
Enhancing the Membranolytic Activity of Chenopodium quinoa Saponins by Fast Microwave Hydrolysis

 ,
,  , ,
, ,  , and
, and 
Abstract

1. Introduction
2. Results and Discussion
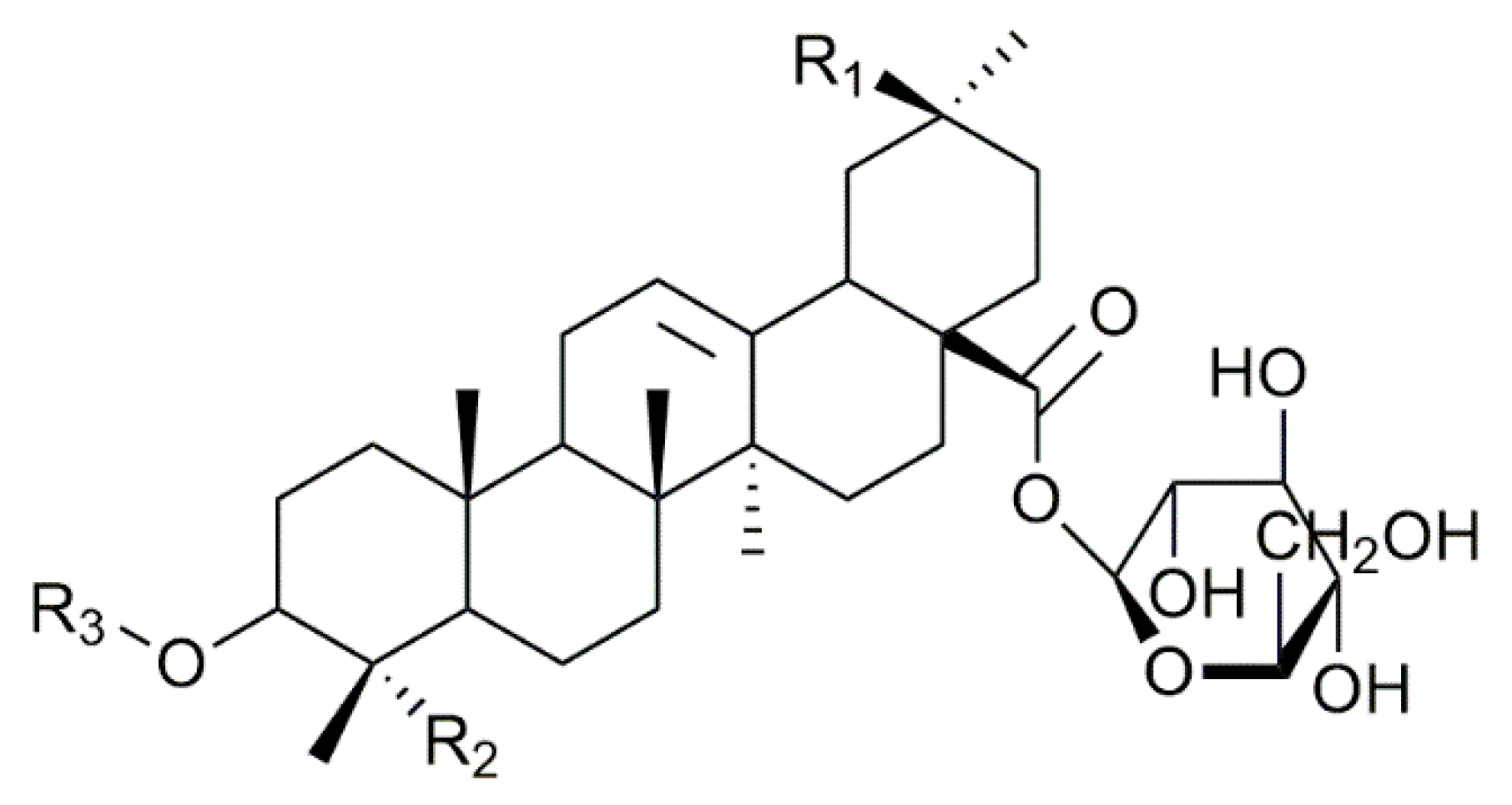
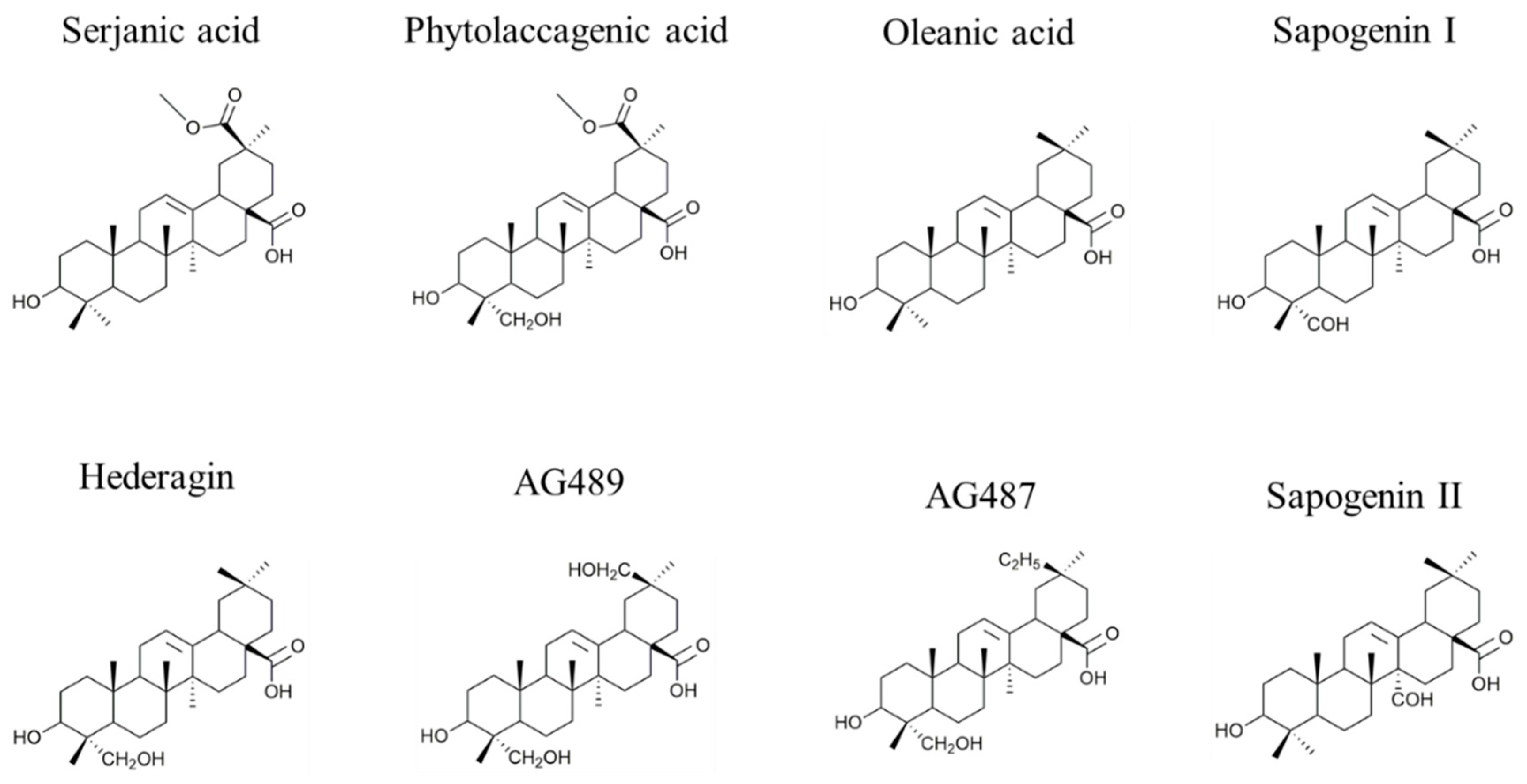
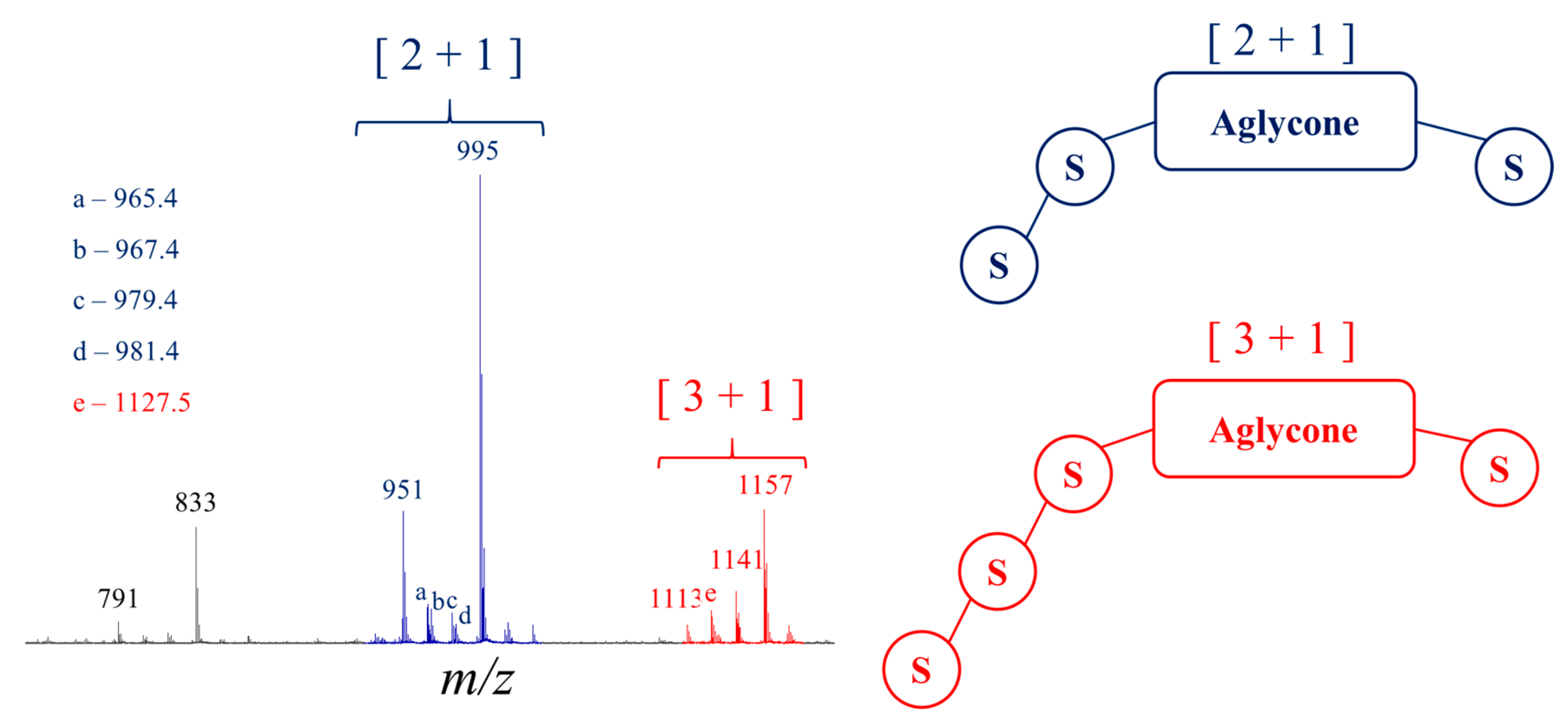
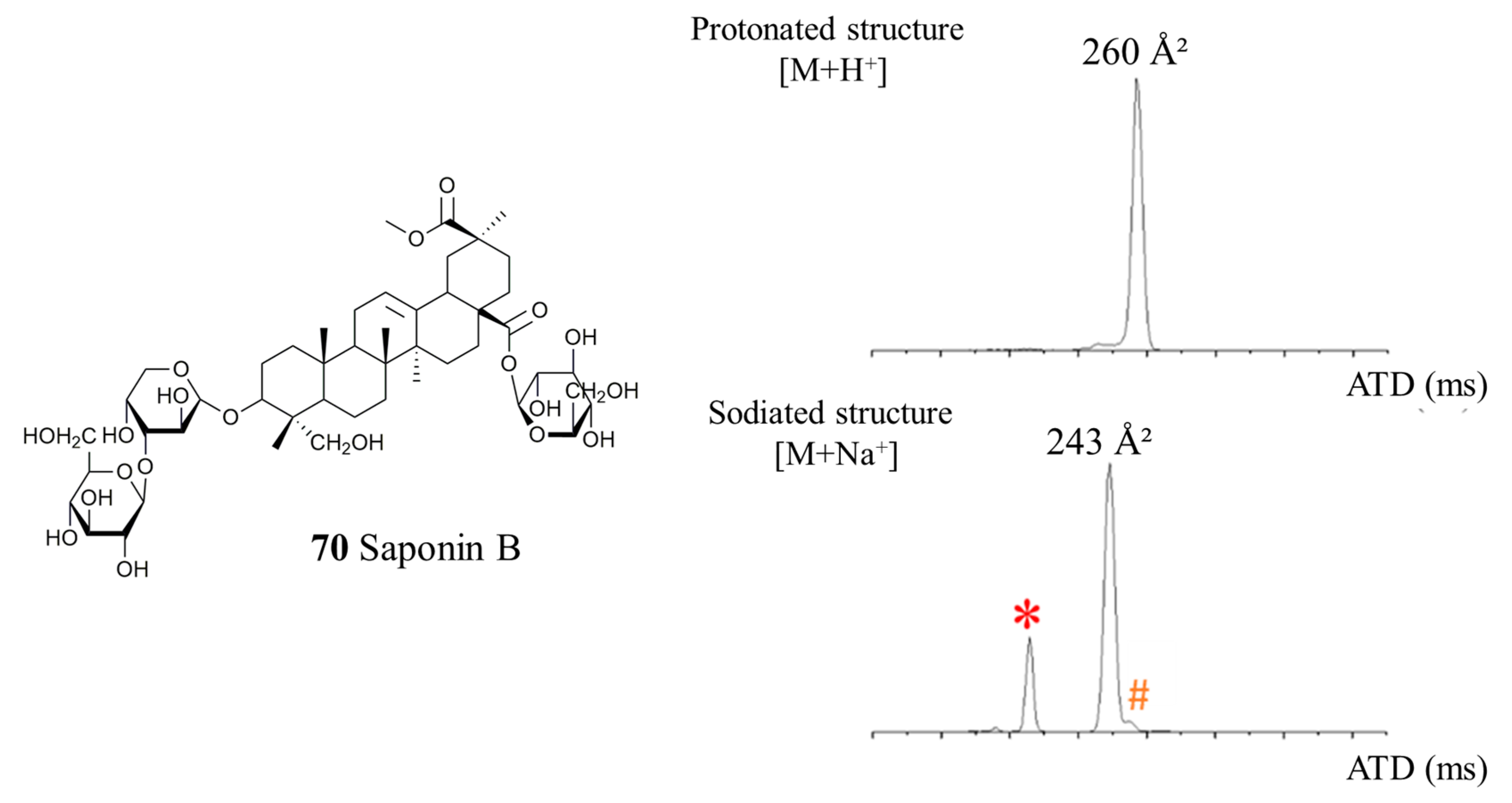
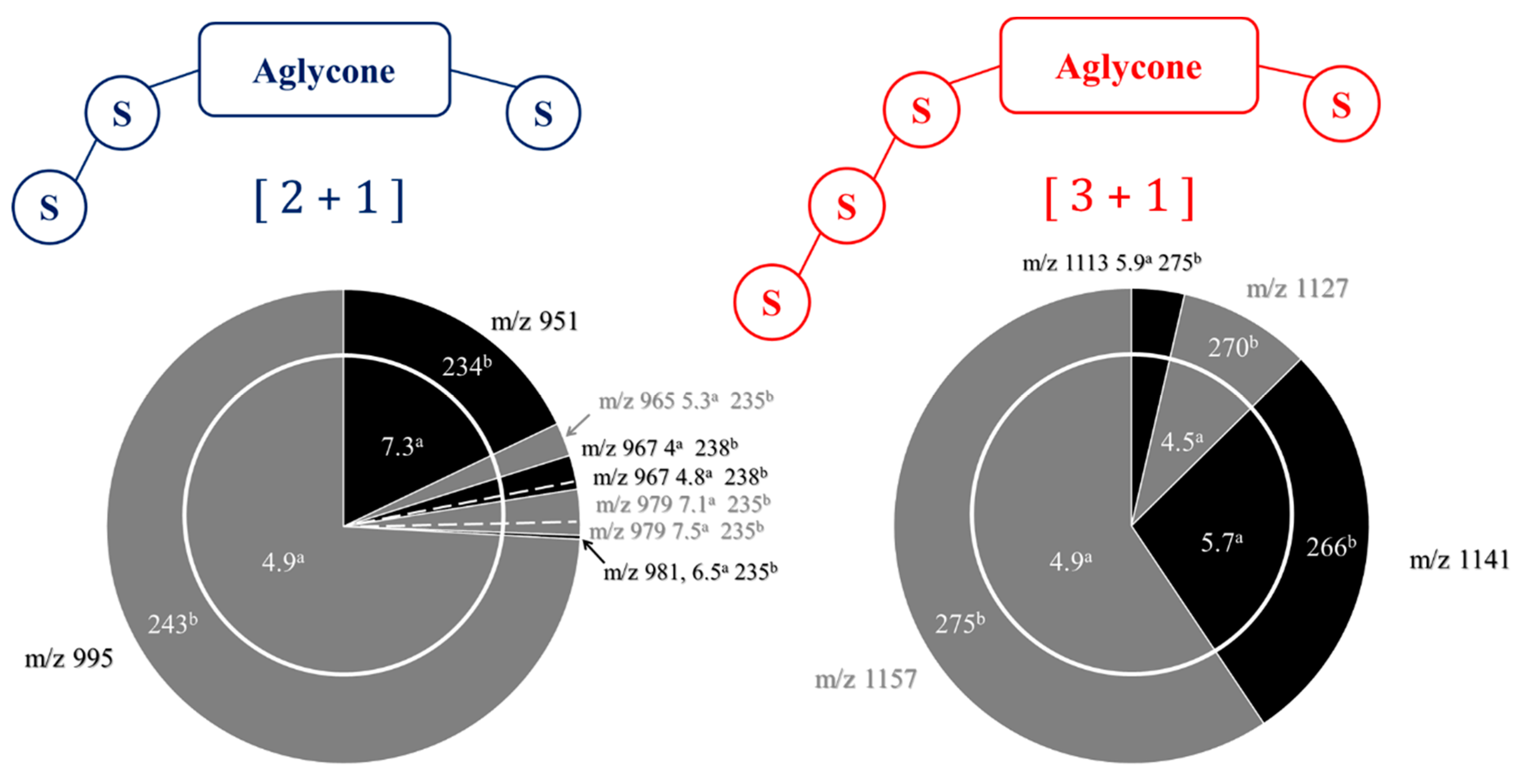
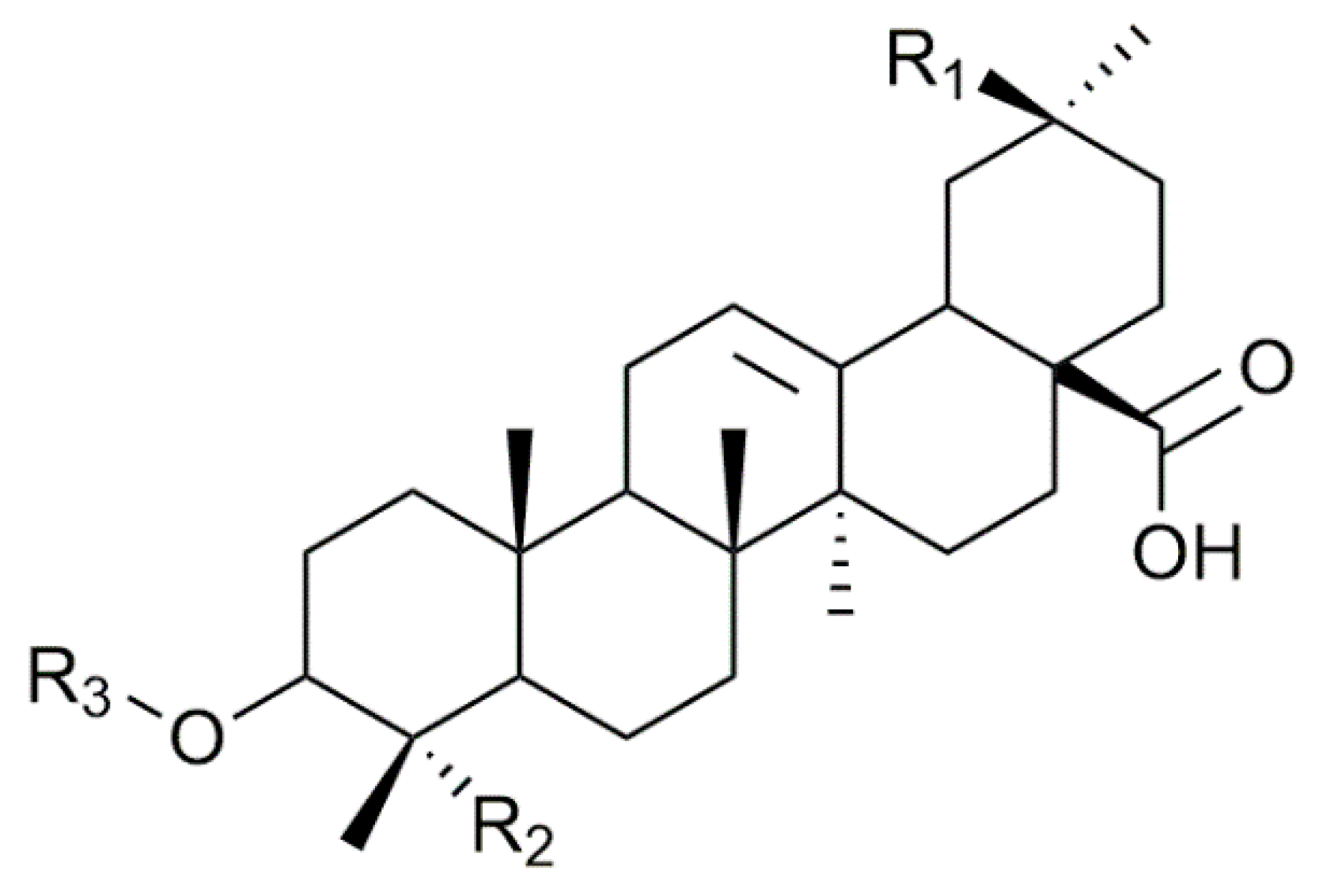
2.1. Saponin Identification and Quantification in the Quinoa Extract (QE)
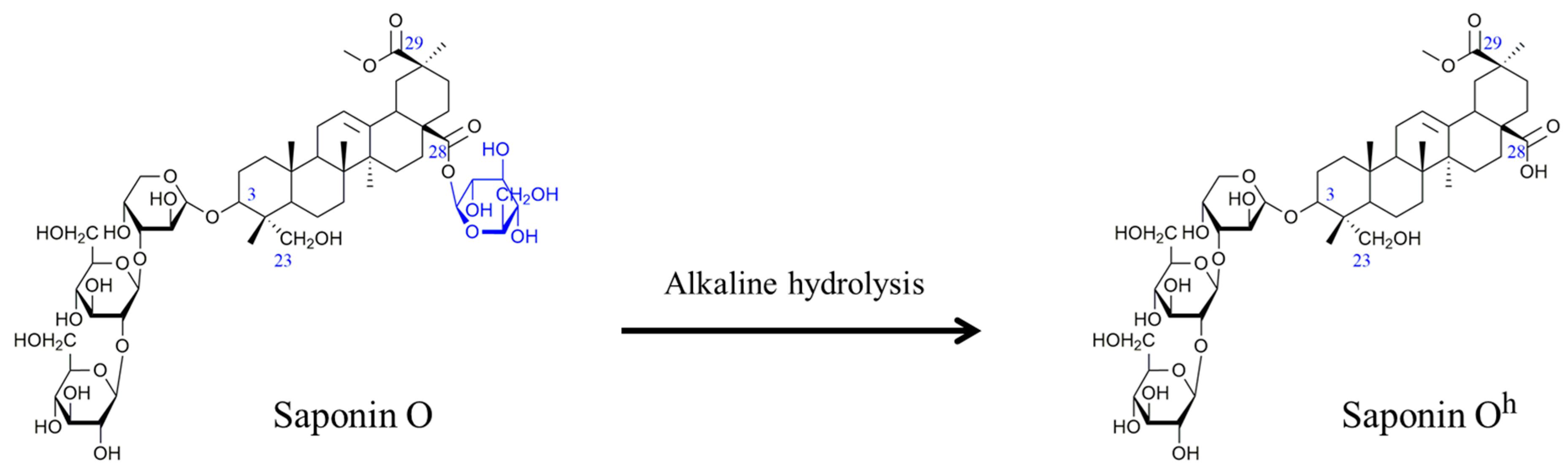
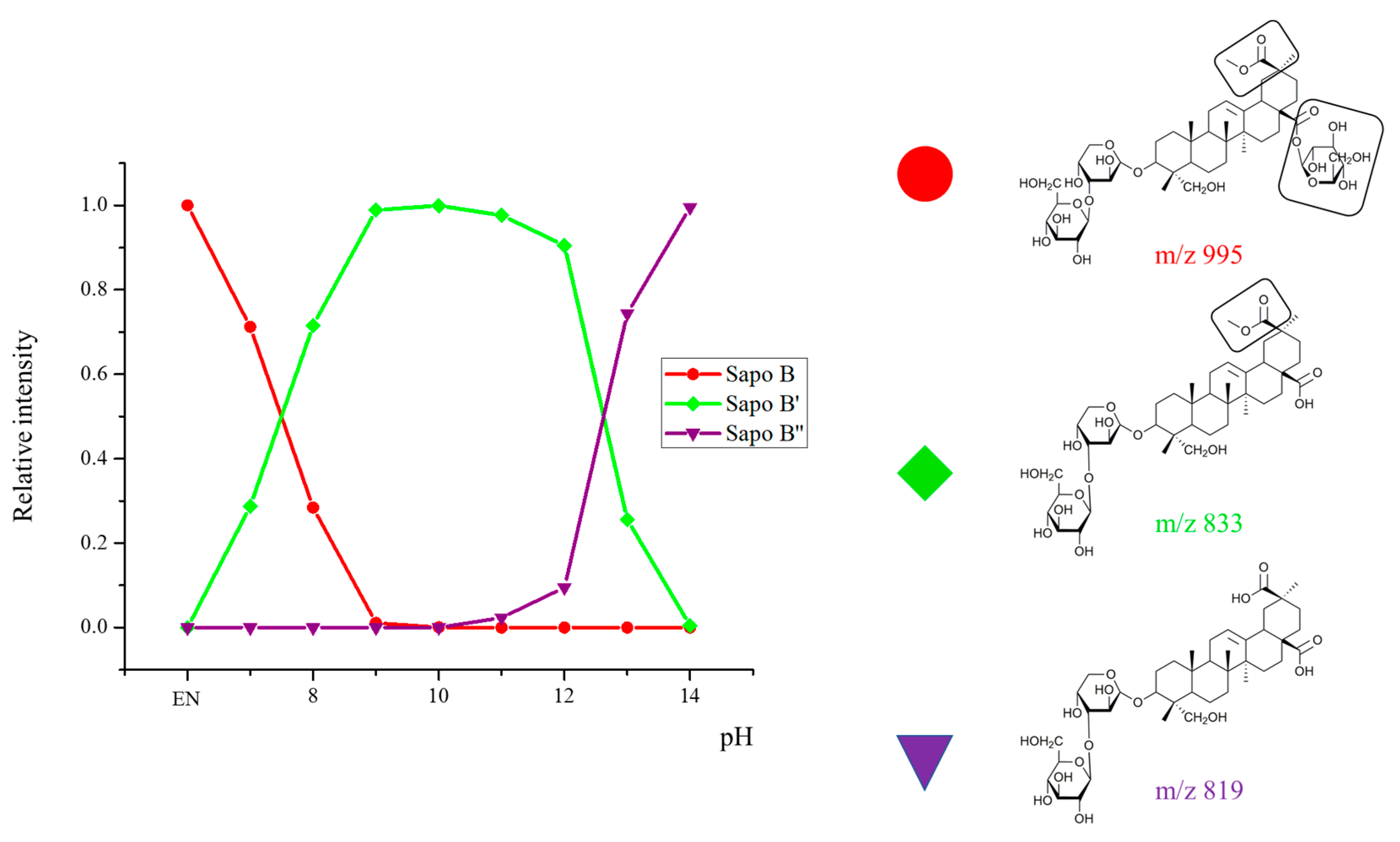
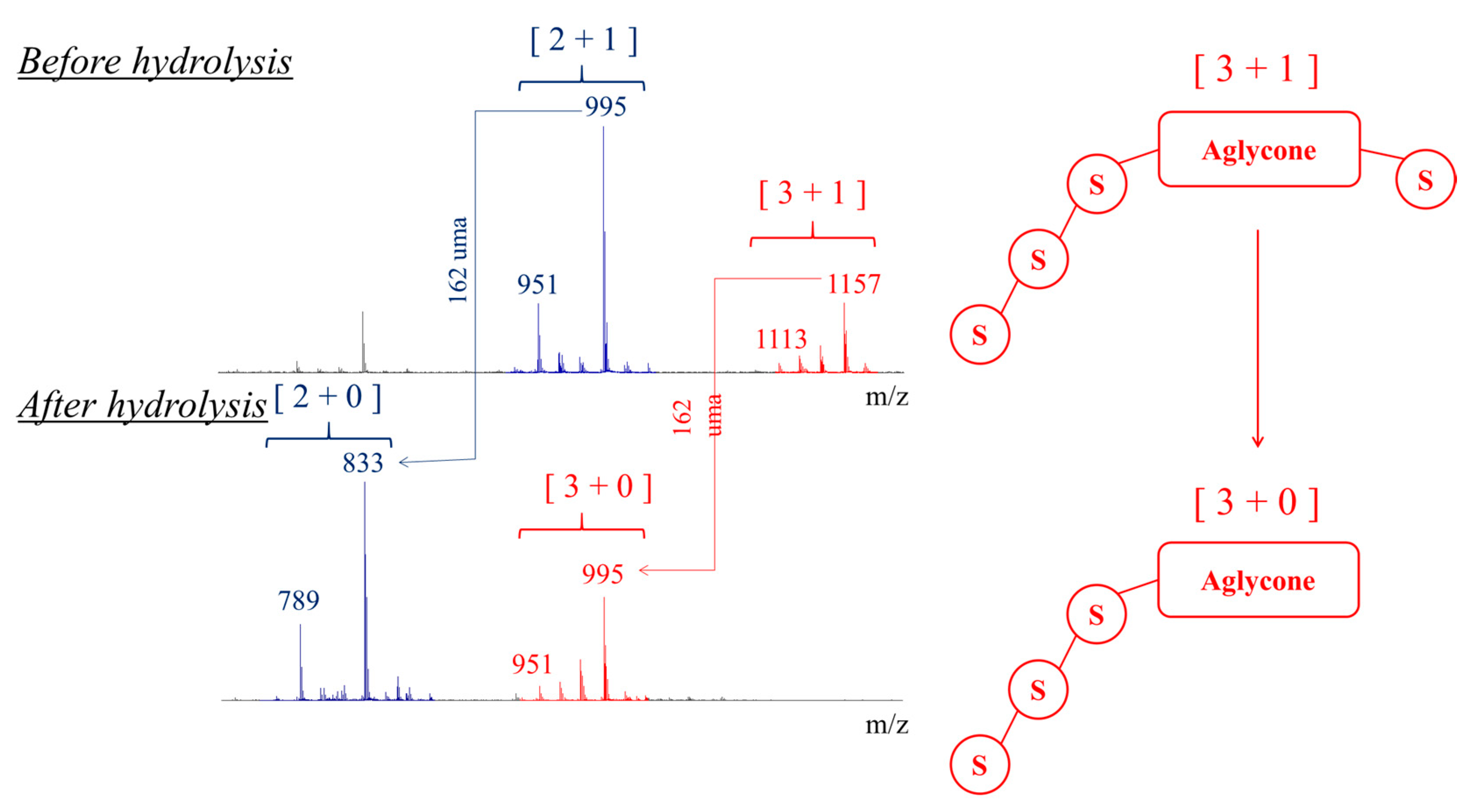
2.2. Selectivity of the Microwave-Assisted Hydrolysis of Saponins
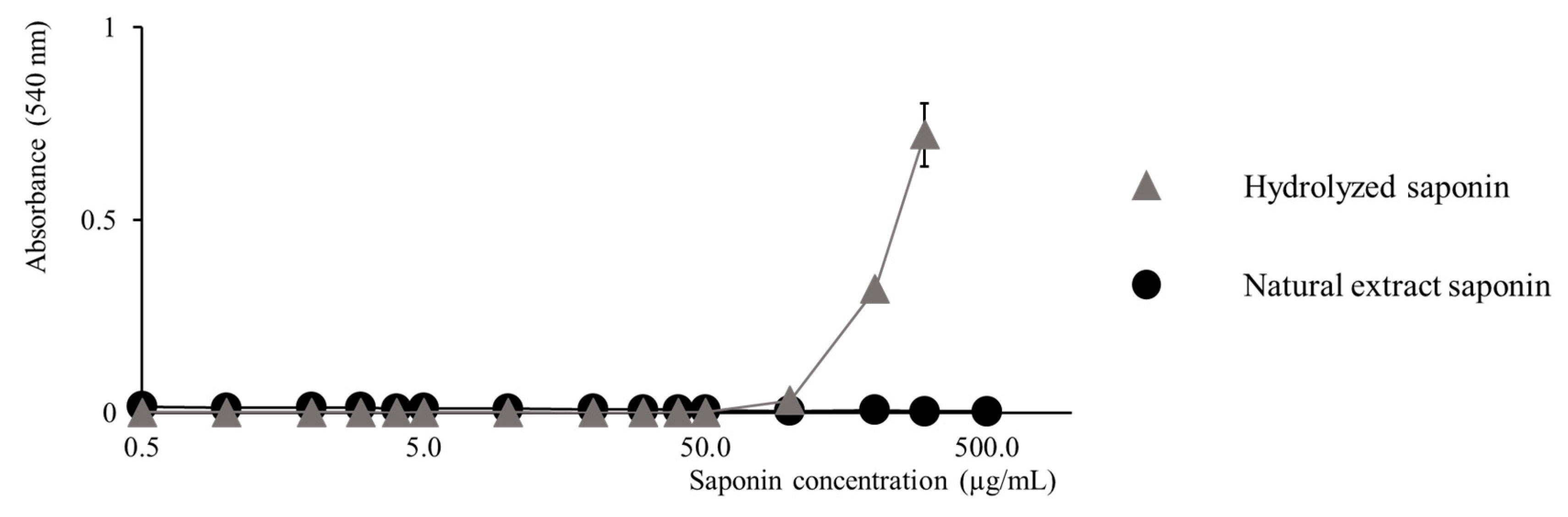
2.3. Activity Modulation–Hemolytic Activity Assay
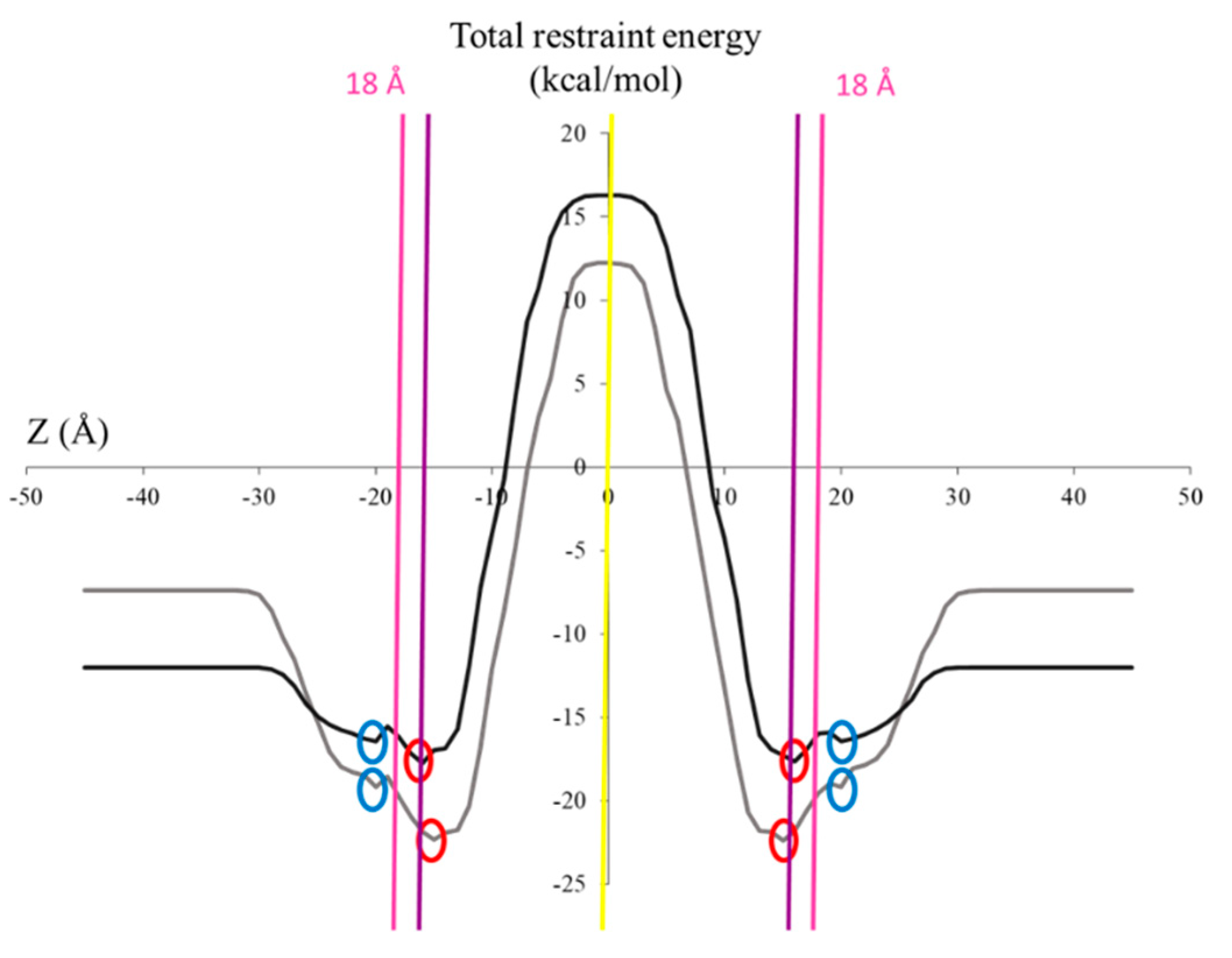
2.4. Activity Modulation–In Silico Evaluation of the Mono- vs. Bidesmosidic Saponin Activities
3. Conclusions
4. Materials and Methods
Chemicals, Plant Sampling and Saponin Extractions
Chemicals
Saponin Extraction from Quinoa Husks
Microwave Hydrolysis
Mass Spectrometry Analysis
LC-MS Analyses
Semi-Quantitative Analysis
Hemolytic Activity
In silico Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaskank, K.; Abbay, K.P. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World. J. 2013, 2013, 1–16. [Google Scholar]
- Arpita, R. A Review on the Alkaloids an Important Therapeutic Compound from Plants. Int. J. Plant Biotechnol. 2017, 3, 1–9. [Google Scholar]
- Podolak, I.; Galanty, A.; Sobolewska, D. Saponins as cytotoxic agents: A review. Phytochem. Rev. 2010, 9, 425–474. [Google Scholar] [CrossRef]
