
Mimicking of Phase I Metabolism Reactions of Molindone by HLM and Photocatalytic Methods with the Use of UHPLC-MS/MS



Abstract
1. Introduction
2. Results and Discussion
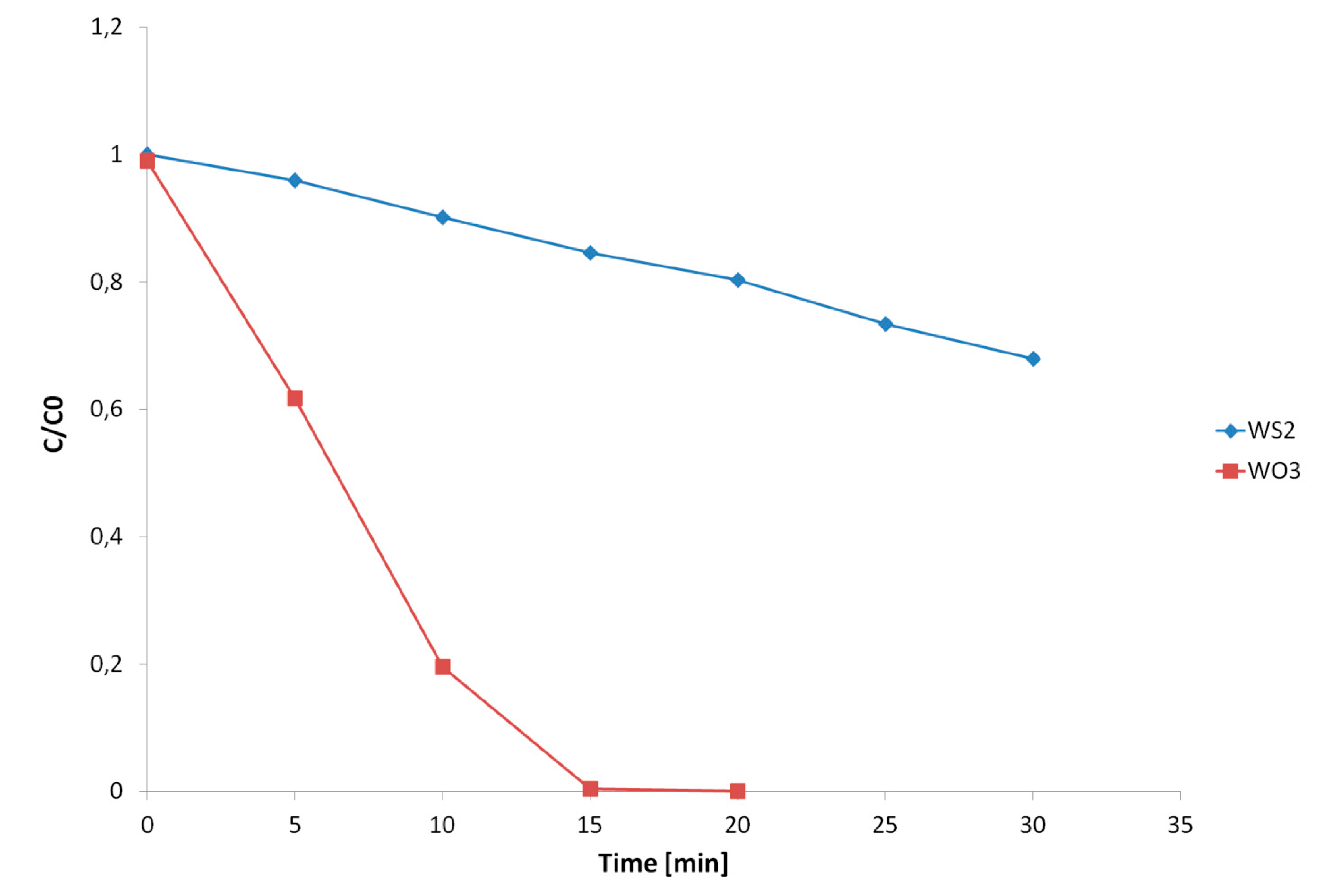
2.1. Photocatalytic Degradation Kinetics
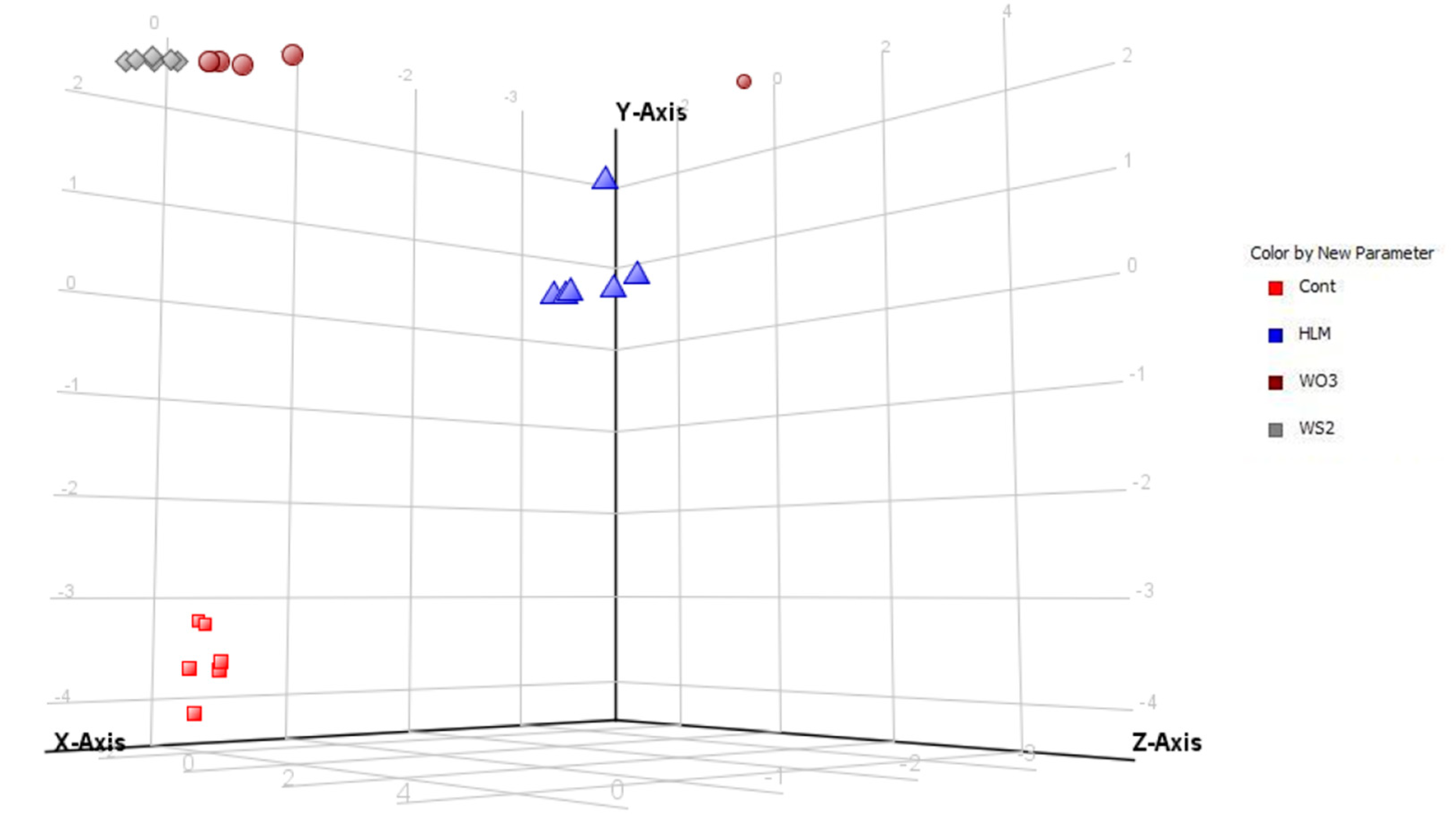
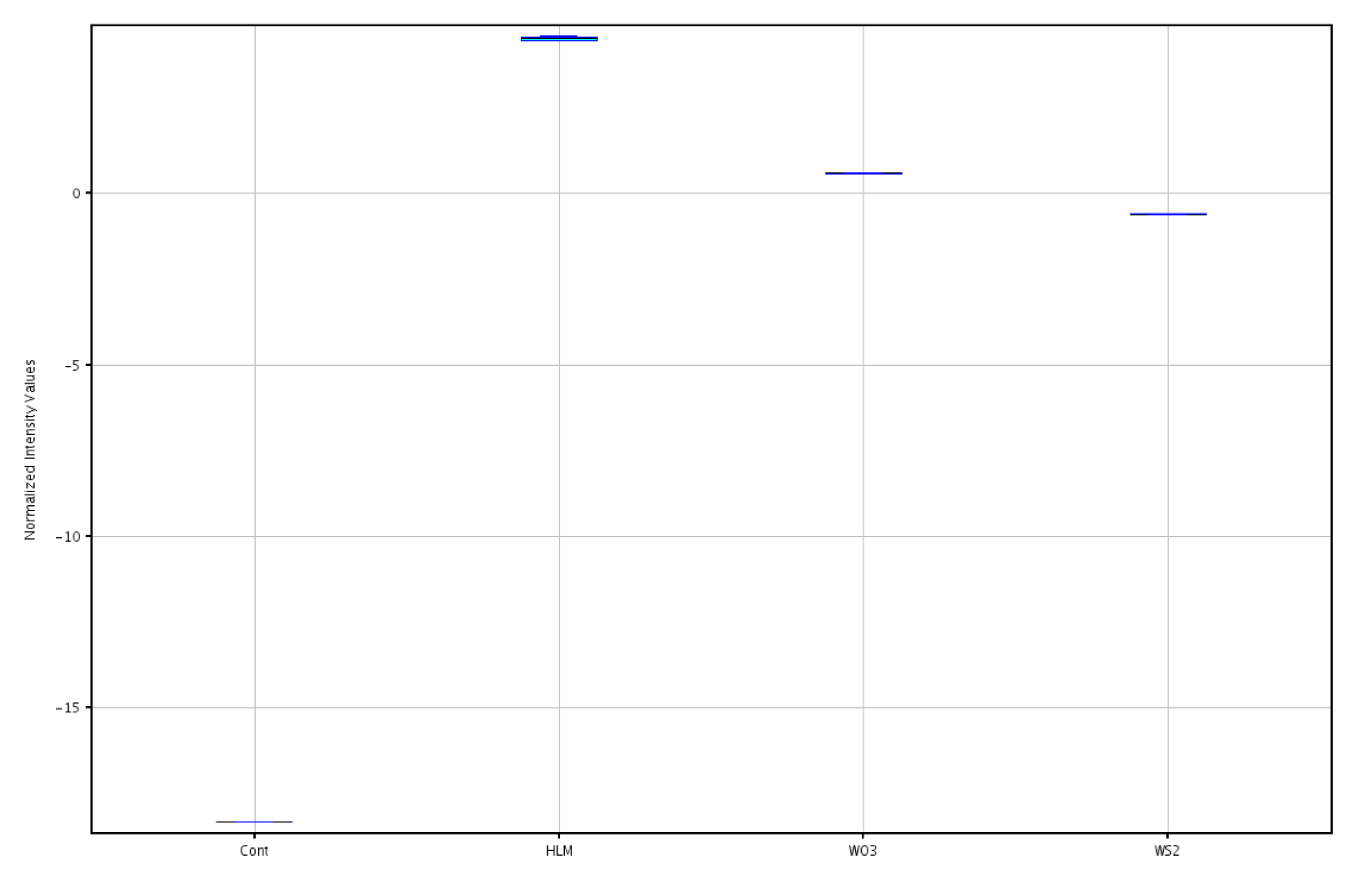
