
Beyond Typical Electrolytes for Energy Dense Batteries

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
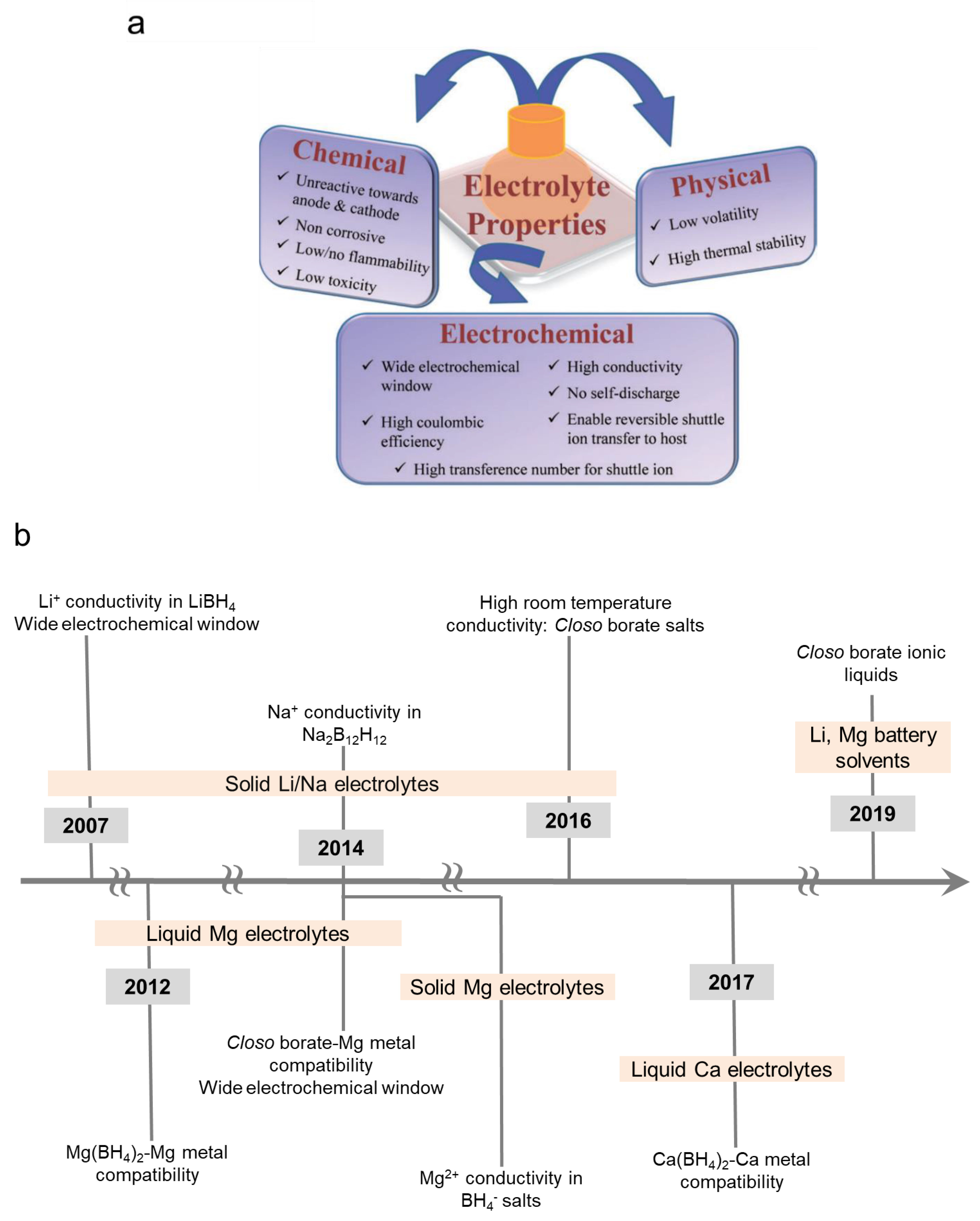
2. Explanation of the Review’s Structure
3. Solid-State Hydride Electrolytes for Monovalent Batteries
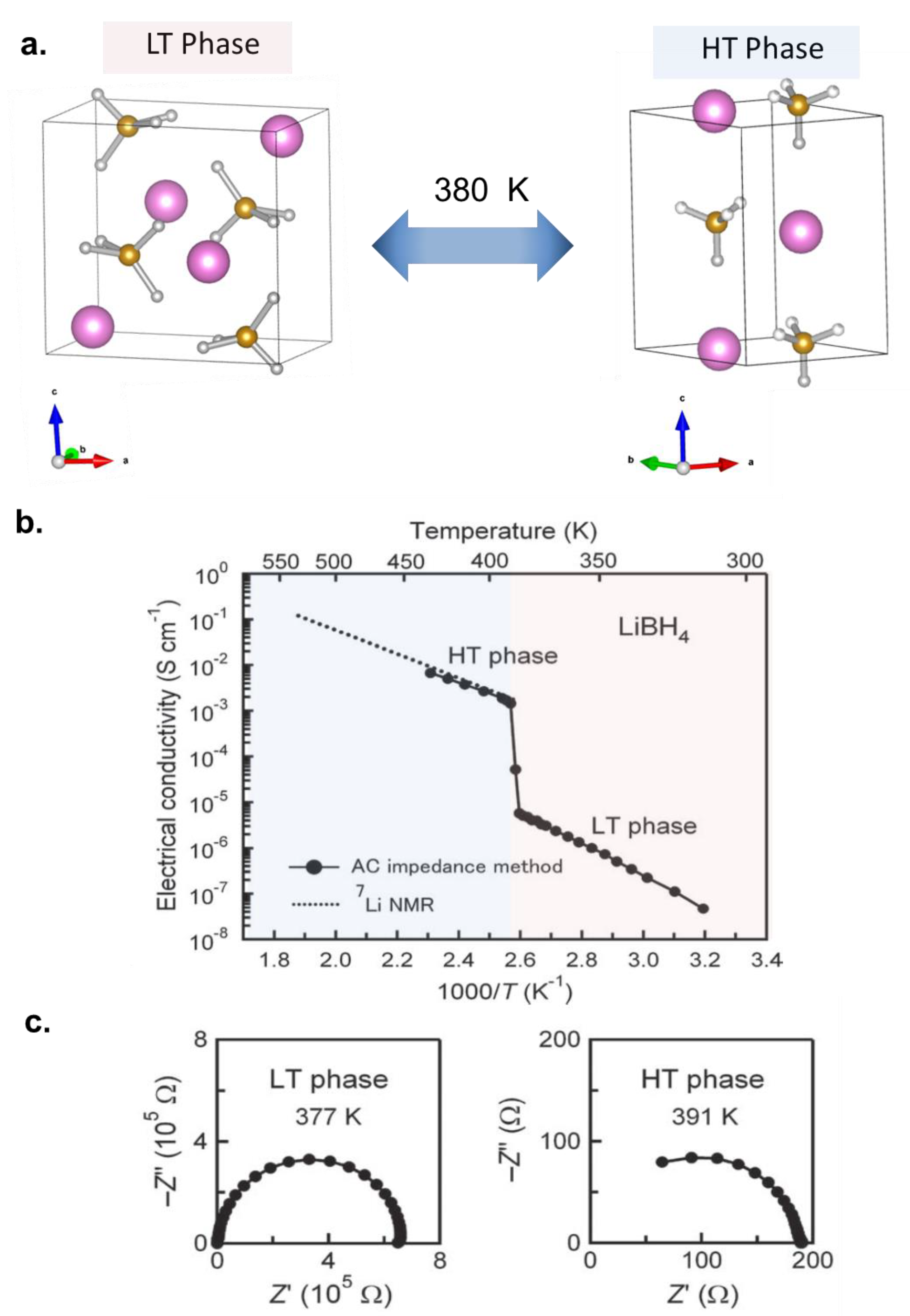
3.1. Development of Highly Conductive Solid Electrolytes
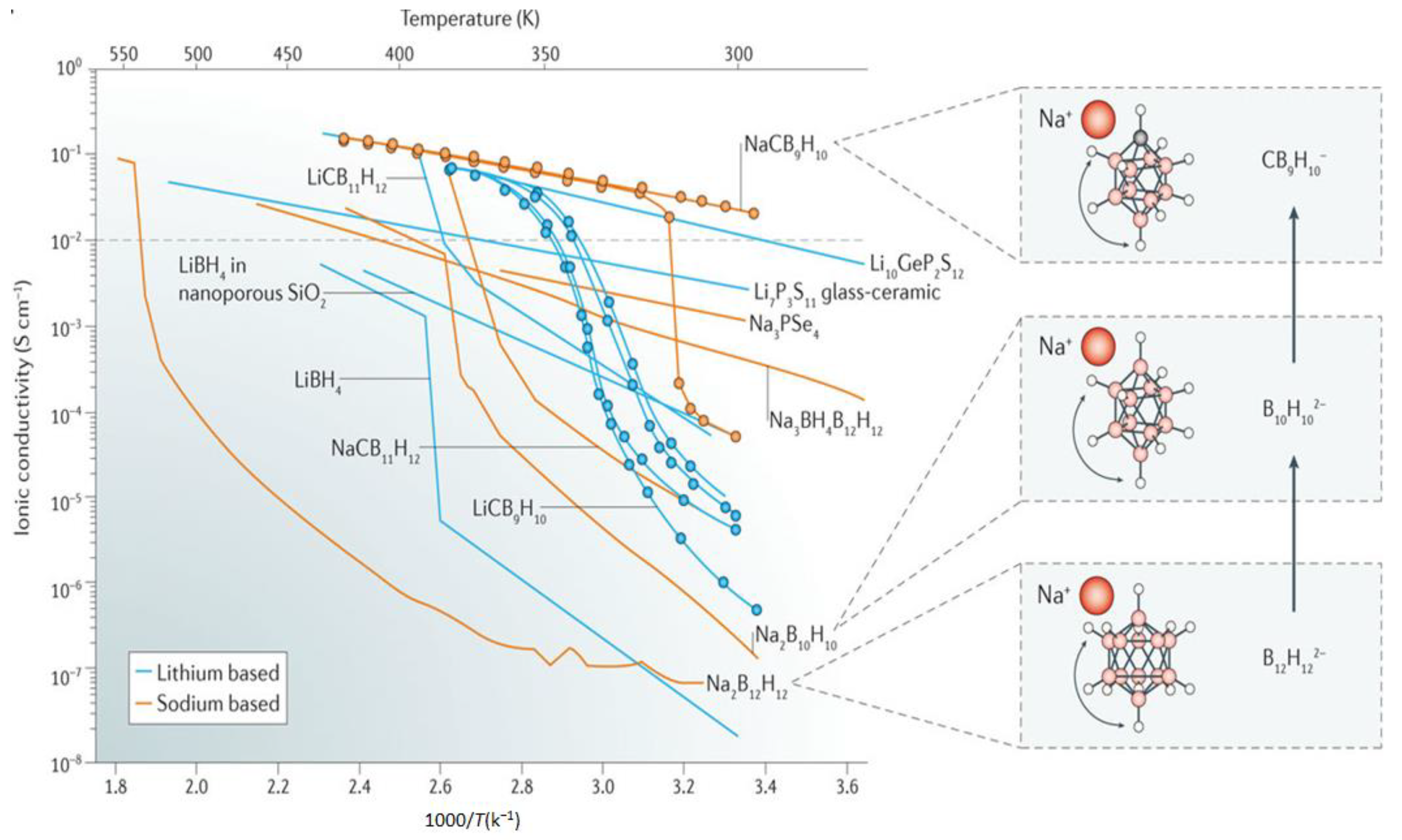
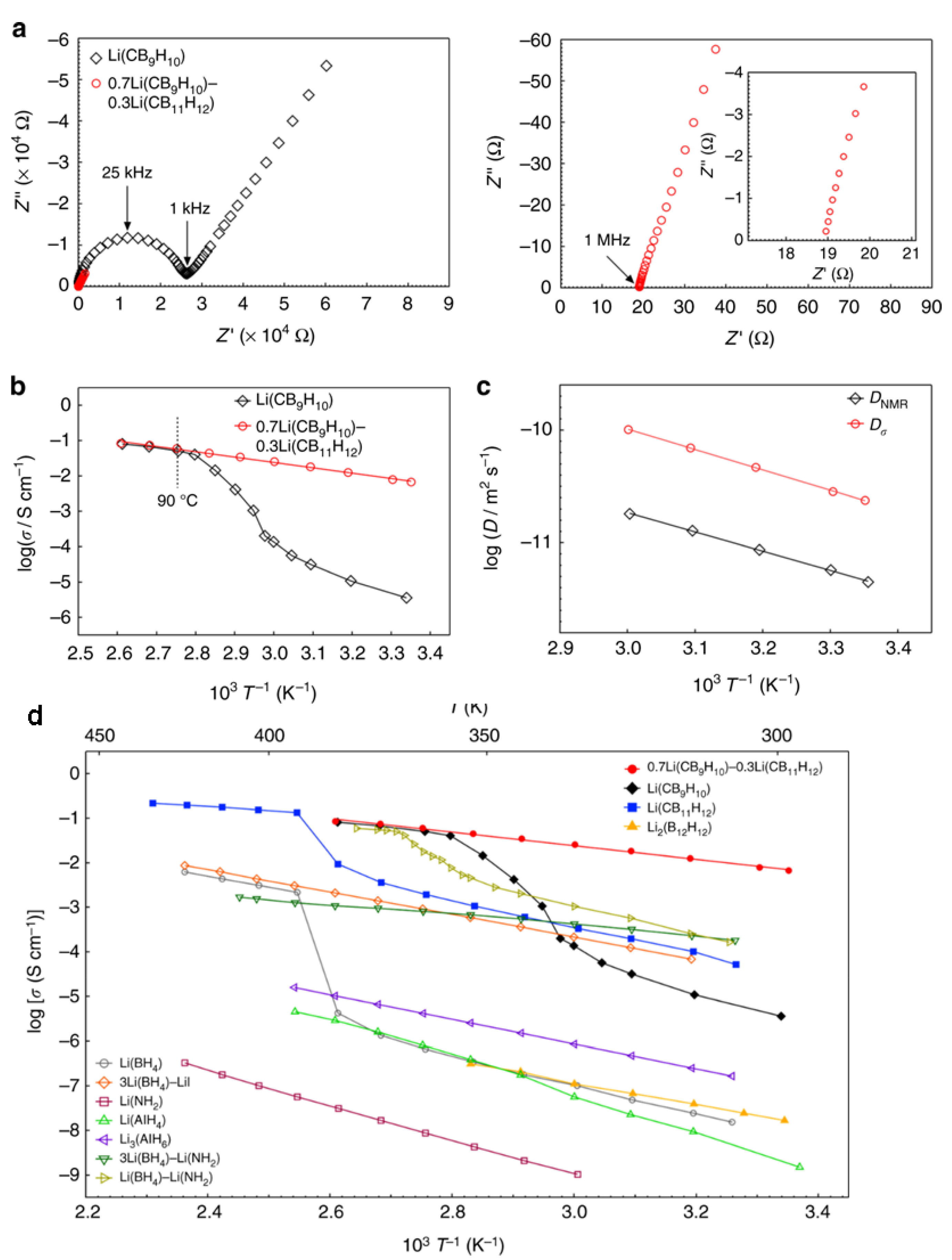
3.2. Towards Achieving High Li+, Na+ Mobilities: Closo-Borates
4. Electrolytes for Multivalent Batteries
4.1. Liquid Borohydride Electrolytes: Achieving Compatibility with Mg Metal
4.2. Electrolytes with Wide Electrochemical Window: Closo-Borate Salts
4.3. Hydride Interfaces in Multivalent Batteries: Significance and Nature
4.4. Solid-State Electrolytes
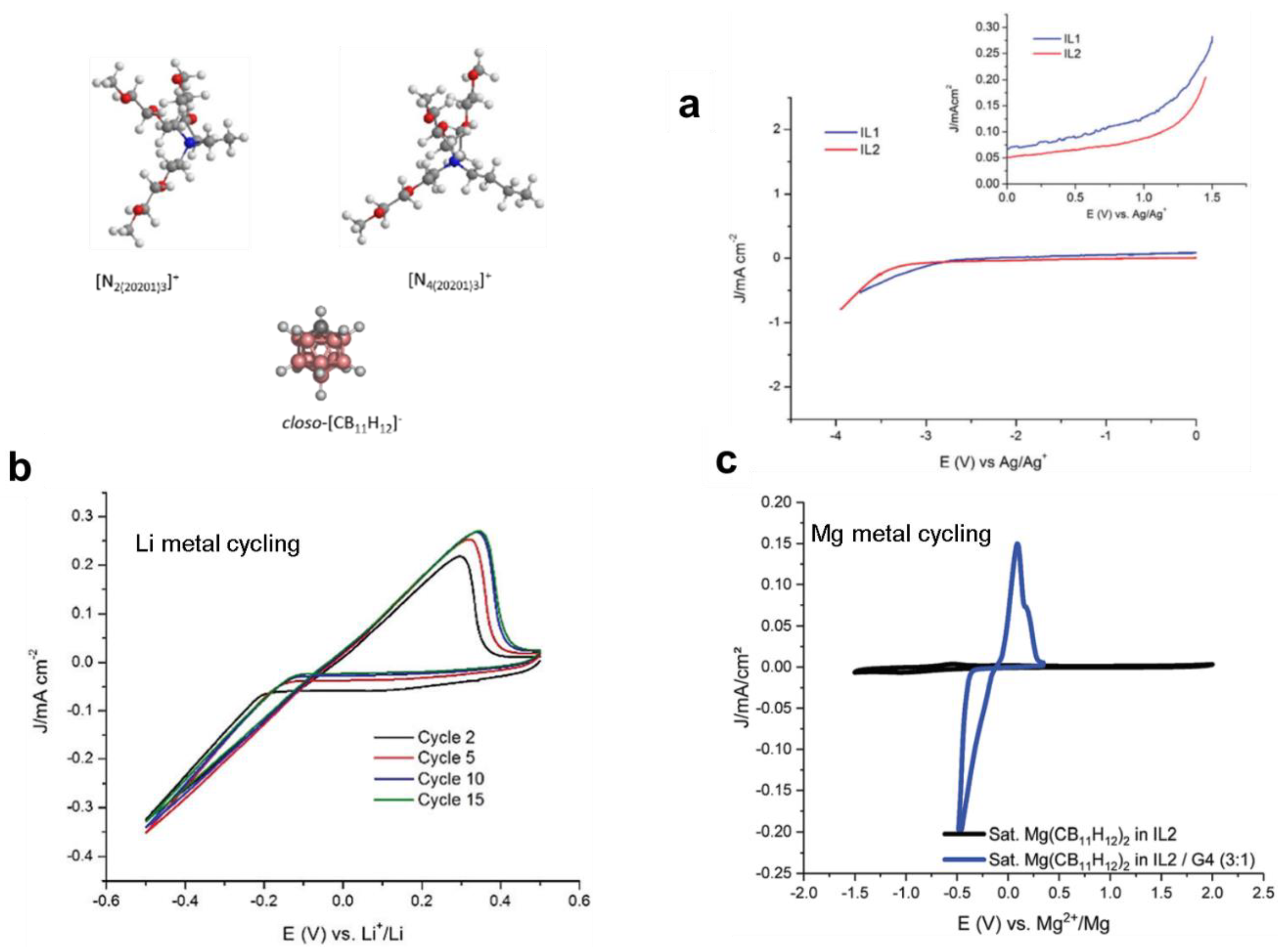
5. Hydrides’ Potential as Stable Solvents: Ionic Liquid Electrolytes
6. Demonstrations of Hydride Electrolytes in Batteries
7. Conclusions
Funding
Conflicts of Interest
References
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, Y.; Lotsch, B.; Hu, Y.-S.; Li, H.; Janek, J.; Nazar, L.F.; Nan, C.-W.; Maier, J.; Armand, M.; et al. New horizons for inorganic solid state ion conductors. Energy Environ. Sci. 2018, 11, 1945–1976. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, L.; Yu, Y.; Sun, J. Progress of enhancing the safety of lithium ion battery from the electrolyte aspect. Nano Energy 2019, 55, 93–114. [Google Scholar] [CrossRef]
- Kato, Y.; Hori, S.; Saito, T.; Suzuki, K.; Hirayama, M.; Mitsui, A.; Yonemura, M.; Iba, H.; Kanno, R. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 2016, 1, 16030. [Google Scholar] [CrossRef]
- Mohtadi, R.; Mizuno, F. Magnesium batteries: Current state of the art, issues and future perspectives. Beilstein J. Nanotechnol. 2014, 5, 1291–1311. [Google Scholar] [CrossRef]
- Ponrouch, A.; Palacin, M.R. On the road toward calcium-based batteries. Curr. Opin. Electrochem. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Mohtadi, R.; Orimo, S.-I. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef]
- Tutusaus, O.; Mohtadi, R. Paving the Way towards Highly Stable and Practical Electrolytes for Rechargeable Magnesium Batteries. ChemElectroChem 2015, 2, 51–57. [Google Scholar] [CrossRef]
- Boukamp, B.A.; Huggins, R.A. Ionic-Conductivity in Lithium Imide. Phys. Lett. A 1979, 72, 464–466. [Google Scholar] [CrossRef]
- Matsuo, M.; Nakamori, Y.; Orimo, S.; Maekawa, H.; Takamura, H. Lithium superionic conduction in lithium borohydride accompanied by structural transition. Appl. Phys. Lett. 2007, 91, 224103. [Google Scholar] [CrossRef]
- Nakamori, Y.; Orimo, S.; Tsutaoka, T. Dehydriding reaction of metal hydrides and alkali borohydrides enhanced by microwave irradiation. Appl. Phys. Lett. 2006, 88, 112104. [Google Scholar] [CrossRef]
- Matsuo, M.; Orimo, S. Lithium Fast-Ionic Conduction in Complex Hydrides: Review and Prospects. Adv. Energy Mater. 2011, 1, 161–172. [Google Scholar] [CrossRef]
- Buchter, F.; Lodziana, Z.; Mauron, P.; Remhof, A.; Friedrichs, O.; Borgschulte, A.; Zuttel, A.; Sheptyakov, D.; Strassle, T.; Ramirez-Cuesta, A.J. Dynamical properties and temperature induced molecular disordering of LiBH4 and LiBD4. Phys. Rev. B 2008, 78, 094302. [Google Scholar] [CrossRef]
- Remhof, A.; Lodziana, Z.; Martelli, P.; Friedrichs, O.; Zuttel, A.; Skripov, A.V.; Embs, J.P.; Strassle, T. Rotational motion of BH4 units in MBH4 (M=Li,Na,K) from quasielastic neutron scattering and density functional calculations. Phys. Rev. B 2010, 81, 214304. [Google Scholar] [CrossRef]
