
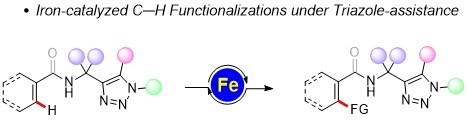
Iron-Catalyzed C–H Functionalizations under Triazole-Assistance



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
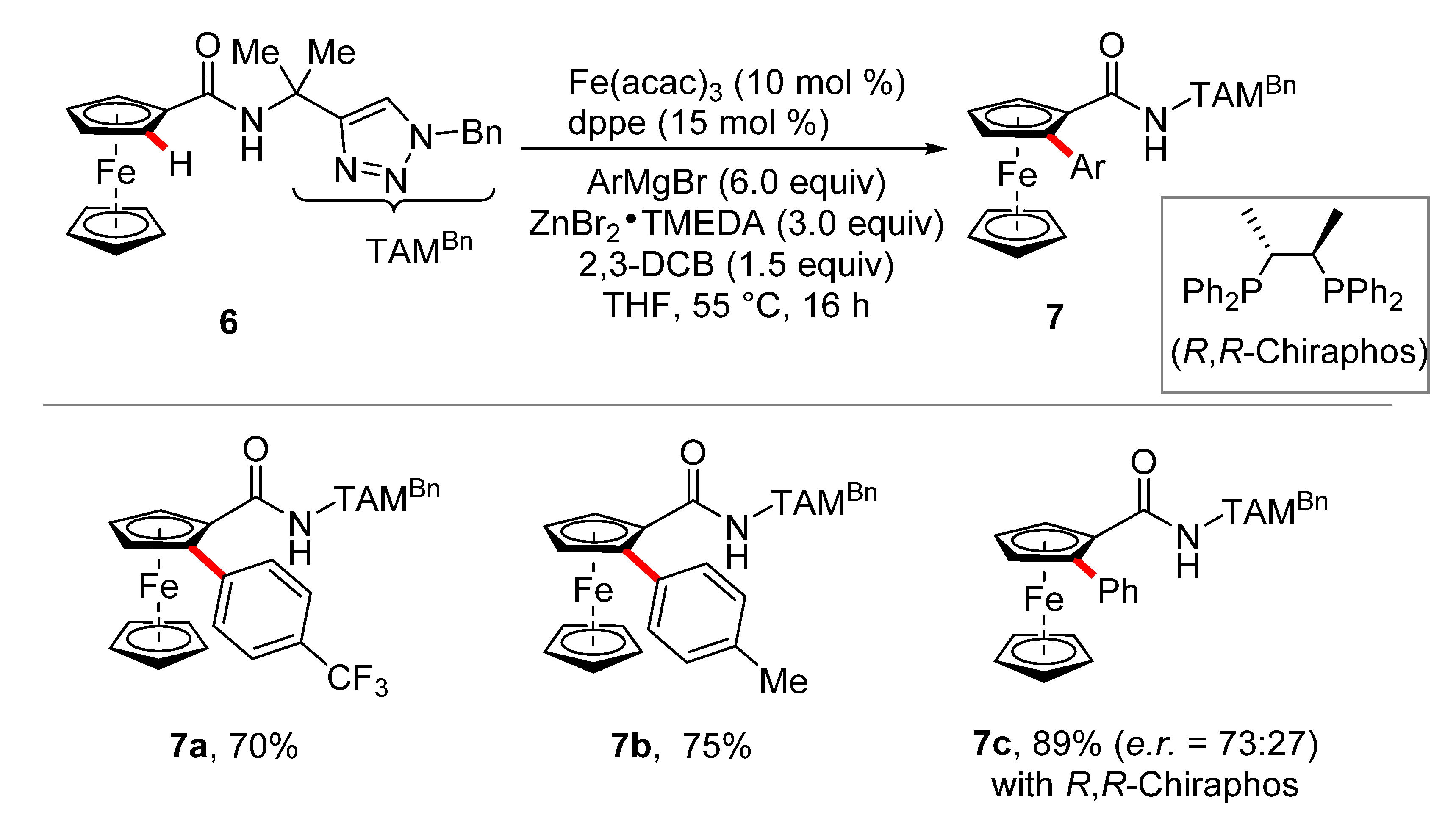
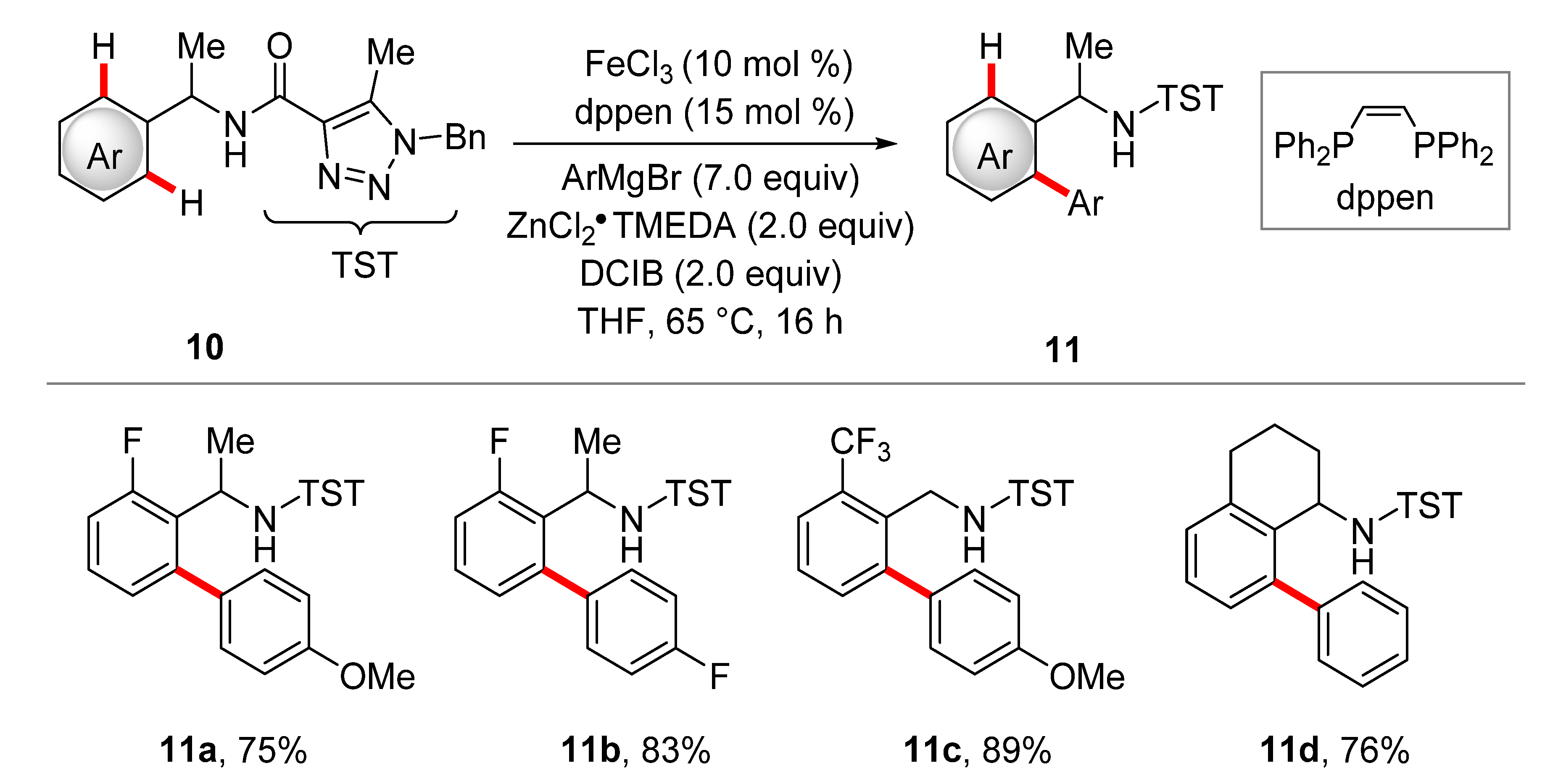
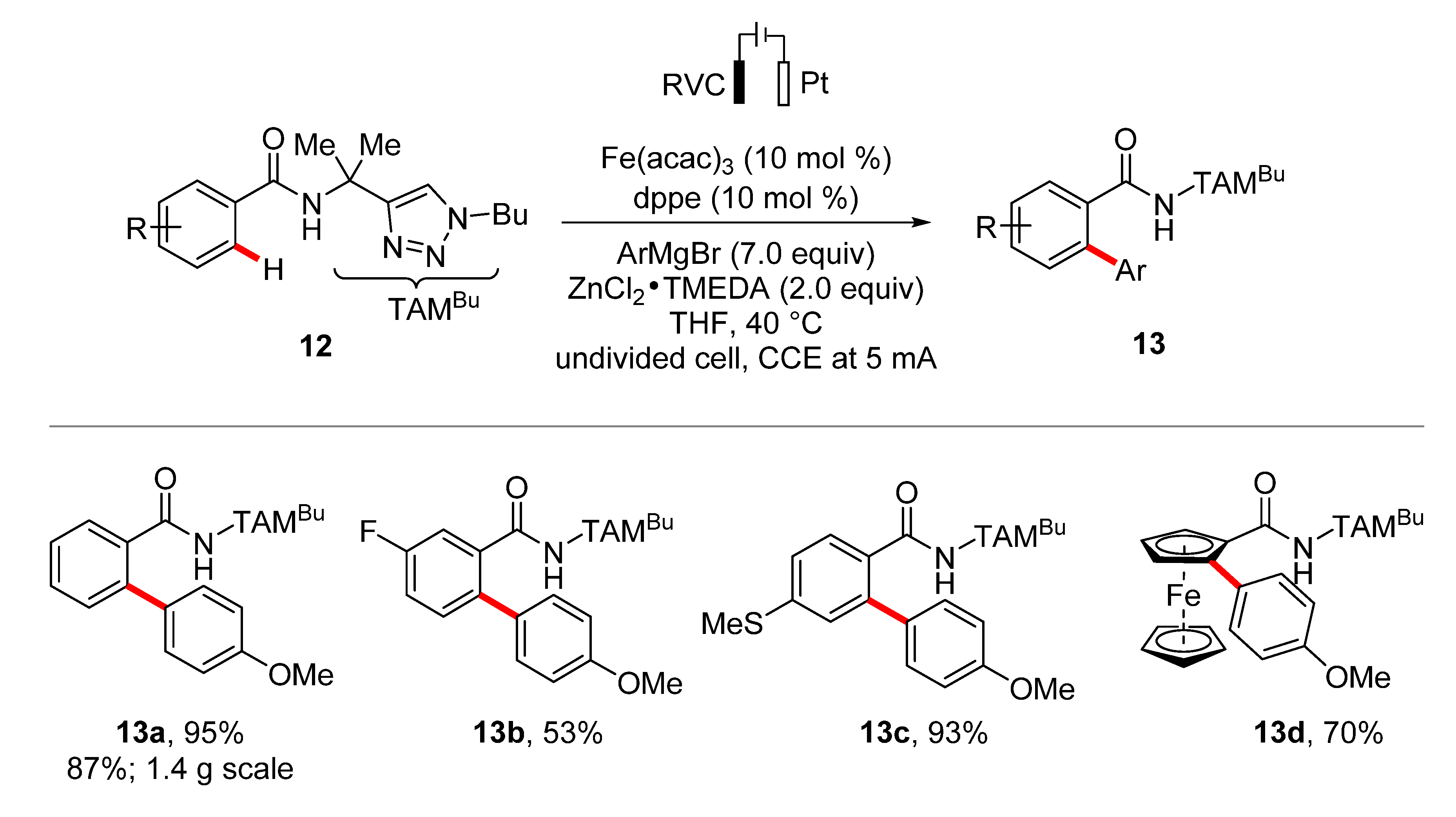
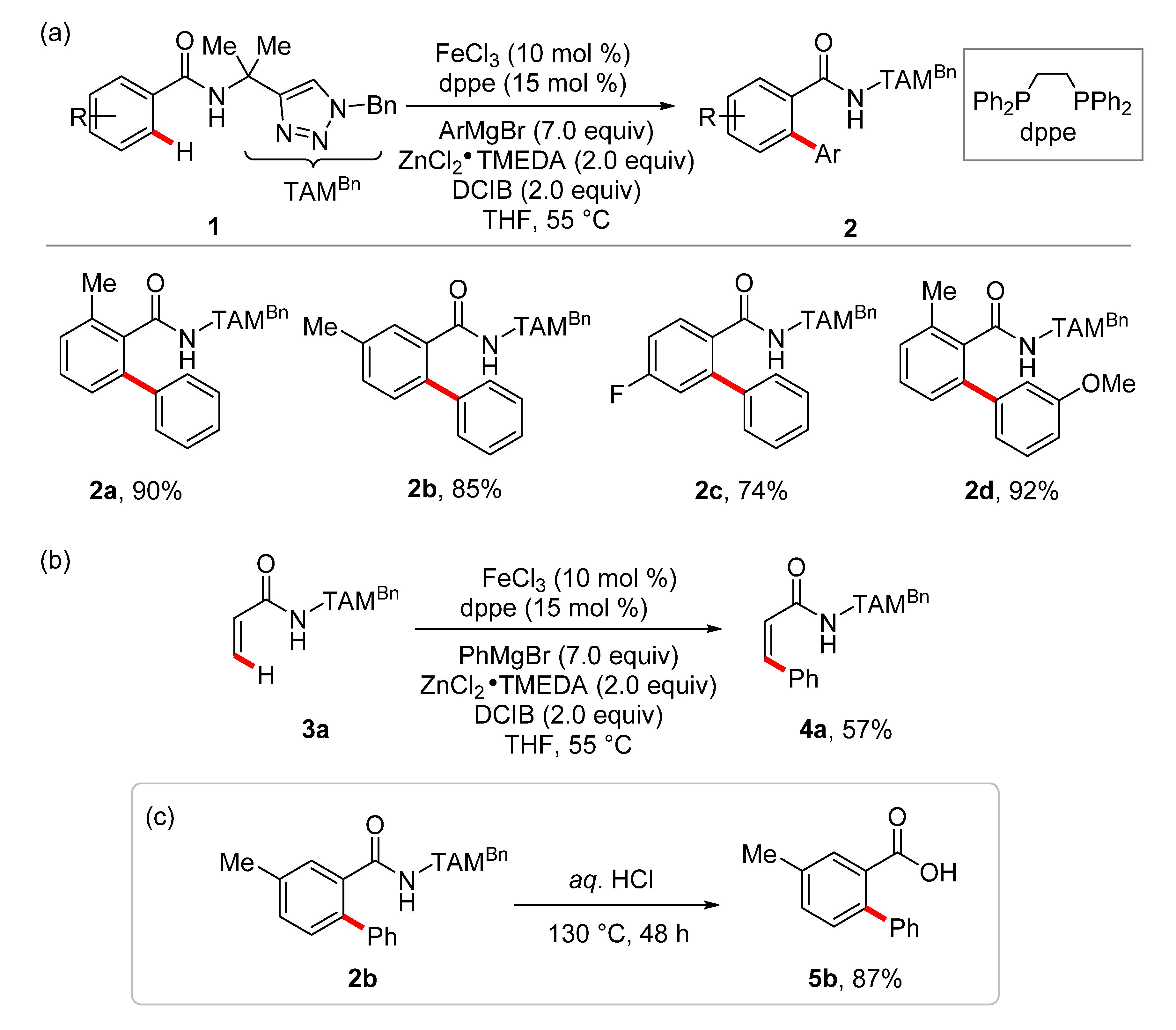
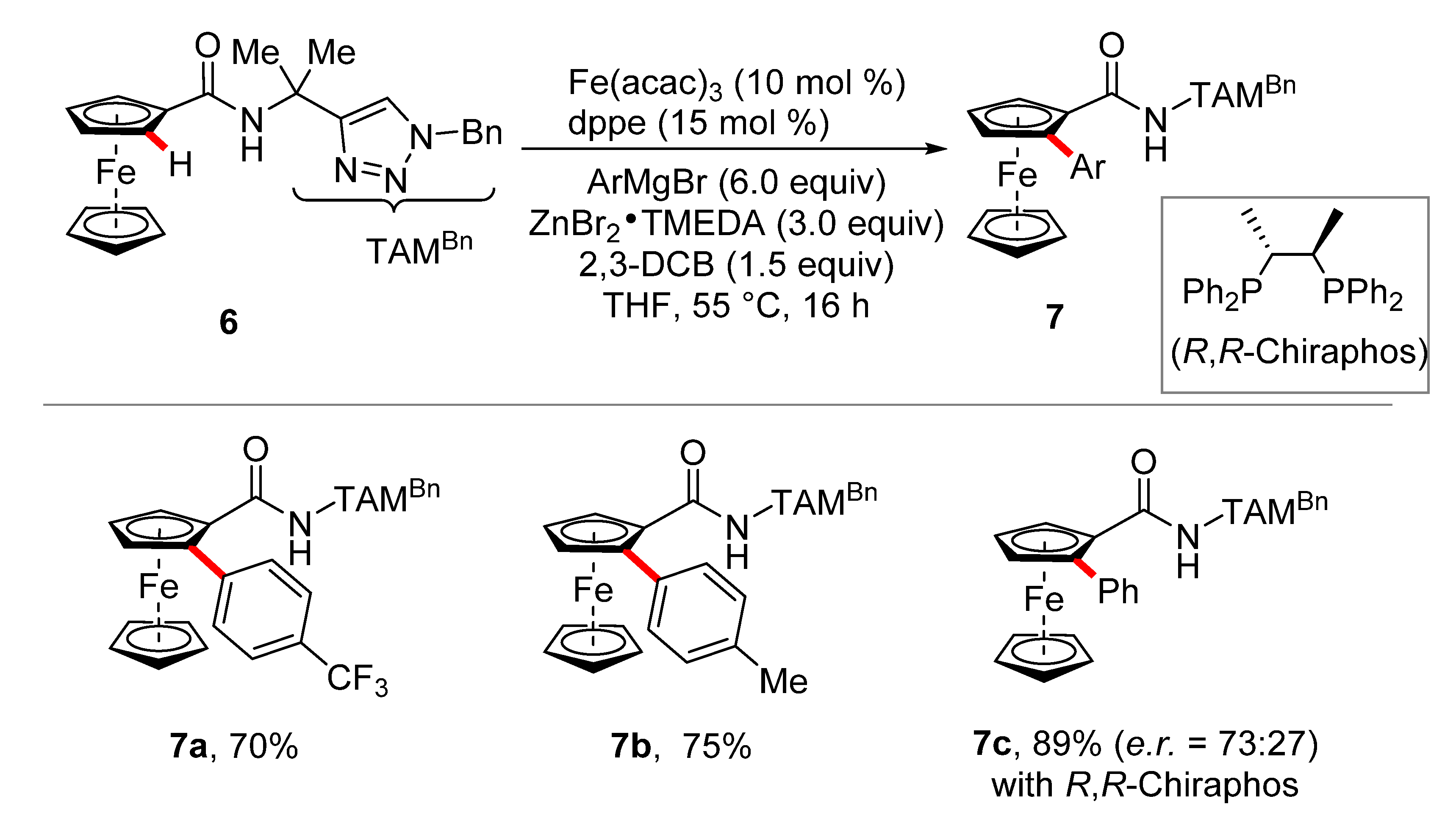
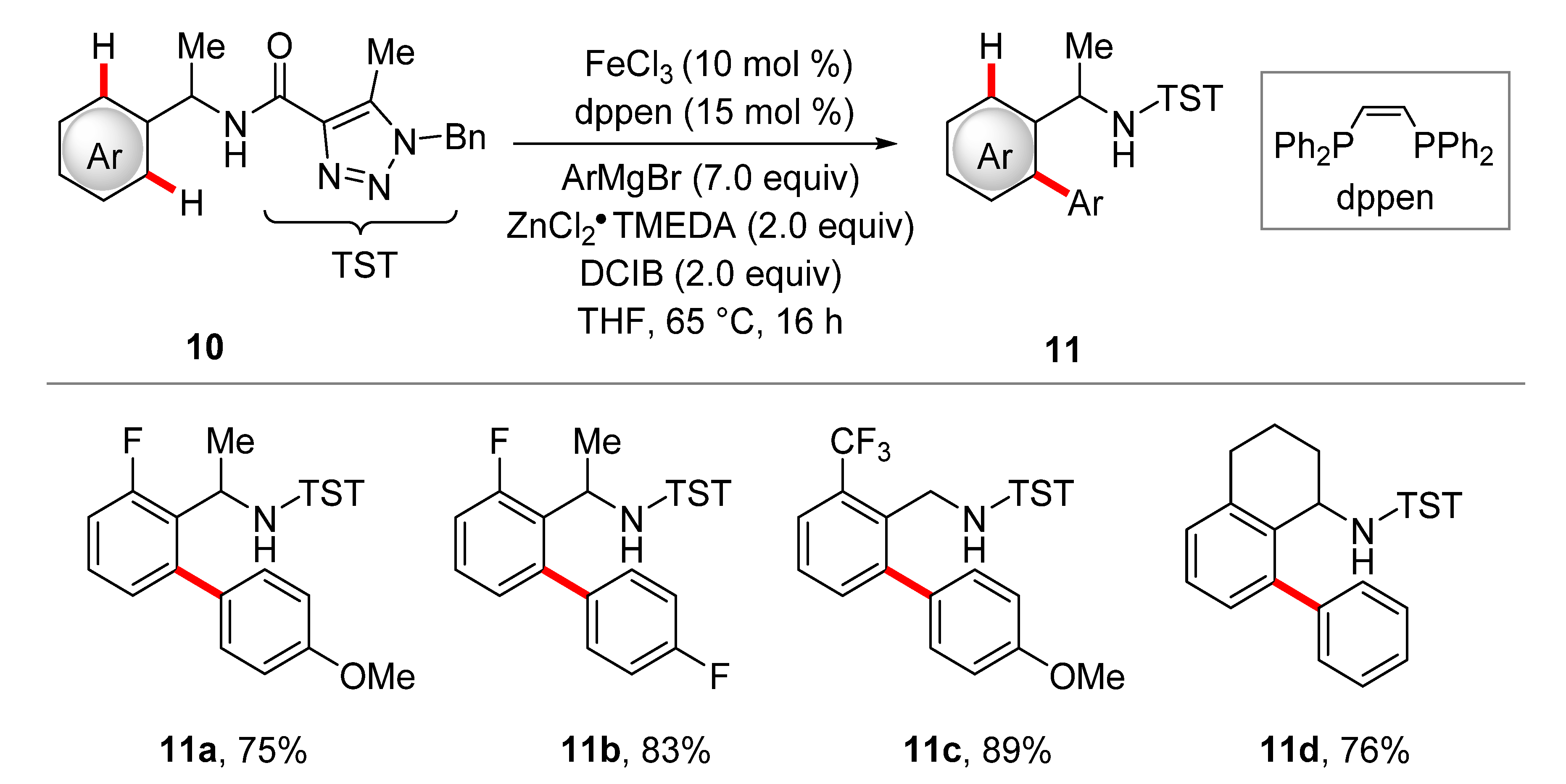
2. Iron-Catalyzed C–H Arylations
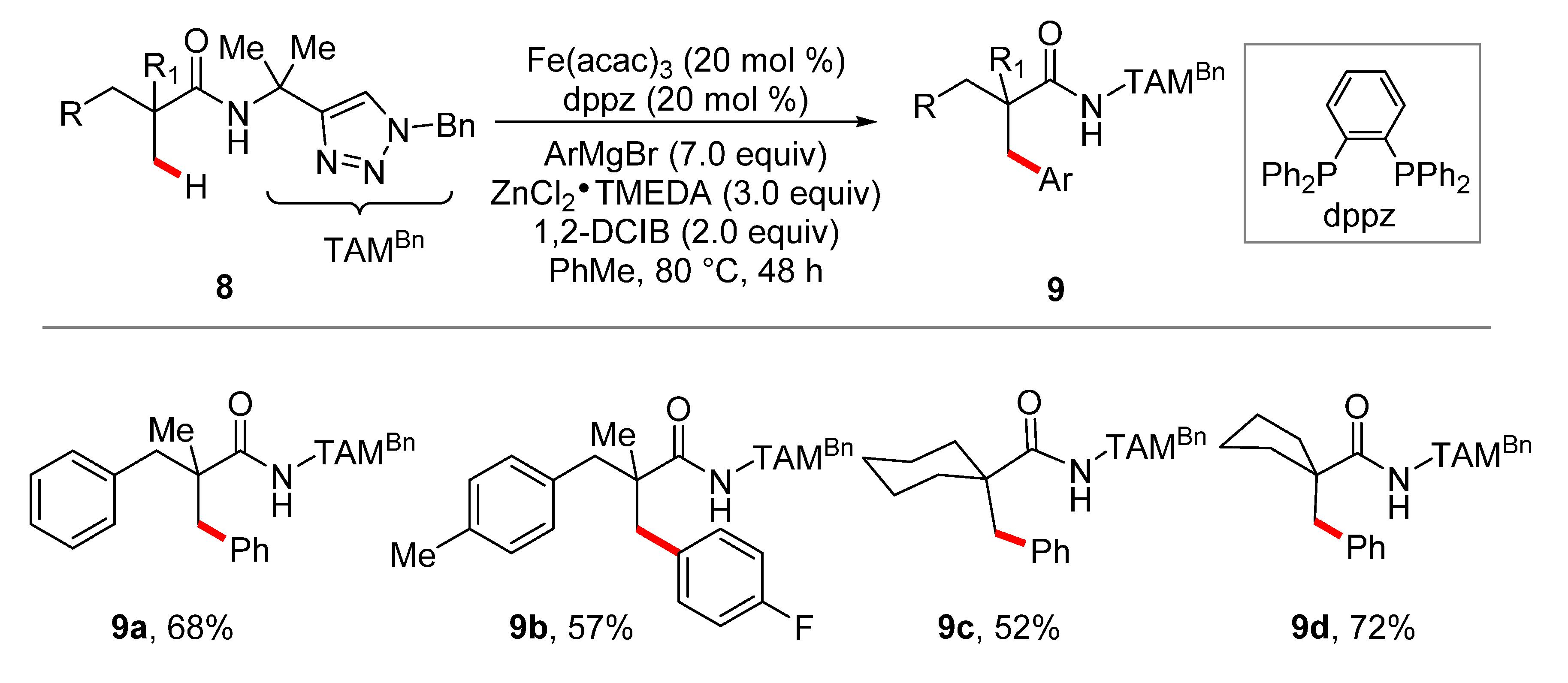
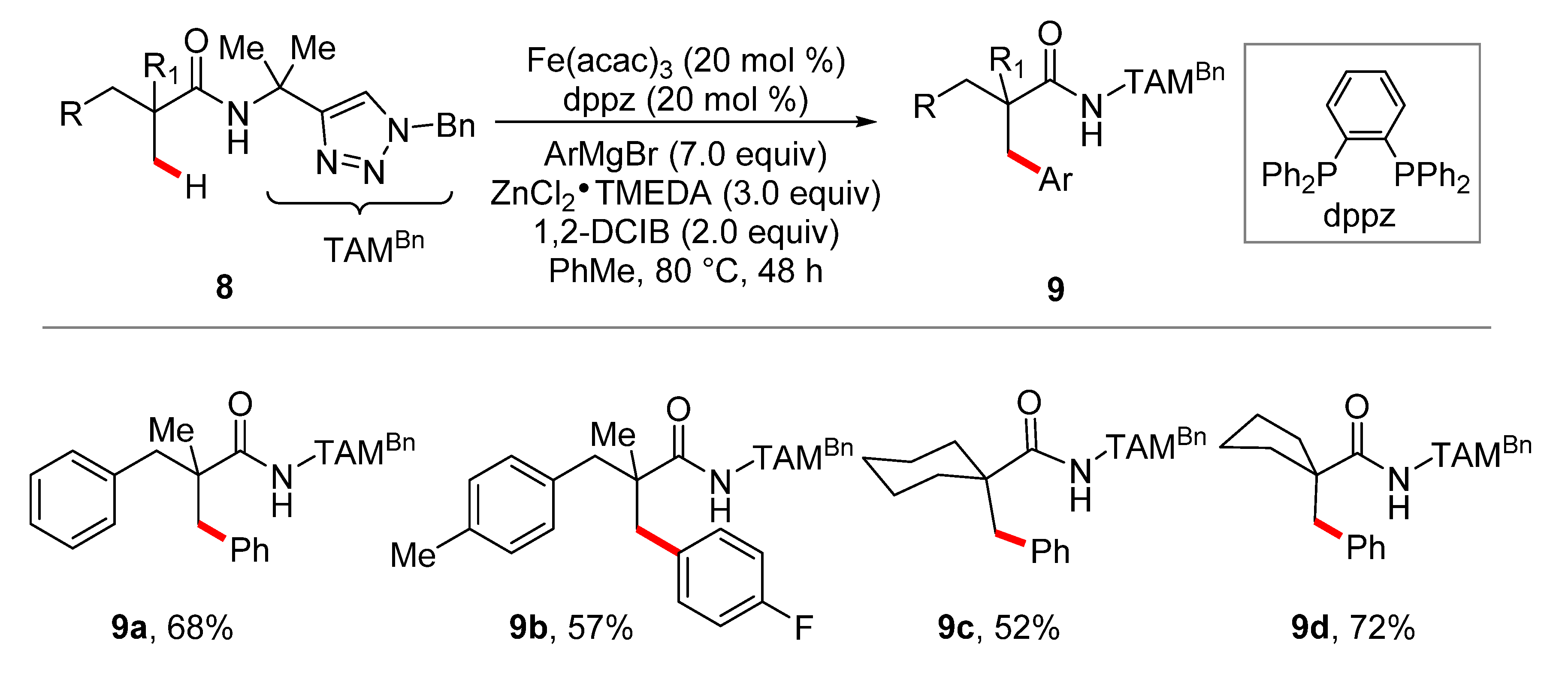
3. Iron-Catalyzed C–H Alkylations/Allylations
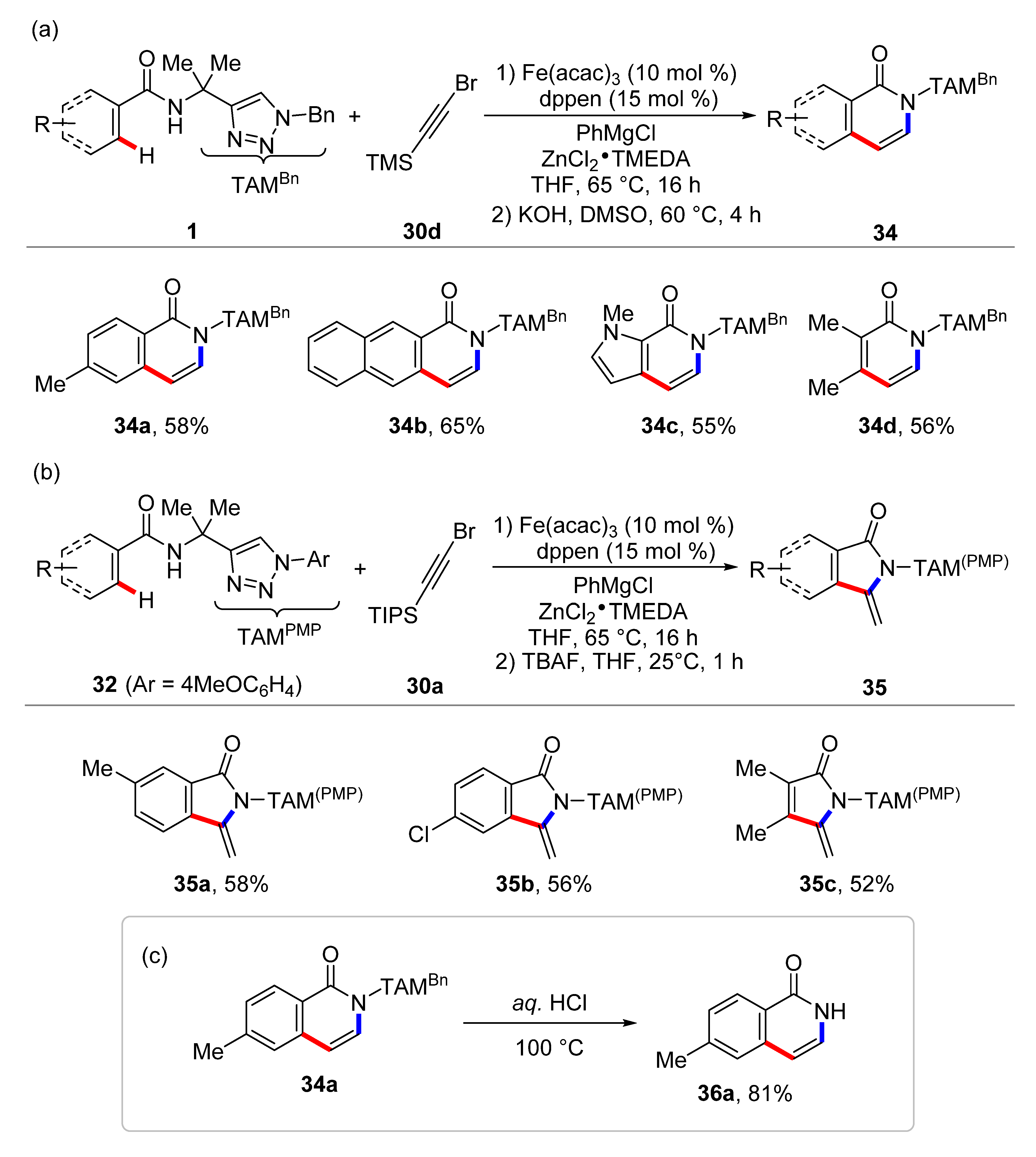
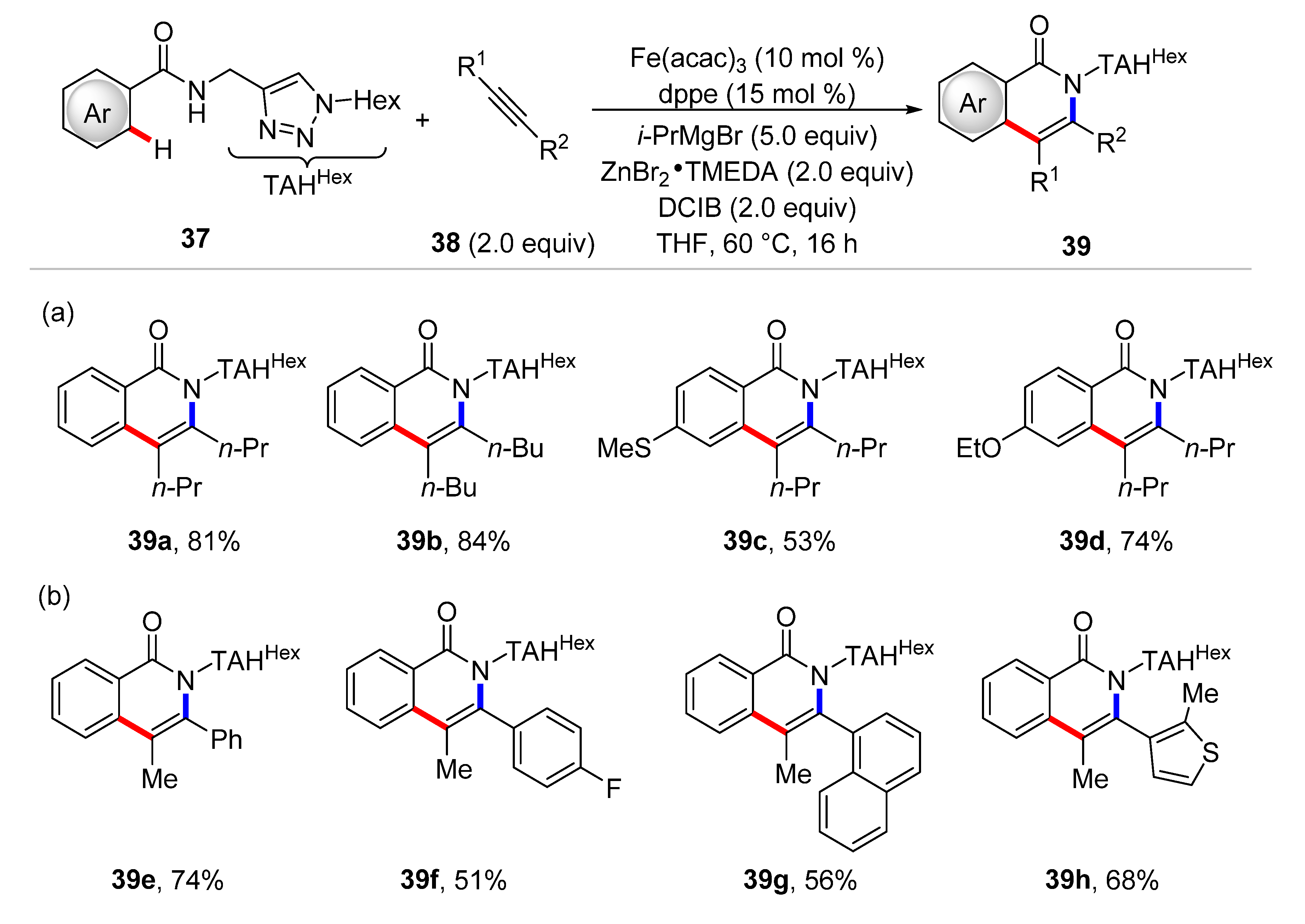
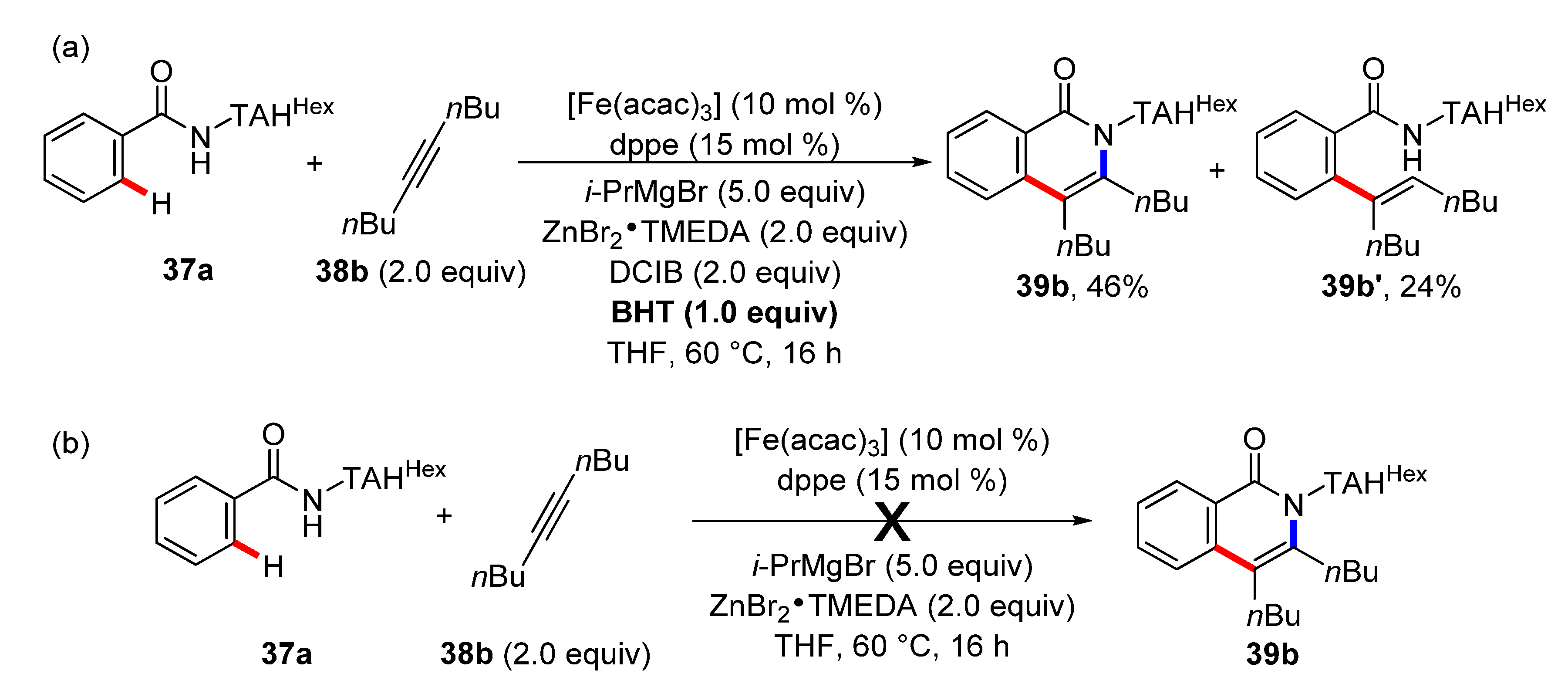
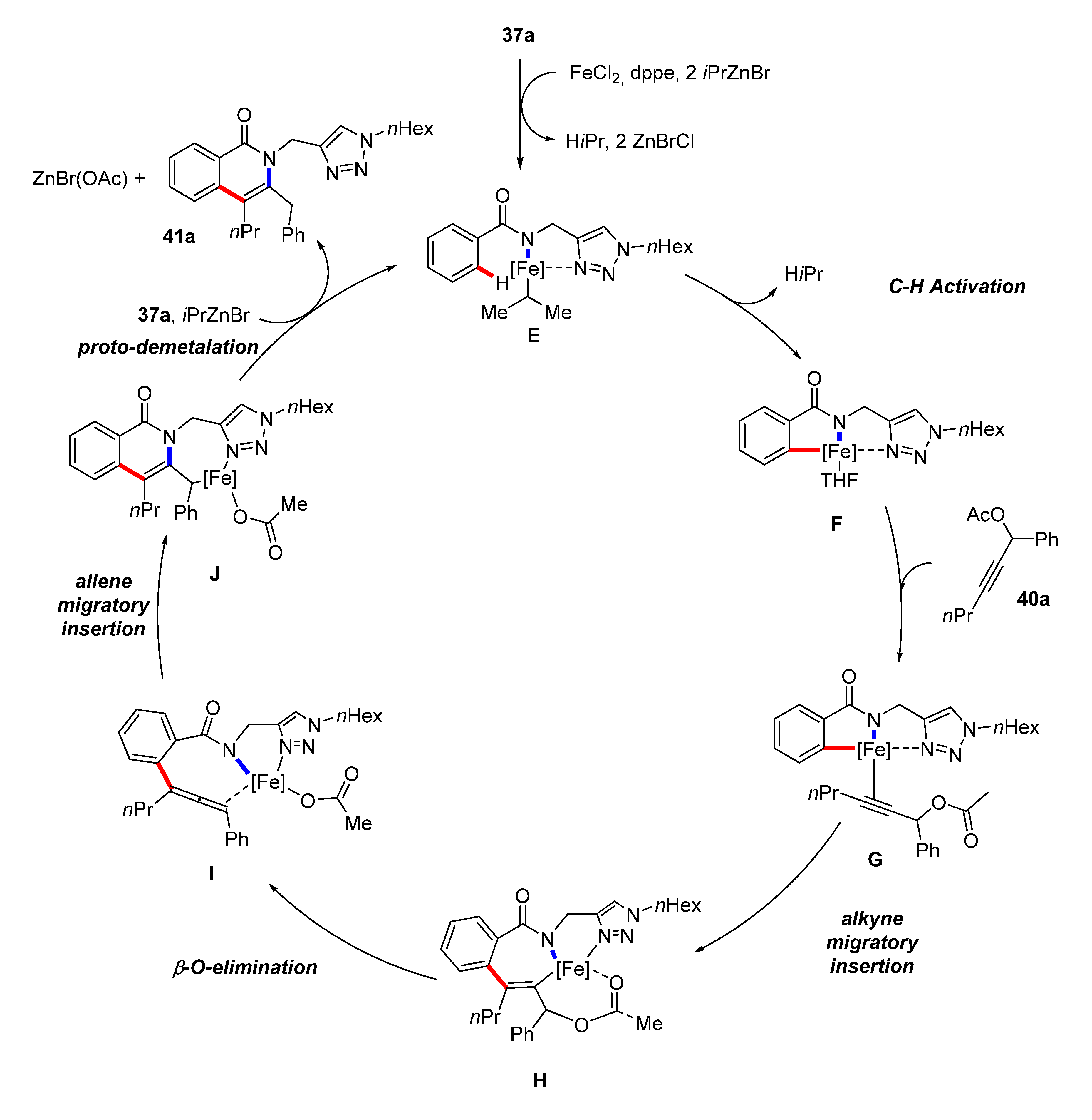
4. Iron-Catalyzed C–H Functionalizations with Alkynes and Allenes
5. Conclusions
Funding
Conflicts of Interest
References
- Kharasch, M.S.; Fields, E.K. Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides. J. Am. Chem. Soc. 1941, 63, 2316–2320. [Google Scholar] [CrossRef]
- Yamamura, M.; Moritami, I.; Murahashi, S. The reaction of σ-vinylpalladium complexes with alkyllithiums. Stereospecific syntheses of olefins from vinyl halides and alkyllithiums. J. Organomet. Chem. 1975, 91, 39–42. [Google Scholar] [CrossRef]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [Green Version]
- Tietze, L.F.; Düfert, A. Multiple Pd-catalyzed reactions in the synthesis of natural products, drugs, and materials. Pure Appl. Chem. 2010, 82, 1375–1392. [Google Scholar] [CrossRef]
- Rodríguez-Padrón, D.; Puente-Santiago, A.R.; Balu, A.M.; Muñoz-Batista, M.J.; Luque, R. Environmental Catalysis: Present and Future. Chem. Cat. Chem. 2019, 11, 18–38. [Google Scholar] [CrossRef]