- Segal, R.; Schlösser, E. Role of glycosidases in the membranolytic, antifungal action of saponins. Arch. Microbiol. 1974, 104, 147–150. [Google Scholar] [CrossRef]
- Lorent, J.H.; Quetin-Leclercq, J.; Mingeot-Leclercq, M.P. The amphiphilic nature of saponins and their effects on artificial and biological membranes and potential consequences for red blood and cancer cells. Org. Biomol. Chem. 2014, 12, 8803–8822. [Google Scholar] [CrossRef]
- Madl, T.; Sterk, H.; Mittelbach, M.; Rechberger, G.N. Tandem Mass Spectrometric Analysis of a Complex Triterpene Saponin Mixture of Chenopodium quinoa. J. Am. Soc. Mass Spectrom. 2006, 17, 795–806. [Google Scholar] [PubMed]
- Bahrami, Y.; Zhang, W.; M. M. Franco, C. Distribution of Saponins in the Sea Cucumber Holothuria lessoni; the Body Wall versus the Viscera, and Their Biological Activities. Mar. Drugs 2018, 16, 423. [Google Scholar] [CrossRef] [PubMed]
- Caulier, G.; Mezali, K.; Soualili, D.L.; Decroo, C.; Demeyer, M.; Eeckhaut, I.; Gerbaux, P.; Flammang, P. Chemical characterization of saponins contained in the body wall and the Cuvierian tubules of the sea cucumber Holothuria (Platyperona) sanctori (Delle Chiaje, 1823). Biochem. Syst. Ecol. 2016, 68, 119–127. [Google Scholar] [CrossRef]
- Demeyer, M.; De Winter, J.; Caulier, G.; Eeckhaut, I.; Flammang, P.; Gerbaux, P. Molecular diversity and body distribution of saponins in the sea star Asterias rubens by mass spectrometry. Comp. Biochem. Physiol. B 2014, 168, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, T. On the poisonous substance contained in holothurians. Seto Mar. Biol. Lab. 1995, 4, 183–203. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Minale, L.; Riccio, R. Polyoxygenated Steroids of Marine. Orig. Chem. Rev. 1993, 93, 1839–1895. [Google Scholar] [CrossRef]