- Asakura, R.; Duchêne, L.; Kühnel, R.-S.; Remhof, A.; Hagemann, H.; Battaglia, C. Electrochemical Oxidative Stability of Hydroborate-Based Solid-State Electrolytes. ACS Appl. Energy Mater. 2019, 2, 6924–6930. [Google Scholar] [CrossRef]
- Maekawa, H.; Matsuo, M.; Takamura, H.; Ando, M.; Noda, Y.; Karahashi, T.; Orimo, S.I. Halide-Stabilized LiBH4, a Room-Temperature Lithium Fast-Ion Conductor. J. Am. Chem. Soc. 2009, 131, 894–895. [Google Scholar] [CrossRef]
- Oguchi, H.; Matsuo, M.; Hummelshoj, J.S.; Vegge, T.; Norskov, J.K.; Sato, T.; Miura, Y.; Takamura, H.; Maekawa, H.; Orimo, S. Experimental and computational studies on structural transitions in the LiBH4-LiI pseudobinary system. Appl. Phys. Lett. 2009, 94, 141912. [Google Scholar] [CrossRef]
- Miyazaki, R.; Karahashi, T.; Kumatani, N.; Noda, Y.; Ando, M.; Takamura, H.; Matsuo, M.; Orimo, S.; Maekawa, H. Room temperature lithium fast-ion conduction and phase relationship of LiI stabilized LiBH4. Solid State Ion. 2011, 192, 143–147. [Google Scholar] [CrossRef]
- Matsuo, M.; Remhof, A.; Martelli, P.; Caputo, R.; Ernst, M.; Miura, Y.; Sato, T.; Oguchi, H.; Maekawa, H.; Takamura, H.; et al. Complex Hydrides with (BH4)(-) and (NH2)(-) Anions as New Lithium Fast-Ion Conductors. J. Am. Chem. Soc. 2009, 131, 16389–16391. [Google Scholar] [CrossRef]
- Yan, Y.; Kühnel, R.-S.; Remhof, A.; Duchêne, L.; Reyes, E.C.; Rentsch, D.; Łodziana, Z.; Battaglia, C. A Lithium Amide-Borohydride Solid-State Electrolyte with Lithium-Ion Conductivities Comparable to Liquid Electrolytes. Adv. Energy Mater. 2017, 7, 1700294. [Google Scholar] [CrossRef]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.-S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a New Lithium-Ion Conductor and Hydrogen Storage Material with Isolated Tetranuclear Anionic Clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- GharibDoust, S.P.; Brighi, M.; Sadikin, Y.; Ravnsbaek, D.B.; Cerny, R.; Skibsted, J.; Jensen, T.R. Synthesis, Structure, and Li-Ion Conductivity of LiLa(BH4)(3)X, X = Cl, Br, I.J. Phys. Chem. C 2017, 121, 19010–19021. [Google Scholar] [CrossRef]
- Lee, Y.S.; Filinchuk, Y.; Lee, H.S.; Suh, J.Y.; Kim, J.W.; Yu, J.S.; Cho, Y.W. On the Formation and the Structure of the First Bimetallic Borohydride Borate, LiCa3(BH4)(BO3)(2). J. Phys. Chem. C 2011, 115, 10298–10304. [Google Scholar] [CrossRef]
- Didelot, E.; Cerny, R. Ionic conduction in bimetallic borohydride borate, LiCa3(BH4)(BO3)(2). Solid State Ion. 2017, 305, 16–22. [Google Scholar] [CrossRef]
- Ngene, P.; Adelhelm, P.; Beale, A.M.; de Jong, K.P.; de Jongh, P.E. LiBH4/SBA-15 Nanocomposites Prepared by Melt Infiltration under Hydrogen Pressure: Synthesis and Hydrogen Sorption Properties. J. Phys. Chem. C 2010, 114, 6163–6168. [Google Scholar] [CrossRef]
- Blanchard, D.; Nale, A.; Sveinbjornsson, D.; Eggenhuisen, T.M.; Verkuijlen, M.H.; Vegge, T.; Kentgens, A.P.M.; de Jongh, P.E. Nanoconfined LiBH4 as a Fast Lithium Ion Conductor. Adv. Funct. Mater. 2015, 25, 184–192. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Liu, X.F.; Majzoub, E.H.; Vajo, J.J.; Gross, A.F. Dynamical Perturbations of Tetrahydroborate Anions in LiBH4 due to Nanoconfinement in Controlled-Pore Carbon Scaffolds. J. Phys. Chem. C 2013, 117, 17983–17995. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, Y.S.; Choi, D.J.; Chae, K.H.; Oh, K.H.; Cho, Y.W. Enhanced Li Ion Conductivity in LiBH4-Al2O3 Mixture via Interface Engineering. J. Phys. Chem. C 2017, 121, 26209–26215. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Colon-Mercado, H.R.; Ward, P.A.; Peters, B.; Giri, S.; Zhou, J.; Greenway, S.; Compton, R.N.; Jena, P.; Zidan, R. Experimental and Theoretical Analysis of Fast Lithium Ionic Conduction in a LiBH4-C-60 Nanocomposite. J. Phys. Chem. C 2014, 118, 21755–21761. [Google Scholar] [CrossRef]