2.2. Multivariate Comparison of Human Liver Microsome (HLM) Metabolites and Photocatalytic Products
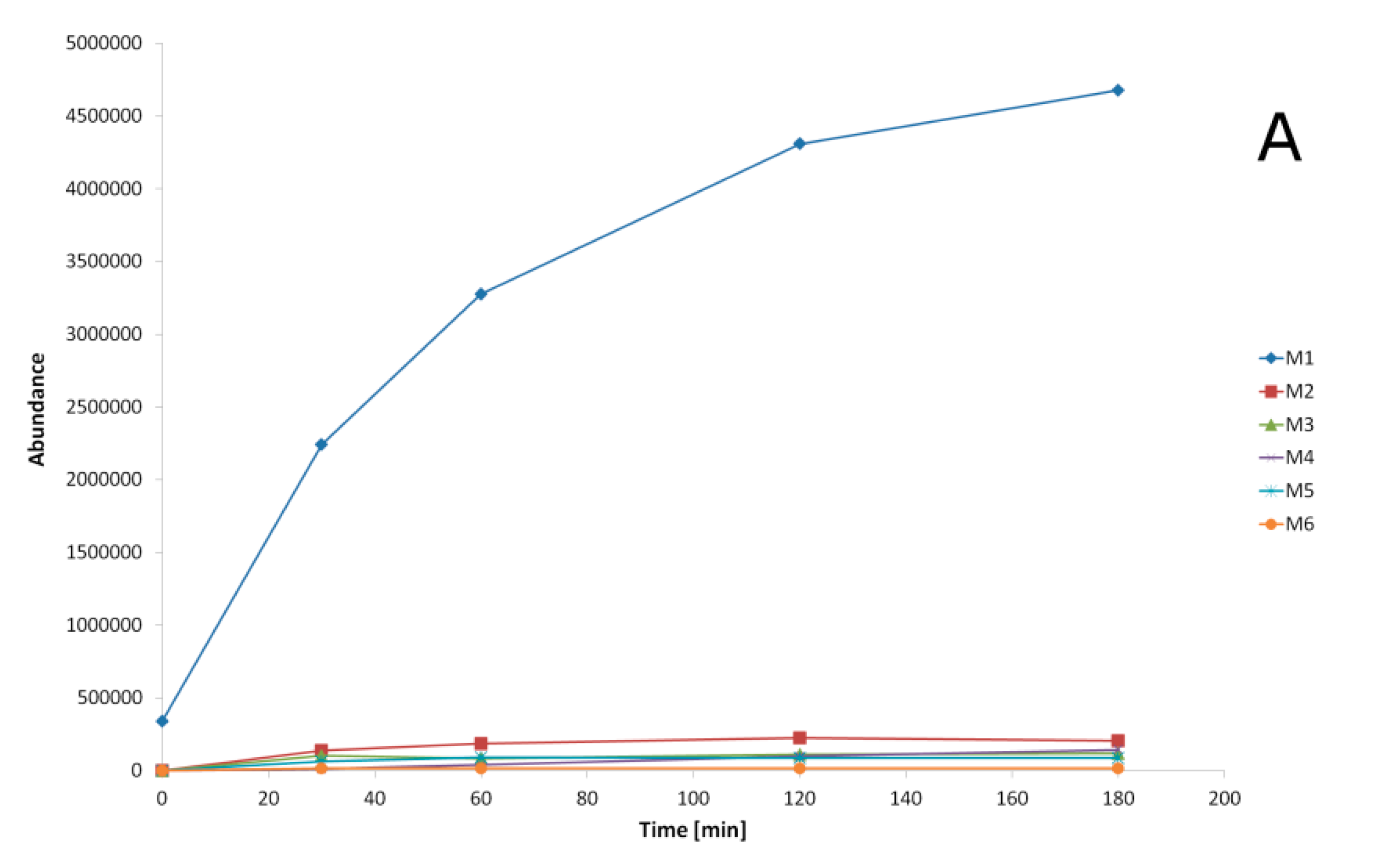
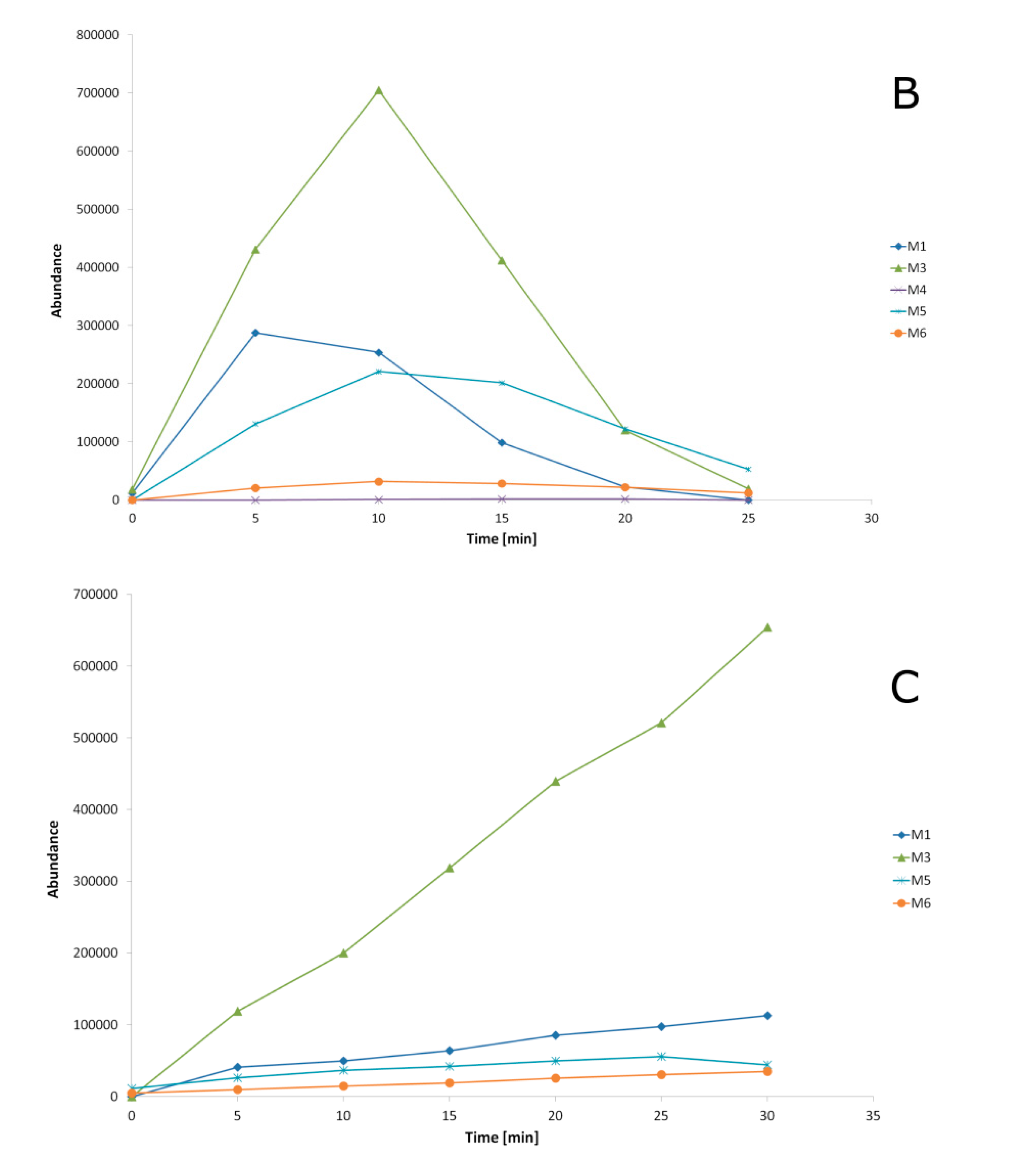
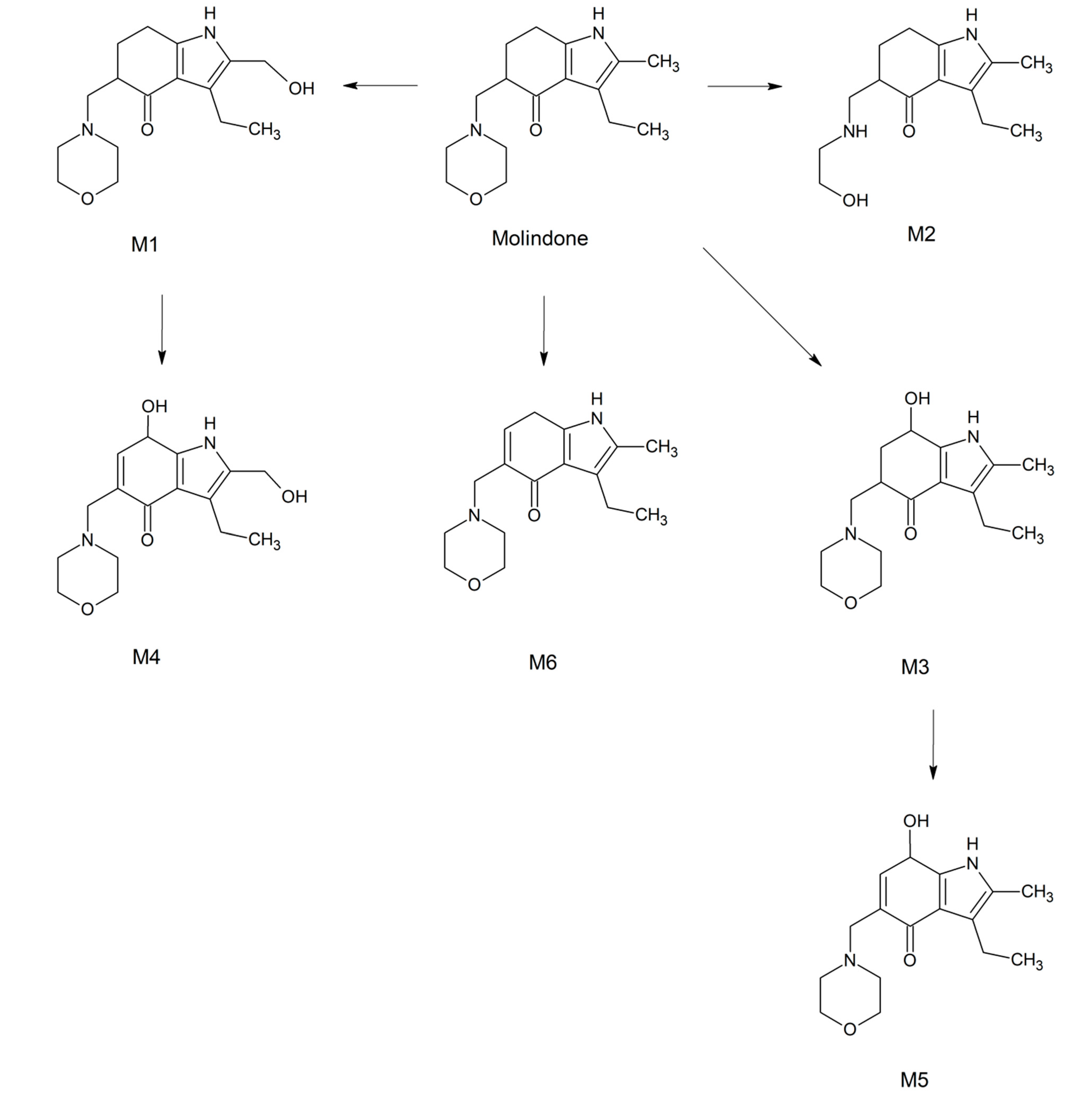
2.3. Metabolites Identification
2.4. Transformation Pathways
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Photocatalytic Experiments