- Mostafa, A.; Sudisha, J.; El-Sayed, M.; Ito, S.; Ikeda, T.; Yamauchi, N.; Shigyo, M. Aginoside saponin, a potent antifungal compound, and secondary metabolite analyses from Allium nigrum L. Phytochem. Lett. 2013, 6, 274–280. [Google Scholar] [CrossRef]
- Keukens, E.A.J.; de Vije, T.; Vanden Boom, C.; de Waard, P.; Plasman, H.H.; Thiel, F.; Chupin, V.; Jongen, W.M.F.; de Kruijff, B. Molecular basis of glycoalkaloid induced membrane disruption. Biochim. Biophys. Acta 1995, 1240, 216. [Google Scholar] [CrossRef]
- Lorent, J.H.; Le Duff, C.; Quetin-Leclercq, J.; Mingeot-Leclercq, M.P. Induction of highly curved structures in relation to membrane permeabilization and budding by the triterpenoid saponins, α- and δ-Hederin. J. Biol. Chem. 2013, 288, 14000–14017. [Google Scholar]
- Böttger, S.; Melzig, M.F. The influence of saponins on cell membrane cholesterol. Bioorg. Med. Chem. 2013, 21, 7118–7124. [Google Scholar] [CrossRef]
- Lorent, J.H.; Lins, L.; Domenech, O.; Quentin-Leclercq, J.; Brasseur, R.; Mingeot-Leclercq, M.P. Domain formation and permeabilization induced by the saponin α-Hederin and its aglycone hederagenin in a cholesterol-containing bilayer. Langmuir 2014, 16, 4556–4569. [Google Scholar] [CrossRef]
- Armah, C.N.; Mackie, A.R.; Roy, C.; Price, K.; Osbourn, A.E.; Bowyer, P.; Ladha, S. The Membrane-Permeabilizing Effect of Avenacin A-1 Involves the Reorganization of Bilayer Cholesterol. Biophys. J. 1999, 76, 281–290. [Google Scholar] [CrossRef]
- Claereboudt, E.J.S.; Eeckhaut, I.; Lins, L.; Deleu, M. How different sterols contribute to saponin tolerant plasma membranes in sea cucumbers. Sci. Rep. 2018, 8, 1–11. [Google Scholar]