- Unemoto, A.; Wu, H.; Udovic, T.J.; Matsuo, M.; Ikeshoji, T.; Orimo, S. Fast lithium-ionic conduction in a new complex hydride-sulphide crystalline phase. Chem. Commun. 2016, 52, 564–566. [Google Scholar] [CrossRef]
- Yamauchi, A.; Sakuda, A.; Hayashi, A.; Tatsumisago, M. Preparation and ionic conductivities of (100-x)(0.75Li(2)S center dot 0.25P(2)S(5))center dot xLiBH(4) glass electrolytes. J. Power Sources 2013, 244, 707–710. [Google Scholar] [CrossRef]
- El Kharbachi, A.; Hu, Y.; Yoshida, K.; Vajeeston, P.; Kim, S.; Sorby, M.H.; Orimo, S.; Fjellvag, H.; Hauback, B.C. Lithium ionic conduction in composites of Li(BH4)(0.75)I-0.25 and amorphous 0.75Li(2)S center dot 0.25P(2)S(5) for battery applications. Electrochim. Acta 2018, 278, 332–339. [Google Scholar] [CrossRef]
- Sveinbjörnsson, D.; Blanchard, D.; Myrdal, J.S.G.; Younesi, R.; Viskinde, R.; Riktor, M.D.; Norby, P.; Vegge, T. Ionic conductivity and the formation of cubic CaH2 in the LiBH4–Ca(BH4)2 composite. J. Solid State Chem. 2014, 211, 81–89. [Google Scholar] [CrossRef][Green Version]
- Xiang, M.; Zhang, Y.; Zhan, L.; Zhu, Y.; Guo, X.; Chen, J.; Wang, Z.; Li, L. Study on xLiBH4-NaBH4 (x = 1.6, 2.3, and 4) composites with enhanced lithium ionic conductivity. J. Alloy. Compd. 2017, 729, 936–941. [Google Scholar] [CrossRef]
- López-Aranguren, P.; Berti, N.; Dao, A.H.; Zhang, J.; Cuevas, F.; Latroche, M.; Jordy, C. An all-solid-state metal hydride–Sulfur lithium-ion battery. J. Power Sources 2017, 357, 56–60. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Zhu, Y.; Guo, X.; Chen, J.; Li, L. Ternary LiBH4-NaBH4-MgH2 composite as fast ionic conductor. Solid State Ion. 2018, 324, 109–113. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Lin, H.; Zhu, Y.; Guo, X.; Chen, J.; Li, L. LiBH4-NaX (X = Cl, I) composites with enhanced lithium ionic conductivity. J. Alloy. Compd. 2018, 764, 307–313. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Song, T.; Miyaoka, H.; Shinzato, K.; Miyaoka, H.; Ichikawa, T.; Shi, S.; Zhang, X.; Isobe, S.; et al. Ammonia, a Switch for Controlling High Ionic Conductivity in Lithium Borohydride Ammoniates. Joule 2018, 2, 1522–1533. [Google Scholar] [CrossRef]
- Matsuo, M.; Kuromoto, S.; Sato, T.; Oguchi, H.; Takamura, H.; Orimo, S. Sodium ionic conduction in complex hydrides with [BH4](-) and [NH2](-) anions. Appl. Phys. Lett. 2012, 100, 203904. [Google Scholar] [CrossRef]
- Unemoto, A.; Matsuo, M.; Orimo, S. Complex Hydrides for Electrochemical Energy Storage. Adv. Funct. Mater. 2014, 24, 2267–2279. [Google Scholar] [CrossRef]
- Matsuo, M.; Oguchi, H.; Sato, T.; Takamura, H.; Tsuchida, E.; Ikeshoji, T.; Orimo, S. Sodium and magnesium ionic conduction in complex hydrides. J. Alloy. Compd. 2013, 580, S98–S101. [Google Scholar] [CrossRef]
- Oguchi, H.; Matsuo, M.; Sato, T.; Takamura, H.; Maekawa, H.; Kuwano, H.; Orimo, S. Lithium-ion conduction in complex hydrides LiAlH(4) and Li(3)AlH(6). J. Appl. Phys. 2010, 107, 096104. [Google Scholar] [CrossRef]
- Li, W.; Wu, G.T.; Xiong, Z.T.; Feng, Y.P.; Chen, P. Li+ ionic conductivities and diffusion mechanisms in Li-based imides and lithium amide. Phys. Chem. Chem. Phys. 2012, 14, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Rijssenbeek, J.; Gao, Y.; Hanson, J.; Huang, Q.; Jones, C.; Toby, B. Crystal structure determination and reaction pathway of amide-hydride mixtures. J. Alloy. Compd. 2008, 454, 233–244. [Google Scholar] [CrossRef]