- Kim, D.-S.; Park, W.-J.; Jun, C.-H. Metal–Organic Cooperative Catalysis in C–H and C–C Bond Activation. Chem. Rev. 2017, 117, 8977–9015. [Google Scholar] [CrossRef]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Basu, D.; Kumar, S.; Sudhir V, S.; Bandichhor, R. Transition metal catalyzed C–H activation for the synthesis of medicinally relevant molecules: A Review. J. Chem. Sci. 2018, 130, 71. [Google Scholar] [CrossRef] [Green Version]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C−H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef]
- Sherry, B.D.; Fürstner, A. The Promise and Challenge of Iron-Catalyzed Cross Coupling. Acc. Chem. Res. 2008, 41, 1500–1511. [Google Scholar] [CrossRef]
- Moselage, M.; Li, J.; Ackermann, L. Cobalt-Catalyzed C–H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Liu, W.; Ackermann, L. Manganese-catalyzed C–H Activation. ACS Catal. 2016, 6, 3743–3752. [Google Scholar] [CrossRef]
- Kaplan, J.; Ward, D.M. The Essential Nature of Iron Usage and Regulation. Curr. Biol. 2013, 23, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes to Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C−O Bond Activation: Opportunity for Sustainable Catalysis. Chem SusChem 2017, 19, 3964–3981. [Google Scholar] [CrossRef]
- Guðmundsson, A.; Bäckvall, J.-E. On the Use of Iron in Organic Chemistry. Molecules 2020, 25, 1349. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Kochi, J.K. Vinylation of Grignard reagents. Catalysis by Iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. [Google Scholar] [CrossRef]
- Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. Iron-Catalyzed Direct Arylation through Directed C–H Bond Activation. J. Am. Chem. Soc. 2008, 18, 5858–5859. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C–H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Yoshikai, N. Iron-Catalyzed C−C Bond Formation via Chelation-Assisted C–H Activation. Isr. J. Chem. 2017, 57, 11171130. [Google Scholar] [CrossRef]