- Yoshikawa, M.; Harada, E.; Murakami, T.; Matsuda, T.; Wariishi, N.; Yamahara, T.; Murakami, N.; Kitagawa, I. Escins Ia, Ib, IIa, IIb, and IIIa, Bioactive triterpene oligoglycosides from the seeds of Aesculus hippocastanum L.: Their inhibitory effects on ethanol absorption and hypoglycemic activity on glucose tolerance test. Chem. Pharm. Bull. 1994, 42, 1357–1359. [Google Scholar]
- Van Dyck, S.; Gerbaux, P.; Flammang, P. Qualitative and quantitative saponin contents in five sea cucumbers from the Indian ocean. Mar. Drugs 2010, 8, 173–189. [Google Scholar] [CrossRef]
- Giles, K.; Williams, J.P.; Campuzano, I. Enhancements in travelling wave ion mobility resolution. Rapid Commun. Mass Spectrom. 2011, 25, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Troc, A.; Zimnicka, M.; Danikiewicz, W. Separation of catechin epimers by complexation using ion mobility mass spectrometry. J. Mass Spectrom. 2015, 50, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Decroo, C.; Colson, E.; Demeyer, M.; Lemaur, V.; Caulier, G.; Eeckhaut, I.; Gerbaux, P. Tackling saponin diversity in marine animals by mass spectrometry: Data acquisition and integration. Anal. Bioanal. Chem. 2017, 409, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Decroo, C.; Colson, E.; Lemaur, V.; Caulier, G.; De Winter, J.; Cabrera-Barjas, G.; Gerbaux, P. Ion mobility mass spectrometry of saponin ions. Rapid Commun. Mass Spectrom. 2018, 33, 1–12. [Google Scholar]
- Colson, E.; Decroo, C.; Cooper-Sheperd, D.; Caulier, G.; Henoumont, C.; Laurent, S.; Flammang, P.; Palmer, M.; Claereboudt, J.; Gerbaux, P. Discrimination of regioisomeric and stereoisomeric saponins from Aesculus hippocastanum seeds by ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2228–2237. [Google Scholar] [CrossRef]
