2015, 379, 1649–1653. [Google Scholar] [CrossRef]
- Taylor, C.D.; Hernandez, S.C.; Francis, M.F.; Schwartz, D.S.; Ray, A.K. Hydrogen trapping in δ-Pu: Insights from electronic structure calculations. J. Phys. Condens. Matter 2013, 25, 265001. [Google Scholar] [CrossRef]
- Li, S.; Ao, B.; Ye, X.; Qiu, R.; Gao, T. New insights into the crystal structures of plutonium hydrides from first-principles calculations. J. Phys. Chem. C 2018, 122, 10103–10112. [Google Scholar] [CrossRef]
- Bing-Yun, A.; Peng, S.; Yong, G.; Tao, G. Abnormal lattice contraction of plutonium hydrides studied by first-principles calculations. Chin. Phys. B 2013, 22, 037103. [Google Scholar]
- Yang, X.; Yang, Y.; Lu, Y.; Sun, Z.; Hussain, S.; Zhang, P. First-principles GGA + U calculation investigating the hydriding and diffusion properties of hydrogen in PuH2+x, 0 ≤ x ≤ 1. Int. J. Hydrog. Energy 2018, 43, 13632–13638. [Google Scholar] [CrossRef]
- Zheng, J.-J.; Wang, B.-T.; Di Marco, I.; Li, W.-D. Electronic structure and phase stability of plutonium hydrides: Role of Coulomb repulsion and spin-orbital coupling. Int. J. Hydrog. Energy 2014, 39, 13255–13265. [Google Scholar] [CrossRef]
- Oetting, F.L. The chemical thermodynamic properties of nuclear materials III. Plutonium mononitride. J. Chem. Thermodyn. 1978, 10, 941–948. [Google Scholar] [CrossRef]
- Kurosaki, K.; Yano, K.; Yamada, K.; Uno, M.; Yamanaka, S. A molecular dynamics study on plutonium mononitride. J. Alloy Compd. 2000, 313, 242–247. [Google Scholar] [CrossRef]
- Zhang, Y.; Lan, J.; Zhuo, Z.; Ge, C.; Zhou, Z.; Chai, Z.; Shi, W. Theoretical study on stability, mechanical and thermodynamic properties of (Pu, Zr)N. J. Nucl. Mater. 2019, 516, 264–270. [Google Scholar] [CrossRef]
- Clark, S.; Segall, M.; Pickard, C.; Hasnip, P.; Probert, M.; Refson, K.; Payne, M. First principles methods using CASTEP. Z. Krist. -Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hamann, D.R.; Schlüter, M.; Chiang, C. Norm-Conserving Pseudopotentials. Phys. Rev. Lett. 1979, 43, 1494–1497. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tegner, B.E.; Molinari, M.; Kerridge, A.; Parker, S.C.; Kaltsoyannis, N. Water Adsorption on AnO2 {111}, {110}, and {100} Surfaces (An = U and Pu): A Density Functional Theory + U Study. J. Phys. Chem. C 2017, 121, 1675–1682. [Google Scholar] [CrossRef]
- Chen, J.-L.; Kaltsoyannis, N. Computational Study of Plutonium–Americium Mixed Oxides (Pu0.92Am0.08O2–x); Water Adsorption on {111}, {110}, and {100} Surfaces. J. Phys. Chem. C 2020, 124, 6646–6658. [Google Scholar] [CrossRef]
- Rák, Z.; Ewing, R.C.; Becker, U. Hydroxylation-induced surface stability of AnO2 (An=U, Np, Pu) from first-principles. Surf. Sci. 2013, 608, 180–187. [Google Scholar] [CrossRef]
- Mulford, R.N.R.; Sturdy, G.E. The plutonium-hydrogen system. II. solid solution of hydrogen in plutonium dihydride1. J. Am. Chem. Soc. 1956, 78, 3897–3901. [Google Scholar] [CrossRef]
- Ao, B.Y.; Wang, X.L.; Shi, P.; Chen, P.H.; Ye, X.Q.; Lai, X.C.; Gao, T. First-principles LDA+U calculations investigating the lattice contraction of face-centered cubic Pu hydrides. J. Nucl. Mater. 2012, 424, 183–189. [Google Scholar] [CrossRef]
- Ao, B.; Qiu, R.; Lu, H.; Chen, P. Differences in the existence states of hydrogen in UO2 and PuO2 from DFT + U calculations. J. Phys. Chem. C 2016, 120, 18445–18451. [Google Scholar] [CrossRef]