- Cera, G.; Ackermann, L. Iron-Catalyzed C–H Functionalization Processes. Top. Curr. Chem. 2016, 374, 57. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition Metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [Green Version]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1877. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp2)−H and C(sp3)−H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Doba, T.; Matsubara, T.; Ilies, L.; Shang, R.; Nakamura, E. Homocoupling-free iron-catalysed twofold C–H activation/cross-couplings of aromatics via transient connection of reactants. Nat. Catal. 2019, 2, 400–406. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Matsumoto, A.; Nakamura, E. β-Arylation of Carboxamides via Iron-Catalyzed C(sp3)–H Bond Activation. J. Am. Chem. Soc. 2013, 135, 6030–6032. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Born, R. Palladium-Catalyzed Direct Arylations of 1,2,3-Triazoles with Aryl Chlorides using Conventional Heating. Adv. Synth. Catal. 2008, 350, 741–748. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R.; Althammer, A. Assisted Ruthenium-Catalyzed C–H Bond Activation: Carboxylic Acids as Cocatalysts for Generally Applicable Direct Arylations in Apolar Solvents. Org. Lett. 2008, 10, 2299–2302. [Google Scholar] [CrossRef]
- Al Mamari, H.H.; Diers, E.; Ackermann, L. Triazole-Assisted Ruthenium-Catalyzed C−H Arylation of Aromatic Amides. Chem. Eur. J. 2014, 20, 9739–9743. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Guerrero, I.; Correa, A. Metal-Catalyzed C−H Functionalization Processes with “Click”-Triazole Assistance. Eur. J. Org. Chem. 2018, 6034–6049. [Google Scholar] [CrossRef]
- Ye, X.; He, Z.; Ahmed, T.; Weise, K.; Akhmedov, N.G.; Petersen, J.L.; Shi, X. 1,2,3-Triazoles as versatile directing group for selective sp2 and sp3 C–H activation: Cyclization vs. substitution. Chem. Sci. 2013, 4, 3712–3716. [Google Scholar] [CrossRef]
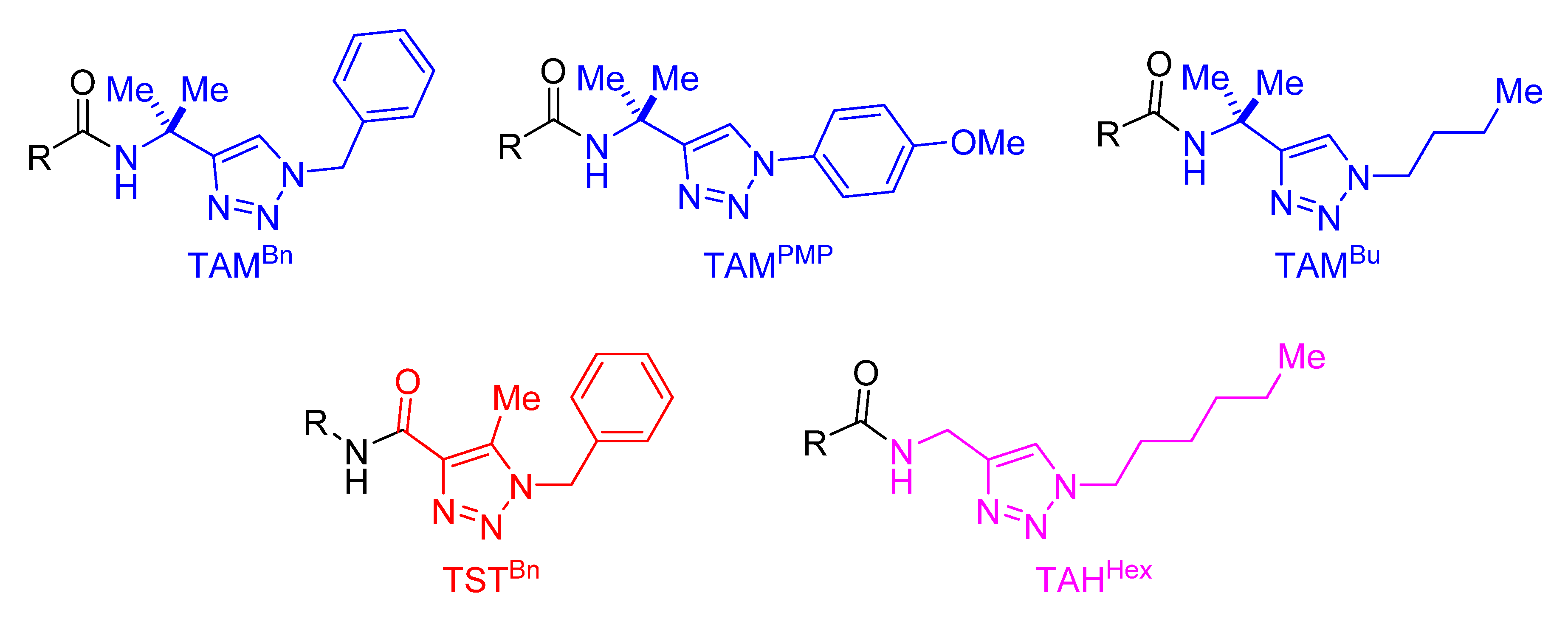
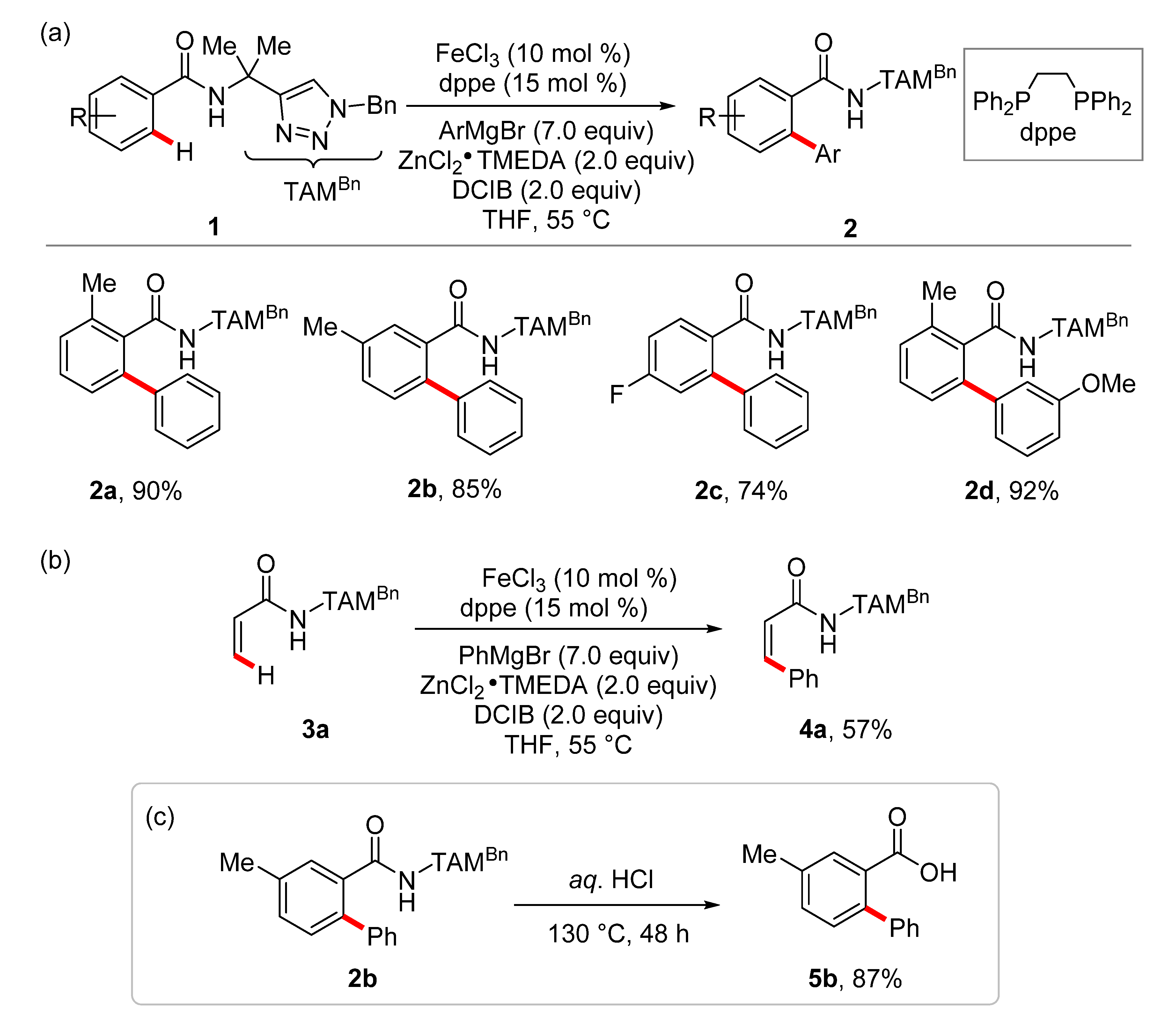
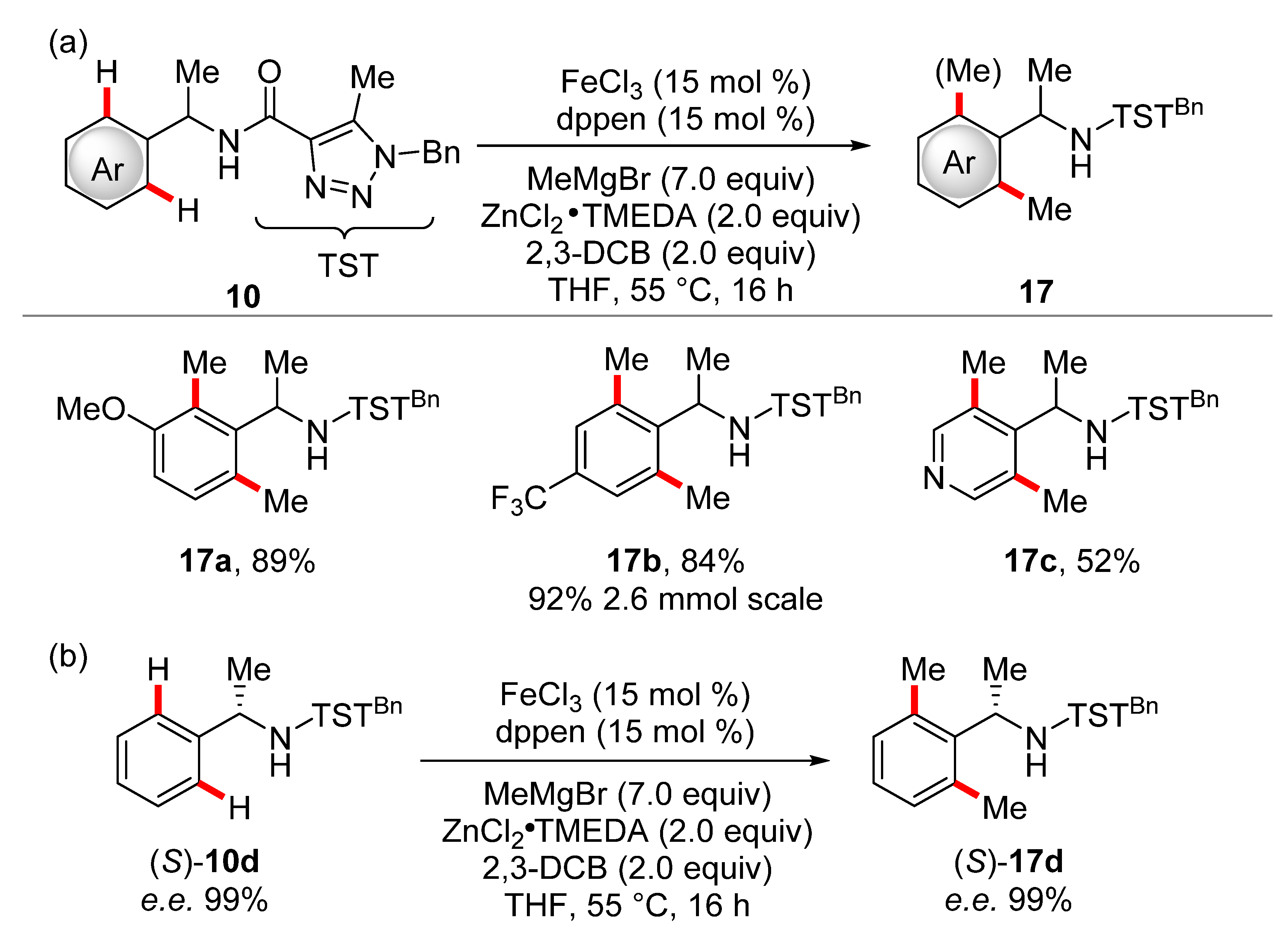