- Sun, X.; Yang, X.; Xue, P.; Zhang, Z.; Ren, G. Improved antibacterial effects of alkali-transformed saponin from quinoa husks against halitosis-related bacteria. BMC Complement. Altern. Med. 2019, 19, 46. [Google Scholar] [CrossRef]
- Kuljanabhagavad, T.; Thongphasuk, P.; Chamulitrat, W.; Wink, M. Triterpene saponins from Chenopodium quinoa Wild. Phytochemistry 2008, 69, 1919–1926. [Google Scholar] [CrossRef]
- Castillo-Ruiz, M.; Canon-Jones, H.; Schlotterbeck, T.; Lopez, M.A.; Tomas, A.; San Martin, R. Safety and efficacy of quinoa (Chenopodium quinoa) saponins derived molluscicide to control of Pomacea maculata in rice fields in the Ebro Delta, Spain. J. Crops Prot. 2018, 111, 42–49. [Google Scholar] [CrossRef]
- Estrada, A.; Katselis, G.; Laarved, B.; Barl, B. Isolation and evaluation of immunological adjuvant activities of saponins from Polygala senega L. Comp. Immunol. Microbiol. Infect. Dis. 2000, 23, 27–40. [Google Scholar] [CrossRef]
- Stuardo, M.; San Martin, R. Antifungal properties of quinoa (Chenopodium quinoa Willd) alkali treated saponins against Botrytis cinereal. Ind. Crops Prod. 2008, 27, 296–302. [Google Scholar] [CrossRef]
- Duez, Q.; Chirot, F.; Liénard, R.; Josse, T.; Choi, C.M.; Coulembier, O.; Dugourd, P.; Cornil, J.; Gerbaux, P.; De Winter, J. Polymers for Traveling Wave Ion Mobility Spectrometry Calibration. J. Am. Soc. Mass Spectrom. 2017, 28, 2483–2491. [Google Scholar] [CrossRef]
- Martens, J.; Koppen, V.; Berden, G.; Cuyckens, F. Oomens, Combined Liquid Chromatography-Infrared Ion Spectroscopy for Identification of Regioisomeric Drug Metabolites. J. Anal. Chem. 2017, 89, 4359–4362. [Google Scholar] [CrossRef]