- Wu, G.T.; Xiong, Z.T.; Liu, T.; Liu, Y.F.; Hu, J.J.; Chen, P.; Feng, Y.P.; Wee, A.T.S. Synthesis and characterization of a new ternary imide-Li2Ca(NH)(2). Inorg. Chem. 2007, 46, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.; Remhof, A.; Borgschulte, A.; Ackermann, R.; Strassle, T.; Embs, J.P.; Ernst, M.; Matsuo, M.; Orimo, S.I.; Zuttel, A. Rotational Motion in LiBH4/LiI Solid Solutions. J. Phys. Chem. A 2011, 115, 5329–5334. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Ley, M.B.; Jensen, T.R.; Filinchuk, Y. Nuclear Magnetic Resonance Studies of BH4 Reorientations and Li Diffusion in LiLa(BH4)(3)Cl. J. Phys. Chem. C 2013, 117, 14965–14972. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Ley, M.B.; Jensen, T.R.; Cho, Y.W. Lithium Ion Disorder and Conduction Mechanism in LiCe(BH4)3Cl. J. Phys. Chem. C 2016, 120, 19035–19042. [Google Scholar] [CrossRef]
- Jansen, M. Volume Effect or Paddle-Wheel Mechanism—Fast Alkali-Metal Ionic-Conduction in Solids with Rotationally Disordered Complex Anions. Angew. Chem. Int. Ed. Engl. 1991, 30, 1547–1558. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Wu, H.; Skripov, A.V. Evolution of the Reorientational Motions of the Tetrahydroborate Anions in Hexagonal LiBH4-Lil Solid Solution by High-Q Quasielastic Neutron Scattering. J. Phys. Chem. C 2013, 117, 12010–12018. [Google Scholar] [CrossRef]
- Orimo, S.-I.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex Hydrides for Hydrogen Storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Paskevicius, M.; Pitt, M.P.; Brown, D.H.; Sheppard, D.A.; Chumphongphan, S.; Buckley, C.E. First-order phase transition in the Li2B12H12 system. Phys. Chem. Chem. Phys. 2013, 15, 15825–15828. [Google Scholar] [CrossRef] [PubMed]
- Skripov, A.V.; Babanova, O.A.; Soloninin, A.V.; Stavila, V.; Verdal, N.; Udovic, T.J.; Rush, J.J. Nuclear Magnetic Resonance Study of Atomic Motion in A2B12H12 (A = Na, K, Rb, Cs): Anion Reorientations and Na+ Mobility. J. Phys. Chem. C 2013, 117, 25961–25968. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Unemoto, A.; Verdal, N.; Stavila, V.; Skripov, A.V.; Rush, J.J.; Takamura, H.; Orimo, S. Sodium superionic conduction in Na2B12H12. Chem. Commun. 2014, 50, 3750–3752. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Tang, W.S.; Wu, H.; Stavila, V.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Rush, J.J.; et al. Exceptional Superionic Conductivity in Disordered Sodium Decahydro-closo-decaborate. Adv. Mater. 2014, 26, 7622–7626. [Google Scholar] [CrossRef]
- Varley, J.B.; Kweon, K.; Mehta, P.; Shea, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Understanding Ionic Conductivity Trends in Polyborane Solid Electrolytes from Ab Initio Molecular Dynamics. ACS Energy Lett. 2017, 2, 250–255. [Google Scholar] [CrossRef]
- Kweon, K.E.; Varley, J.B.; Shea, P.; Adelstein, N.; Mehta, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Structural, Chemical, and Dynamical Frustration: Origins of Superionic Conductivity in closo-Borate Solid Electrolytes. Chem. Mater. 2017, 29, 9142–9153. [Google Scholar] [CrossRef]
- Sadikin, Y.; Brighi, M.; Schouwink, P.; Cerny, R. Superionic Conduction of Sodium and Lithium in Anion-Mixed Hydroborates Na3BH4B12H12 and (Li0.7Na0.3)(3)BH4B12H12. Adv. Energy Mater. 2015, 5, 1501016. [Google Scholar] [CrossRef]
- He, L.; Li, H.-W.; Nakajima, H.; Tumanov, N.; Filinchuk, Y.; Hwang, S.-J.; Sharma, M.; Hagemann, H.; Akiba, E. Synthesis of a Bimetallic Dodecaborate LiNaB12H12 with Outstanding Superionic Conductivity. Chem. Mater. 2015, 27, 5483–5486. [Google Scholar] [CrossRef]