- Gu, Q.; Al Mamari, H.H.; Graczyk, K.; Diers, E.; Ackermann, L. Iron-Catalyzed C(sp2)−H and C(sp3)−H Arylation by Triazole Assistance. Angew. Chem. Int. Ed. 2014, 53, 3868–3871. [Google Scholar] [CrossRef]
- Ilies, L.; Asako, S.; Nakamura, E. Iron-Catalyzed Stereospecific Activation of Olefinic C–H Bonds with Grignard Reagent for Synthesis of Substituted Olefins. J. Am. Chem. Soc. 2011, 133, 7672–7675. [Google Scholar] [CrossRef] [PubMed]
- Schmiel, D.; Butenschön, H. Directed Iron-Catalyzed ortho-Alkylation and Arylation: Toward the Stereoselective Catalytic Synthesis of 1,2-Disubstituted Planar-Chiral Ferrocene Derivatives. Organometallics 2017, 36, 4979–4989. [Google Scholar] [CrossRef]
- Baudoin, O. Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds. Chem. Soc. Rev. 2011, 40, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Roane, J.; Dieu Tran, L. Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon–Hydrogen Bonds. Acc. Chem. Res. 2015, 48, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilies, L.; Itabashi, Y.; Shang, R.; Nakamura, E. Iron/Zinc-Co-catalyzed Directed Arylation and Alkenylation of C(sp3)–H Bonds with Organoborates. ACS Catal. 2017, 7, 89–92. [Google Scholar] [CrossRef]
- Shen, Z.; Cera, G.; Haven, T.; Ackermann, L. Tri-Substituted Triazole-Enabled C–H Activation of Benzyl and Aryl Amines by Iron Catalysis. Org. Lett. 2017, 19, 3795–3798. [Google Scholar] [CrossRef]
- Zhu, C.; Stangier, M.; Oliveira, J.C.A.; Massignan, L.; Ackermann, L. Iron-Electrocatalyzed C−H Arylations: Mechanistic Insights into Oxidation-Induced Reductive Elimination for Ferraelectrocatalysis. Chem. Eur. J. 2019, 25, 16382–16389. [Google Scholar] [CrossRef] [Green Version]
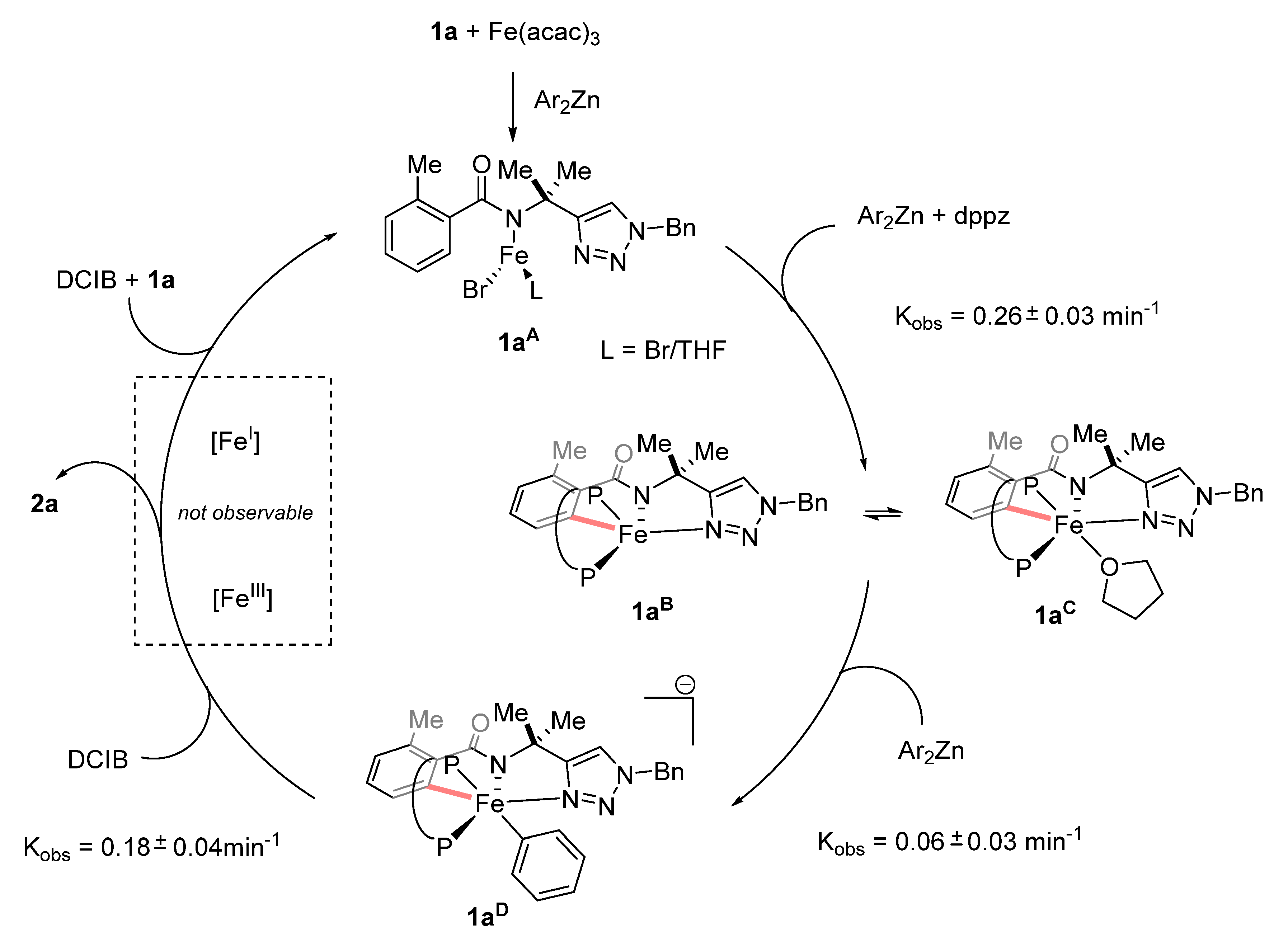
- Boddie, T.E.; Carpenter, S.H.; Baker, T.M.; DeMuth, J.C.; Cera, G.; Brennessel, W.W.; Ackermann, L.; Neidig, M.L. Identification and Reactivity of Cyclometalated Iron(II) Intermediates in Triazole-Directed Iron-Catalyzed C–H Activation. J. Am. Chem. Soc. 2019, 141, 12338–12345. [Google Scholar] [CrossRef]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef]
- Schönherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
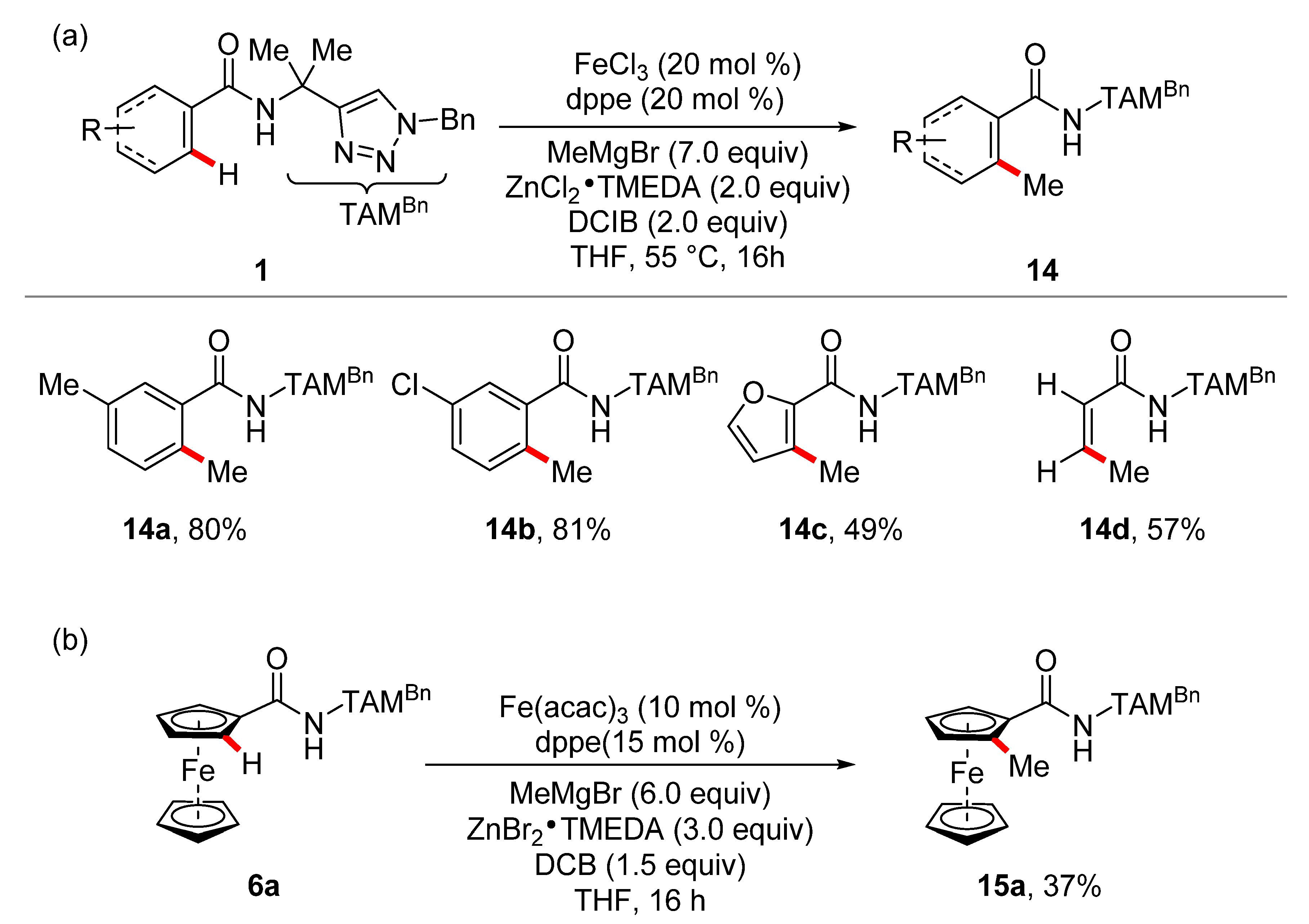
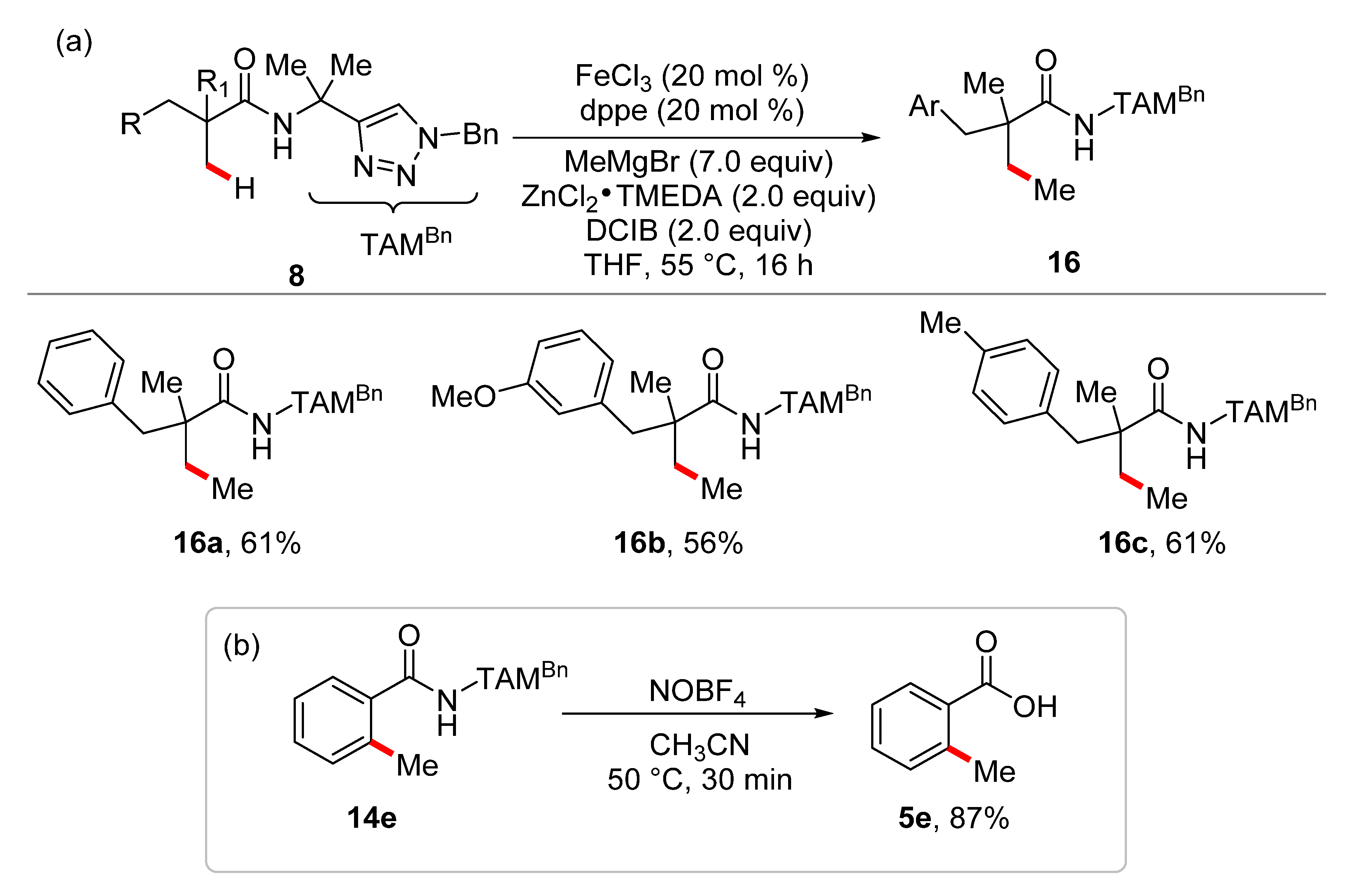
- Graczyk, K.; Haven, T.; Ackermann, L. Iron-Catalyzed C(sp2)–H and C(sp3)–H Methylations of Amides and Anilides. Chem. Eur. J. 2015, 21, 8812–8815. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed Directed C(sp2)–H and C(sp3)–H Functionalization with Trimethylaluminum. J. Am. Chem. Soc. 2015, 137, 7660–7663. [Google Scholar] [CrossRef] [PubMed]
- Cera, G.; Haven, T.; Ackermann, L. Expedient Iron-Catalyzed C−H Allylation/Alkylation by Triazole Assistance with Ample Scope. Angew. Chem. Int. Ed. 2016, 55, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Monks, B.M.; Fruchey, E.R.; Cook, S.P. Iron-Catalyzed C(sp2)−H Alkylation of Carboxamides with Primary Electrophiles. Angew. Chem. Int. Ed. 2014, 53, 11065–11069. [Google Scholar] [CrossRef]
- Fruchey, E.R.; Monks, B.M.; Cook, S.P. A Unified Strategy for Iron-Catalyzed ortho-Alkylation of Carboxamides. J. Am. Chem. Soc. 2014, 136, 13130–13133. [Google Scholar] [CrossRef]
- Noda, D.; Sunada, Y.; Hatakeyama, T.; Nakamura, M.; Nagashima, H. Effect of TMEDA on Iron-Catalyzed Coupling Reactions of ArMgX with Alkyl Halides. J. Am. Chem. Soc. 2009, 131, 6078–6079. [Google Scholar] [CrossRef]
- Asako, S.; Ilies, L.; Nakamura, E. Iron-Catalyzed Ortho-Allylation of Aromatic Carboxamides with Allyl Ethers. J. Am. Chem. Soc. 2013, 135, 17755–17757. [Google Scholar] [CrossRef]
- Asako, S.; Norinder, J.; Ilies, L.; Yoshikai, N.; Nakamura, E. ortho-Allylation of 1-Arylpyrazoles with Allyl Phenyl Ether via Iron-Catalyzed C–H Bond Activation under Mild Conditions. Adv. Synth. Catal. 2014, 356, 1481–1485. [Google Scholar] [CrossRef]