- Gabelica, V.; Shvartsburg, A.A.; Alfonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, J.; Li, L.; Xu, H.; Xing, Y.; Zhao, Y.; Yu, Z. Pharmacokinetic parameters of three active ingredients hederacoside C, hederacoside D, and ɑ-hederin in Hedera helix in rats. J. Sep. Sci. 2016, 39, 3292–3301. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, Q.; Lai, D.; Sun, W.; Zhang, T.; Ito, Y. Separation and purification of triterpene saponins from roots of Radix phytolaccae by high-speed countercurrent chromatography coupled with evaporative light scattering detection. J. Liq. Chromatogr. Relat. Technol. 2010, 33, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Woldemichael, G.M.; Wink, M. Identification and Biological Activities of Triterpenoid Saponins from Chenopodium quinoa. J. Agric. Food Chem. 2001, 49, 2327–2332. [Google Scholar] [CrossRef]
- Ducarme, P.; Rahman, M.; Brasseur, R. IMPALA: A simple restraint field to simulate the biological membrane in molecular structure studies. Proteins 1998, 30, 357–371. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | m/z [M + Na]+ | Δ(m/z) (ppm) | R1 | R2 | Aglycone | R3 | RT (min) | CCS (Ų) [M + Na]+ | CCS (Ų) [M + H]+ | Molar Proportion(%) | Mass Fraction (mg·g−1) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | C47H76O18 | 951.4929 | 2.1 | - CH3 | - CH2OH | Hed | Glc – Ara - | 7.3 | 234 | 254 | 14.9 | 6.1 |
| Unknown-1 | C47H74O19 | 965.4722 | 0.2 | ? | ? | ? | Glc – Ara - | 5.3 | 235 | 254 | 1.9 | 0.8 |
| 19 | C47H76O19 | 967.4878 | 4.9 | - CH2OH | - CH2OH | AG489 | Xyl - Glc - | 4.0 | 238 | 254 | 1.6 | 0.7 |
| 19a | - CH2OH | - CH2OH | AG489 | Xyl - Glc - | 4.8 | 238 | 256 | 0.3 | 0.1 | |||