- Duchene, L.; Kuhnel, R.S.; Rentsch, D.; Remhof, A.; Hagemann, H.; Battaglia, C. A highly stable sodium solid-state electrolyte based on a dodeca/deca-borate equimolar mixture. Chem. Commun. 2017, 53, 4195–4198. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sato, T.; Unemoto, A.; Matsuo, M.; Ikeshoji, T.; Udovic, T.J.; Orimo, S. Fast sodium ionic conduction in Na2B10H10-Na2B12H12 pseudo-binary complex hydride and application to a bulk-type all-solid-state battery. Appl. Phys. Lett. 2017, 110, 103901. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Colon-Mercado, H.; Washington Ii, A.L.; Ward, P.A.; Greenway, S.; Missimer, D.M.; Hartman, H.; Velten, J.; Christian, J.H.; Zidan, R. Bi-functional Li2B12H12 for energy storage and conversion applications: Solid-state electrolyte and luminescent down-conversion dye. J. Mater. Chem. A 2015, 3, 22853–22859. [Google Scholar] [CrossRef]
- Tang, W.S.; Yoshida, K.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Dimitrievska, M.; Stavila, V.; Orimo, S.-I.; Udovic, T.J. Stabilizing Superionic-Conducting Structures via Mixed-Anion Solid Solutions of Monocarba-closo-borate Salts. ACS Energy Lett. 2016, 1, 659–664. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Unemoto, A.; Orimo, S.-I.; Udovic, T.J. Stabilizing lithium and sodium fast-ion conduction in solid polyhedral-borate salts at device-relevant temperatures. Energy Storage Mater. 2016, 4, 79–83. [Google Scholar] [CrossRef]
- Kim, S.; Toyama, N.; Oguchi, H.; Sato, T.; Takagi, S.; Ikeshoji, T.; Orimo, S.-I. Fast Lithium-Ion Conduction in Atom-Deficient closo-Type Complex Hydride Solid Electrolytes. Chem. Mater. 2018, 30, 386–391. [Google Scholar] [CrossRef]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.-I.; Udovic, T.J. Unparalleled lithium and sodium superionic conduction in solid electrolytes with large monovalent cage-like anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef]
- Dimitrievska, M.; Shea, P.; Kweon, K.E.; Bercx, M.; Varley, J.B.; Tang, W.S.; Skripov, A.V.; Stavila, V.; Udovic, T.J.; Wood, B.C. Carbon Incorporation and Anion Dynamics as Synergistic Drivers for Ultrafast Diffusion in Superionic LiCB11H12 and NaCB11H12. Adv. Energy Mater. 2018, 8, 1703422. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Zhou, W.; Talin, A.A.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; et al. Liquid-like Ionic Conduction in Solid Lithium and Sodium Monocarba-closo-decaborates near or at Room Temperature. Adv. Energy Mater. 2016, 6, 1502237. [Google Scholar] [CrossRef]
- Kim, S.; Oguchi, H.; Toyama, N.; Sato, T.; Takagi, S.; Otomo, T.; Arunkumar, D.; Kuwata, N.; Kawamura, J.; Orimo, S.-I. A complex hydride lithium superionic conductor for high-energy-density all-solid-state lithium metal batteries. Nat. Commun. 2019, 10, 1081. [Google Scholar] [CrossRef]
- Brighi, M.; Murgia, F.; Łodziana, Z.; Schouwink, P.; Wołczyk, A.; Cerny, R. A mixed anion hydroborate/carba-hydroborate as a room temperature Na-ion solid electrolyte. J. Power Sources 2018, 404, 7–12. [Google Scholar] [CrossRef]
- Tang, W.S.; Dimitrievska, M.; Stavila, V.; Zhou, W.; Wu, H.; Talin, A.A.; Udovic, T.J. Order–Disorder Transitions and Superionic Conductivity in the Sodium nido-Undeca(carba)borates. Chem. Mater. 2017, 29, 10496–10509. [Google Scholar] [CrossRef]
- Yan, Y.; Rentsch, D.; Battaglia, C.; Remhof, A. Synthesis, stability and Li-ion mobility of nanoconfined Li2B12H12. Dalton Trans. 2017, 46, 12434–12437. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.R.S.; Paskevicius, M.; Jørgensen, M.; Jensen, T.R. Halogenated Sodium-closo-Dodecaboranes as Solid-State Ion Conductors. Chem. Mater. 2017, 29, 3423–3430. [Google Scholar] [CrossRef]