- Plietker, B. Regioselective Iron-Catalyzed Allylic Amination. Angew. Chem. Int. Ed. 2006, 45, 6053–6056. [Google Scholar] [CrossRef]
- Plietker, B. A Highly Regioselective Salt-Free Iron-Catalyzed Allylic Alkylation. Angew. Chem. Int. Ed. 2006, 45, 1469–1473. [Google Scholar] [CrossRef]
- Plietker, B.; Dieskau, A.; Möws, K.; Jatsch, A. Ligand-Dependent Mechanistic Dichotomy in Iron-Catalyzed Allylic Substitutions: σ-Allyl versus π-Allyl Mechanism. Angew. Chem. Int. Ed. 2008, 47, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Zell, D.; Oliveira, J.C.A.; Keil, H.; Stalke, D.; Ackermann, L. Asymmetric Iron-Catalyzed C−H Alkylation Enabled by Remote Ligand meta-Substitution. Angew. Chem. Int. Ed. 2017, 56, 14197–14201. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, A.S.; Gevorgyan, V. Formal Inverse Sonogashira Reaction: Direct Alkynylation of Arenes and Heterocycles with Alkynyl Halides. Angew. Chem. Int. Ed. 2010, 49, 2096–2098. [Google Scholar] [CrossRef]
- Negishi, E.; Anastasia, L. Palladium-Catalyzed Alkynylation. Chem. Rev. 2003, 103, 1979–2018. [Google Scholar] [CrossRef] [PubMed]
- Carril, M.; Correa, A.; Bolm, C. Iron-Catalyzed Sonogashira Reactions. Angew. Chem. Int. Ed. 2008, 47, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Xu, C.; Wojtas, L.; Akhmedov, N.G.; Chen, H.; Shi, X. Silver-Free Palladium-Catalyzed sp3 and sp2 C–H Alkynylation Promoted by a 1,2,3-Triazole Amine Directing Group. Org. Lett. 2016, 18, 2970–2973. [Google Scholar] [CrossRef]
- Cera, G.; Haven, T.; Ackermann, L. Iron-Catalyzed C−H Alkynylation through Triazole Assistance: Expedient Access to Bioactive Heterocycles. Chem. Eur. J. 2017, 23, 3577–3582. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Asymmetric Synthesis of Isoquinoline Alkaloids: 2004−2015. Chem. Rev. 2016, 116, 12369–12465. [Google Scholar] [CrossRef]
- Matsubara, T.; Ilies, L.; Nakamura, E. Oxidative C−H Activation Approach to Pyridone and Isoquinolone through an Iron-Catalyzed Coupling of Amides with Alkynes. Chem. Asian J. 2016, 11, 380–384. [Google Scholar] [CrossRef]