| H | C48H76O19 | 979.4878 | 1.5 | - COOCH3 | - CH3 | SA | Glc – Ara - | 7.1 | 235 | NA | 1.6 | 0.7 |
| 70 | - CH3 | - CH3 | OA | Glc – GlcA - | 7.5 | 235 | NA | 0.9 | 0.4 | |||
| Q | C48H78O19 | 981,5035 | 0.8 | - CH3 | - CH2OH | Hed | Glc – Gal - | 6.5 | 235 | 260 | 0.2 | 0.1 |
| B | C48H76O20 | 995.4828 | 3.2 | - COOCH3 | - CH2OH | PA | Glc – Ara - | 4.9 | 243 | 260 | 61.7 | 25.3 |
| 61 | C53H86O23 | 1113.5458 | 1.6 | - CH3 | - CH2OH | Hed | Glc – Glc – Ara - | 5.9 | 275 | 290 | 0.7 | 0.3 |
| Unknown-2 | C53H86O24 | 1127.5251 | 2.1 | ? | ? | ? | Glc – Glc – Ara - | 4.5 | 270 | 290 | 1.7 | 0.7 |
| G | C54H86O24 | 1141.5407 | 3.6 | - COOCH3 | - CH3 | SA | Glc – Glc – Ara - | 5.7 | 266 | 291 | 3.0 | 4.6 |
| Composition | m/z [M + Na]+ | Δ(m/z) (ppm) | R1 | R2 | Aglycone | R3 | RT (min) | CCS (Ų) [M + Na]+ | CCS (Ų) [M + H]+ | Molar Proportion (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ih | C41H66O13 | 789.4401 | 0.8 | - CH3 | - CH2OH | Hed | Glc – Ara - | 10.7 | 224 | 219 | 16.5 |
| Unknown-1h | C41H64O14 | 803.4194 | 0.4 | ? | ? | ? | Glc – Ara - | 7.2 | 225 | 221 | 2.4 |
| 19h | C41H66O14 | 805.4350 | 0.7 | - CH2OH | - CH2OH | AG489 | Xyl - Glc - | 5.6 | 227 | 221 | 2.2 |
| 19ah | - CH2OH | - CH2OH | AG489 | Xyl - Glc - | 5.8 | NA | NA | 0.1 | |||
| Hh | C42H66O14 | 817.4350 | 2.1 | - COOCH3 | - CH3 | SA | Glc – Ara - | 9.4 | 240 | NA | 2.2 |
| 70h | - CH3 | - CH3 | OA | Glc – GlcA - | 13.1 | 235 | NA | 1.0 | |||
| Qh | C42H68O14 | 819.4501 | 0.6 | - CH3 | - CH2OH | Hed | Glc – Gal - | 9.4 | 228 | 222 | 0.1 |
| Bh | C42H66O15 | 833.4299 | 1.1 | - COOCH3 | - CH2OH | PA | Glc – Ara - | 9.9 | 234 | 227 | 61.4 |