- Sharma, M.; Sethio, D.; Lawson Daku, L.M.; Hagemann, H. Theoretical Study of Halogenated B12HnX(12–n)2– (X = F, Cl, Br). J. Phys. Chem. A 2019, 123, 1807–1813. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: An on-going challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Aurbach, D.; Cohen, Y.; Moshkovich, M. The Study of Reversible Magnesium Deposition by In Situ Scanning Tunneling Microscopy. Electrochem. Solid-State Lett. 2001, 4, A113–A116. [Google Scholar] [CrossRef]
- Dong, H.; Tutusaus, Y.L.O.; Mohtadi, R.; Zhang, Y.; Hao, F.; Yao, Y. Directing Mg-storage chemistry in organic polymers towards high-energy Mg batteries. Joule 2018, 3, 782–793. [Google Scholar] [CrossRef]
- Mohtadi, R.; Matsui, M.; Arthur, T.S.; Hwang, S.J. Magnesium borohydride: From hydrogen storage to magnesium battery. Angew. Chem. Int. Ed. 2012, 51, 9780–9783. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, T.; Li, G.; Gu, M.; Nie, Z.; Engelhard, M.; Xiao, J.; Lv, D.; Wang, C.; Zhang, J.-G.; et al. Coordination Chemistry in magnesium battery electrolytes: How ligands affect their performance. Sci. Reports 2013, 3, 3130. [Google Scholar] [CrossRef]
- Tuerxun, F.; Abulizi, Y.; NuLi, Y.N.; Su, S.J.; Yang, J.; Wang, J.L. High concentration magnesium borohydride/tetraglyme electrolyte for rechargeable magnesium batteries. J. Power Sources 2015, 276, 255–261. [Google Scholar] [CrossRef]
- Samuel, D.; Steinhauser, C.; Smith, J.G.; Kaufman, A.; Radin, M.D.; Naruse, J.; Hiramatsu, H.; Siegel, D.J. Ion Pairing and Diffusion in Magnesium Electrolytes Based on Magnesium Borohydride. ACS Appl. Mater. Interfaces 2017, 9, 43755–43766. [Google Scholar] [CrossRef] [PubMed]
- Deetz, J.D.; Cao, F.; Wang, Q.; Sun, H. Exploring the Liquid Structure and Ion Formation in Magnesium Borohydride Electrolyte Using Density Functional Theory. J. Electrochem. Soc. 2018, 165, A61–A70. [Google Scholar] [CrossRef]
- Orikasa, Y.; Masese, T.; Koyama, Y.; Mori, T.; Hattori, M.; Yamamoto, K.; Okado, T.; Huang, Z.-D.; Minato, T.; Tassel, C.; et al. High energy density rechargeable magnesium battery using earth-abundant and non-toxic elements. Sci. Rep. 2014, 4, 5622. [Google Scholar] [CrossRef]
- Sa, N.; Rajput, N.N.; Wang, H.; Key, B.; Ferrandon, M.; Srinivasan, V.; Persson, K.A.; Burrell, A.K.; Vaughey, J.T. Concentration dependent electrochemical properties and structural analysis of a simple magnesium electrolyte: Magnesium bis(trifluoromethane sulfonyl)imide in diglyme. RSC Adv. 2016, 6, 113663–113670. [Google Scholar] [CrossRef]
- Rajput, N.N.; Qu, X.; Sa, N.; Burrell, A.K.; Persson, K.A. The Coupling between Stability and Ion Pair Formation in Magnesium Electrolytes from First-Principles Quantum Mechanics and Classical Molecular Dynamics. J. Am. Chem. Soc. 2015, 137, 3411–3420. [Google Scholar] [CrossRef]
- Hu, J.Z.; Rajput, N.N.; Wan, C.; Shao, Y.; Deng, X.; Jaegers, N.R.; Hu, M.; Chen, Y.; Shin, Y.; Monk, J.; et al. 25Mg NMR and computational modeling studies of the solvation structures and molecular dynamics in magnesium based liquid electrolytes. Nano Energy 2018, 46, 436–446. [Google Scholar] [CrossRef]
- Kar, M.; Ma, Z.; Azofra, L.M.; Chen, K.; Forsyth, M.; MacFarlane, D.R. Ionic liquid electrolytes for reversible magnesium electrochemistry. Chem. Commun. 2016, 52, 4033–4036. [Google Scholar] [CrossRef]
- Watkins, T.; Kumar, A.; Buttry, D.A. Designer Ionic Liquids for Reversible Electrochemical Deposition/Dissolution of Magnesium. J. Am. Chem. Soc. 2016, 138, 641–650. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Z.; Li, J.; Qiao, L.; Lu, C.; Tang, K.; Dong, S.; Ma, J.; Liu, Y.; Zhou, X.; et al. Multifunctional Additives Improve the Electrolyte Properties of Magnesium Borohydride Toward Magnesium–Sulfur Batteries. ACS Appl. Mater. Interfaces 2018, 10, 23757–23765. [Google Scholar] [CrossRef]