- Jung, E.M.; Piizzi, G. gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev. 2015, 105, 1735–1766. [Google Scholar] [CrossRef]
- Cera, G.; Haven, T.; Ackermann, L. Iron-catalyzed C–H/N–H activation by triazole guidance: Versatile alkyne annulation. Chem. Commun. 2017, 53, 6460–6463. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Müller, T.; Oliveira, J.C.A.; Demeshko, S.; Meyer, F.; Ackermann, L. Iron-Catalyzed C−H Activation with Propargyl Acetates: Mechanistic Insights into Iron(II) by Experiment, Kinetics, Mössbauer Spectroscopy, and Computation. Angew. Chem. Int. Ed. 2019, 58, 12874–12878. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Chauhan, R.; Rodrigues, C.A.B.; Maulide, N. Bridging C−H Activation: Mild and Versatile Cleavage of the 8-Aminoquinoline Directing Group. Chem. Eur. J. 2016, 22, 16805–16808. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Parchomyk, T.; Lülf, S.; Demeshko, S.; Meyer, F.; Koszinowski, K.; Ackermann, L. Mössbauer and mass spectrometry support for iron(II) catalysts in enantioselective C–H activation. Dalton Trans. 2019, 48, 5135–5139. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Tang, H.; Chen, K.; Hu, L.; Yao, J.; Shaik, S.; Chen, H. Two-State Reactivity in Low-Valent Iron-Mediated C–H Activation and the Implications for Other First-Row Transition Metals. J. Am. Chem. Soc. 2016, 138, 3715–3730. [Google Scholar] [CrossRef]
- Balcells, D.; Clot, E.; Eisenstein, O. C–H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev. 2010, 110, 749–823. [Google Scholar] [CrossRef]
- Yu, S.; Ma, S. Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses. Angew. Chem. Int. Ed. 2012, 51, 3074–3112. [Google Scholar] [CrossRef]
- Mo, J.; Müller, T.; Oliveira, J.C.A.; Ackermann, L. 1,4-Iron Migration for Expedient Allene Annulations through Iron-Catalyzed C−H/N−H/C−O/C−H Functionalizations. Angew. Chem. Int. Ed. 2018, 57, 7719–7723. [Google Scholar] [CrossRef]