3.3. In Vitro Simulation of Metabolism by Human Liver Microsomes
3.4. Analytical Procedures
3.5. Chemometric Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Krishna, G.; Gopalakrishnan, G.; Goel, S. In vitro and in vivo genotoxicity assessment of the dopamine receptor antagonist molindone hydrochloride. Environ. Mol. Mutagen. 2016, 57, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Krishna, G.; Gopalakrishnan, G.; Goel, S. Toxicity assessment of molindone hydrochloride, a dopamine D2/D5 receptor antagonist in juvenile and adult rats. Toxicol. Mech. Methods 2017, 27, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Stocks, J.D.K.; Taneja, B.; Baroldi, P.L.; Findling, R. A Phase 2a Randomized, Parallel Group, Dose-Ranging Study of Molindone in Children with Attention-Deficit/Hyperactivity Disorder and Persistent, Serious Conduct Problems. J. Child. Adolesc. Psychopharmacol. 2012, 22, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Kapur, S. Psychosis as a State of Aberrant Salience: A Framework Linking Biology, Phenomenology, and Pharmacology in Schizophrenia. Am. J. Psychiatry 2003, 160, 13–23. [Google Scholar] [CrossRef]
- Yu, C.; Gopalakrishnan, G. In Vitro Pharmacological Characterization of SPN-810M (Molindone). Available online: https://www.dovepress.com/in-vitro-pharmacological-characterization-of-spn-810m-molindone-peer-reviewed-article-JEP (accessed on 5 October 2019).