- Hebié, S.; Ngo, H.P.K.; Leprêtre, J.-C.; Iojoiu, C.; Cointeaux, L.; Berthelot, R.; Alloin, F. Electrolyte Based on Easily Synthesized, Low Cost Triphenolate–Borohydride Salt for High Performance Mg(TFSI)2-Glyme Rechargeable Magnesium Batteries. ACS Appl. Mater. Interfaces 2017, 9, 28377–28385. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, X.; Chen, Y.; Jin, L.; Kuss, C.; Bruce, P.G. Plating and stripping calcium in an organic electrolyte. Nat. Mater. 2017, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.J.; Mohtadi, R.; Arthur, T.S.; Mizuno, F.; Zhang, R.; Shirai, S.; Kampf, J.W. Boron Clusters as Highly Stable Magnesium-Battery Electrolytes. Angew. Chem. Int. Ed. 2014, 53, 3173–3177. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, O.; Mohtadi, R.; Arthur, T.S.; Mizuno, F.; Nelson, E.G.; Sevryugina, Y.V. An Efficient Halogen-Free Electrolyte for Use in Rechargeable Magnesium Batteries. Angew. Chem. Int. Ed. 2015, 54, 7900–7904. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.T.; Seguin, T.J.; Lau, K.-C.; Liao, C.; Ingram, B.J.; Persson, K.A.; Zavadil, K.R. Enhanced Stability of the Carba-closo-dodecaborate Anion for High-Voltage Battery Electrolytes through Rational Design. J. Am. Chem. Soc. 2018, 140, 11076–11084. [Google Scholar] [CrossRef]
- Song, J.; Sahadeo, E.; Noked, M.; Lee, S.B. Mapping the Challenges of Magnesium Battery. J. Phys. Chem. Lett. 2016, 7, 1736–1749. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef]
- Arthur, T.S.; Glans, P.-A.; Singh, N.; Tutusaus, O.; Nie, K.; Liu, Y.-S.; Mizuno, F.; Guo, J.; Alsem, D.H.; Salmon, N.J.; et al. Interfacial Insight from Operando XAS/TEM for Magnesium Metal Deposition with Borohydride Electrolytes. Chem. Mater. 2017, 29, 7183–7188. [Google Scholar] [CrossRef]
- Chang, J.; Haasch, R.T.; Kim, J.; Spila, T.; Braun, P.V.; Gewirth, A.A.; Nuzzo, R.G. Synergetic Role of Li+ during Mg Electrodeposition/Dissolution in Borohydride Diglyme Electrolyte Solution: Voltammetric Stripping Behaviors on a Pt Microelectrode Indicative of Mg–Li Alloying and Facilitated Dissolution. ACS Appl. Mater. Interfaces 2015, 7, 2494–2502. [Google Scholar] [CrossRef]
- Tutusaus, O.; Mohtadi, R.; Singh, N.; Arthur, T.S.; Mizuno, F. Study of Electrochemical Phenomena Observed at the Mg Metal/Electrolyte Interface. ACS Energy Lett. 2017, 2, 224–229. [Google Scholar] [CrossRef]
- Singh, N.; Arthur, T.S.; Tutusaus, O.; Li, J.; Kisslinger, K.; Xin, H.L.; Stach, E.A.; Fan, X.; Mohtadi, R. Achieving High Cycling Rates via In Situ Generation of Active Nanocomposite Metal Anodes. ACS Appl. Energy Mater. 2018, 1, 4651–4661. [Google Scholar] [CrossRef]
- Higashi, S.; Miwa, K.; Aoki, M.; Takechi, K. A novel inorganic solid state ion conductor for rechargeable Mg batteries. Chem. Commun. 2014, 50, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Roedern, E.; Kuhnel, R.S.; Remhof, A.; Battaglia, C. Magnesium Ethylenediamine Borohydride as Solid-State Electrolyte for Magnesium Batteries. Sci. Rep. 2017, 7, 46189. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Rajput, N.N.; Hu, J.Z.; Hu, M.; Liu, T.B.; Wei, Z.H.; Gu, M.; Deng, X.C.; Xu, S.C.; Han, K.S.; et al. Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nano Energy 2015, 12, 750–759. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Forsyth, M.; Howlett, P.C.; Kar, M.; Passerini, S.; Pringle, J.M.; Ohno, H.; Watanabe, M.; Yan, F.; Zheng, W.; et al. Ionic liquids and their solid-state analogues as materials for energy generation and storage. Nat. Rev. Mater. 2016, 1, 15005. [Google Scholar] [CrossRef]