- Savrasov, S.Y.; Kotliar, G. Ground State Theory of δ-Pu. Phys. Rev. Lett. 2000, 84, 3670–3673. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. Ab initio study of molecular oxygen adsorption on Pu (111) surface. Int. J. Quantum Chem. 2005, 105, 280–291. [Google Scholar] [CrossRef]
- Atta-Fynn, R.; Ray, A.K. Ab initio full-potential fully relativistic study of atomic carbon, nitrogen, and oxygen chemisorption on the (111) surface of δ-Pu. Phys. Rev. B 2007, 75, 195112. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Wellington, J.P.W.; Kerridge, A.; Austin, J.; Kaltsoyannis, N. Electronic structure of bulk AnO2 (An = U, Np, Pu) and water adsorption on the (111) and (110) surfaces of UO2 and PuO2 from hybrid density functional theory within the periodic electrostatic embedded cluster method. J. Nucl. Mater. 2016, 482, 124–134. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Yeo, S.C.; Han, S.S.; Lee, H.M. Adsorption, dissociation, penetration, and diffusion of N2 on and in bcc Fe: First-principles calculations. Phys. Chem. Chem. Phys. 2013, 15, 5186–5192. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site. | Orientation | Eads (eV) | ED3 (eV) | rd (Å) | rN (Å) |
|---|---|---|---|---|---|
| top | Ori1 | −0.20 | −0.18 | 2.60 | 1.14 |
| Ori2 | −0.21 | −0.18 | 2.58 | 1.15 | |
| Ori3 | −0.49 | −0.16 | 2.50 | 1.13 | |
| ccp | Ori1 | −1.63 | −0.21 | 0.93 | 1.34 |
| Ori2 | −0.94 | −0.19 | 1.48 | 1.25 | |
| Ori3 | −0.54 | −0.19 | 1.37 | 1.19 | |
| hcp | Ori1 | −0.96 | −0.19 | 1.25 | 1.28 |
| Ori2 | −0.84 | −0.18 | 1.56 | 1.24 | |
| Ori3 | −0.31 | −0.18 | 1.79 | 1.15 | |
| bridge | Ori1 | −0.66 | −0.19 | 1.77 | 1.17 |
| Ori2 1 | −0.89 | −0.19 | 1.29 | 1.25 | |
| Ori3 | −0.37 | −0.19 | 1.72 | 1.16 |
| Site | Eads (eV) | rd (Å) | rPu-N (Å) |
|---|---|---|---|
| ccp | −6.85 | 0.45 | 2.23 |
| top | −4.26 | 1.81 | 1.81 |
| bridge | −5.85 | 0.96 | 2.11 |
| hcp | −5.67 | 0.45 | 2.22 |
| Site | Eads (eV) | Edis (eV) | rd-A (Å) | rPu-N-A (Å) | rd-B (Å) | rPu-N-B (Å) |
|---|---|---|---|---|---|---|
| ccp-ccp | −13.60 | −2.13 | 0.45 | 2.23 | 0.46 | 2.23 |
| ccp-bridge 1 | −12.48 | −1.01 | 0.42 | 2.22 | 0.94 | 2.10 |
| ccp-hcp1 | −11.86 | −0.39 | 0.33 | 2.20 | 0.90 | 2.39 |
| ccp-hcp2 | −12.17 | −0.70 | 0.51 | 2.24 | 0.54 | 2.24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zhang, K.; Song, P.; Hu, X.; Mu, J.; Miao, Z.; Zhou, J.; He, H. First-Principles Study of Nitrogen Adsorption and Dissociation on PuH2 (111) Surface. Molecules 2020, 25, 1891. https://doi.org/10.3390/molecules25081891
Wang C, Zhang K, Song P, Hu X, Mu J, Miao Z, Zhou J, He H. First-Principles Study of Nitrogen Adsorption and Dissociation on PuH2 (111) Surface. Molecules. 2020; 25(8):1891. https://doi.org/10.3390/molecules25081891
Chicago/Turabian StyleWang, Changshui, Kai Zhang, Peng Song, Xiaofei Hu, Jinglin Mu, Zhichao Miao, Jin Zhou, and Hui He. 2020. "First-Principles Study of Nitrogen Adsorption and Dissociation on PuH2 (111) Surface" Molecules 25, no. 8: 1891. https://doi.org/10.3390/molecules25081891
APA StyleWang, C., Zhang, K., Song, P., Hu, X., Mu, J., Miao, Z., Zhou, J., & He, H. (2020). First-Principles Study of Nitrogen Adsorption and Dissociation on PuH2 (111) Surface. Molecules, 25(8), 1891. https://doi.org/10.3390/molecules25081891