| 61h | C47H76O18 | 951.4929 | 1.3 | - CH3 | - CH2OH | Hed | Glc – Glc – Ara - | 10.2 | 250 | 253 | 0.4 |
| Unknown-2h | C47H74O19 | 965.4722 | 2.5 | ? | ? | ? | Glc – Glc – Ara - | 7.1 | 252 | 254 | 0.9 |
| Gh | C48H76O19 | 979.4879 | 0.5 | - COOCH3 | - CH3 | SA | Glc – Glc – Ara - | 9.4 | 265 | NA | 2.7 |
| Oh | C48H76O20 | 995.4828 | 0.4 | - COOCH3 | - CH2OH | PA | Glc – Glc – Ara - | 7.6 | 258 | 263 | 10.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colson, E.; Savarino, P.; J.S. Claereboudt, E.; Cabrera-Barjas, G.; Deleu, M.; Lins, L.; Eeckhaut, I.; Flammang, P.; Gerbaux, P. Enhancing the Membranolytic Activity of Chenopodium quinoa Saponins by Fast Microwave Hydrolysis. Molecules 2020, 25, 1731. https://doi.org/10.3390/molecules25071731
Colson E, Savarino P, J.S. Claereboudt E, Cabrera-Barjas G, Deleu M, Lins L, Eeckhaut I, Flammang P, Gerbaux P. Enhancing the Membranolytic Activity of Chenopodium quinoa Saponins by Fast Microwave Hydrolysis. Molecules. 2020; 25(7):1731. https://doi.org/10.3390/molecules25071731
Chicago/Turabian StyleColson, Emmanuel, Philippe Savarino, Emily J.S. Claereboudt, Gustavo Cabrera-Barjas, Magali Deleu, Laurence Lins, Igor Eeckhaut, Patrick Flammang, and Pascal Gerbaux. 2020. "Enhancing the Membranolytic Activity of Chenopodium quinoa Saponins by Fast Microwave Hydrolysis" Molecules 25, no. 7: 1731. https://doi.org/10.3390/molecules25071731
APA StyleColson, E., Savarino, P., J.S. Claereboudt, E., Cabrera-Barjas, G., Deleu, M., Lins, L., Eeckhaut, I., Flammang, P., & Gerbaux, P. (2020). Enhancing the Membranolytic Activity of Chenopodium quinoa Saponins by Fast Microwave Hydrolysis. Molecules, 25(7), 1731. https://doi.org/10.3390/molecules25071731