- Ma, Z.; Forsyth, M.; MacFarlane, D.R.; Kar, M. Ionic liquid/tetraglyme hybrid Mg[TFSI]2 electrolytes for rechargeable Mg batteries. Green Energy Environ. 2018, 4, 146–153. [Google Scholar] [CrossRef]
- Kar, M.; Tutusaus, O.; MacFarlane, D.R.; Mohtadi, R. Novel and versatile room temperature ionic liquids for energy storage. Energy Environ. Sci. 2018, 12, 566–571. [Google Scholar] [CrossRef]
- Takahashi, K.; Hattori, K.; Yamazaki, T.; Takada, K.; Matsuo, M.; Orimo, S.; Maekawa, H.; Takamura, H. All-solid-state lithium battery with LiBH4 solid electrolyte. J. Power Sources 2013, 226, 61–64. [Google Scholar] [CrossRef]
- Unemoto, A.; Ikeshoji, T.; Yasaku, S.; Matsuo, M.; Stavila, V.; Udovic, T.J.; Orimo, S.-I. Stable Interface Formation between TiS2 and LiBH4 in Bulk-Type All-Solid-State Lithium Batteries. Chem. Mater. 2015, 27, 5407–5416. [Google Scholar] [CrossRef]
- Unemoto, A.; Nogami, G.; Tazawa, M.; Taniguchi, M.; Orimo, S. Development of 4V-Class Bulk-Type All-Solid-State Lithium Rechargeable Batteries by a Combined Use of Complex Hydride and Sulfide Electrolytes for Room Temperature Operation. Mater. Trans. 2017, 58, 1063–1068. [Google Scholar] [CrossRef]
- Unemoto, A.; Yasaku, S.; Nogami, G.; Tazawa, M.; Taniguchi, M.; Matsuo, M.; Ikeshoji, T.; Orimo, S.-I. Development of bulk-type all-solid-state lithium-sulfur battery using LiBH4 electrolyte. Appl. Phys. Lett. 2014, 105, 083901. [Google Scholar] [CrossRef]
- Das, S.; Ngene, P.; Norby, P.; Vegge, T.; de Jongh, P.E.; Blanchard, D. All-Solid-State Lithium-Sulfur Battery Based on a Nanoconfined LiBH4 Electrolyte. J. Electrochem. Soc. 2016, 163, A2029–A2034. [Google Scholar] [CrossRef]
- Atsushi, U.; ChunLin, C.; Zhongchang, W.; Motoaki, M.; Tamio, I.; Shin-ichi, O. Pseudo-binary electrolyte, LiBH 4 –LiCl, for bulk-type all-solid-state lithium-sulfur battery. Nanotechnology 2015, 26, 254001. [Google Scholar]
- Duchene, L.; Kuhnel, R.S.; Stilp, E.; Cuervo Reyes, E.; Remhof, A.; Hagemann, H.; Battaglia, C. A sTable 3 V all-solid-state sodium-ion battery based on a closo-borate electrolyte. Energy Environ. Sci. 2017, 10, 2609–2615. [Google Scholar] [CrossRef]
- Duchêne, L.; Kim, D.H.; Song, Y.B.; Jun, S.; Moury, R.; Remhof, A.; Hagemann, H.; Jung, Y.S.; Battaglia, C. Crystallization of closo-borate electrolytes from solution enabling infiltration into slurry-casted porous electrodes for all-solid-state batteries. Energy Storage Mater. 2019, 26, 543–549. [Google Scholar] [CrossRef]
- Shao, Y.; Gu, M.; Li, X.; Nie, Z.; Zuo, P.; Li, G.; Liu, T.; Xiao, J.; Cheng, Y.; Wang, C.; et al. Highly Reversible Mg Insertion in Nanostructured Bi for Mg Ion Batteries. Nano Lett. 2014, 14, 255–260. [Google Scholar] [CrossRef]
- Singh, R.; Kumari, P.; Rathore, R.K.; Shinzato, K.; Ichikawa, T.; Verma, A.S.; Saraswat, V.K.; Awasthi, K.; Jain, A.; Kumar, M. LiBH4 as solid electrolyte for Li-ion batteries with Bi2Te3 nanostructured anode. Int. J. Hydrog. Energy 2018, 43, 21709–21714. [Google Scholar] [CrossRef]










© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohtadi, R. Beyond Typical Electrolytes for Energy Dense Batteries. Molecules 2020, 25, 1791. https://doi.org/10.3390/molecules25081791
Mohtadi R. Beyond Typical Electrolytes for Energy Dense Batteries. Molecules. 2020; 25(8):1791. https://doi.org/10.3390/molecules25081791
Chicago/Turabian StyleMohtadi, Rana. 2020. "Beyond Typical Electrolytes for Energy Dense Batteries" Molecules 25, no. 8: 1791. https://doi.org/10.3390/molecules25081791
APA StyleMohtadi, R. (2020). Beyond Typical Electrolytes for Energy Dense Batteries. Molecules, 25(8), 1791. https://doi.org/10.3390/molecules25081791