- Davies, D.L.; Macgregor, S.A.; McMullin, C.L. Computational Studies of Carboxylate-Assisted C–H Activation and Functionalization at Group 8–10 Transition Metal Centers. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef]
- Zell, D.; Bursch, M.; Müller, V.; Grimme, S.; Ackermann, L. Full Selectivity Control in Cobalt(III)-Catalyzed C−H Alkylations by Switching of the C−H Activation Mechanism. Angew. Chem. Int. Ed. 2017, 56, 10378–10382. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Carrow, B.P. Oligothiophene Synthesis by a General C–H Activation Mechanism: Electrophilic Concerted Metalation–Deprotonation (eCMD). ACS Catal. 2019, 9, 6821–6836. [Google Scholar] [CrossRef]
- Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. Synthesis of Anthranilic Acid Derivatives through Iron-Catalyzed Ortho Amination of Aromatic Carboxamides with N-Chloroamines. J. Am. Chem. Soc. 2014, 136, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Yoshigoe, Y.; Kuninobu, Y. Iron-Catalyzed ortho-Selective C–H Borylation of 2-Phenylpyridines and Their Analogs. Org. Lett. 2017, 19, 3450–3453. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzi, M.; Cera, G. Iron-Catalyzed C–H Functionalizations under Triazole-Assistance. Molecules 2020, 25, 1806. https://doi.org/10.3390/molecules25081806
Lanzi M, Cera G. Iron-Catalyzed C–H Functionalizations under Triazole-Assistance. Molecules. 2020; 25(8):1806. https://doi.org/10.3390/molecules25081806
Chicago/Turabian StyleLanzi, Matteo, and Gianpiero Cera. 2020. "Iron-Catalyzed C–H Functionalizations under Triazole-Assistance" Molecules 25, no. 8: 1806. https://doi.org/10.3390/molecules25081806
APA StyleLanzi, M., & Cera, G. (2020). Iron-Catalyzed C–H Functionalizations under Triazole-Assistance. Molecules, 25(8), 1806. https://doi.org/10.3390/molecules25081806