- Van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Howes, O.D.; Kapur, S. The Dopamine Hypothesis of Schizophrenia: Version III—The Final Common Pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.E.; Berney, S.; Mishra, R.; Sulser, F. The relative role of dopamine and norepinephrine receptor blockade in the action of antipsychotic drugs: Metoclopramide, thiethylperazine, and molindone as pharmacological tools. Psychopharmacology 1979, 64, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Sugerman, A.A.; Herrmann, J. Molindone: An indole derivative with antipsychotic activity. Clin. Pharmacol. Ther. 1967, 8, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Glazer, W.M.; Hafez, H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr. Res. 1990, 3, 315–320. [Google Scholar] [CrossRef]
- Koller, W.; Curtin, J.; Fields, J. Molindone compared to haloperidol in a guinea-pig model of tardive dyskinesia. Neuropharmacology 1984, 23, 1191–1194. [Google Scholar] [CrossRef]
- Flammia, D.D.; Bateman, H.R.; Saady, J.J.; Christensen, E.D. Tissue Distribution of Molidone in a Multidrug Overdose. J. Anal. Toxicol. 2004, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.C.; Banta, L.E.; Ehrlich, D.W. Molindone and Hepatotoxicity. Drug Intell. Clin. Pharm. 1985, 19, 744–746. [Google Scholar] [CrossRef] [PubMed]
- McCue, R.E.; Unuigbe, F.E.; Charles, R.A.; Orendain, G.C.M.; Waheed, R. Treatment of Morbidly Obese Psychotic Patients with Molindone: Three Case Reports. J. Clin. Psychiatry 2009, 70, 1606–1607. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, A.-M.; Fenton, M.; Kleijnen, J.; Lewis, R. Molindone for schizophrenia and severe mental illness. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.N.; Surapaneni, S. Role of drug metabolism in drug discovery and development. Med. Res. Rev. 2001, 21, 397–411. [Google Scholar] [CrossRef]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of Hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef]
- David, S.; Hamilton, J.P. Drug-induced Liver Injury. US Gastroenterol. Hepatol. Rev. 2010, 6, 73–80. [Google Scholar]
- Njoku, D.B. Drug-Induced Hepatotoxicity: Metabolic, Genetic and Immunological Basis. Int. J. Mol. Sci. 2014, 15, 6990–7003. [Google Scholar] [CrossRef]
- Asha, S.; Vidyavathi, M. Role of human liver microsomes in in vitro metabolism of drugs—A review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Choi, Y.J.; Oh, S.J.; Chi, Y.H.; Paik, S.H.; Lee, K.H.; Jung, J.-K.; Ryu, C.S.; Kim, K.-B.; Kim, D.-H.; et al. Characterization of fimasartan metabolites in human liver microsomes and human plasma. Xenobiotica 2016, 46, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Freudenberger, C.; Weinz, C. In Vitro Metabolism of Rivaroxaban, an Oral, Direct Factor Xa Inhibitor, in Liver Microsomes and Hepatocytes of Rats, Dogs, and Humans. Drug Metab. Dispos. 2009, 37, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Metabolism Study and Biological Evaluation of Bosentan Derivatives|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S0223523416304834?token=4357C7C17B76634A86BC9152B3E97D3394109F6110507C92AA2991D7FBF0CD73A3888B5CCEF74E2E0E969858250D04B1 (accessed on 28 October 2019).
- Zhuo, X.; Zheng, N. Drug Metabolism | Metabolite Isolation and Identification. In Encyclopedia of Analytical Science, 3rd ed.; Worsfold, P., Poole, C., Townshend, A., Miró, M., Eds.; Academic Press: Oxford, UK, 2019; pp. 232–239. ISBN 978-0-08-101984-9. [Google Scholar]
- Baranczewski, P.; Stańczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar] [PubMed]
- Morgan, S.; Grootendorst, P.; Lexchin, J.; Cunningham, C.; Greyson, D. The cost of drug development: A systematic review. Health Policy 2011, 100, 4–17. [Google Scholar] [CrossRef]
- Calza, P.; Pelizzetti, E.; Brussino, M.; Baiocchi, C. Ion trap tandem mass spectrometry study of dexamethasone transformation products on light activated TiO2 surface. J. Am. Soc. Mass Spectrom. 2001, 12, 1286–1295. [Google Scholar] [CrossRef]
- Calza, P.; Pazzi, M.; Medana, C.; Baiocchi, C.; Pelizzetti, E. The photocatalytic process as a tool to identify metabolitic products formed from dopant substances: The case of buspirone. J. Pharm. Biomed. Anal. 2004, 35, 9–19. [Google Scholar] [CrossRef]
- Medana, C.; Calza, P.; Giancotti, V.; Bello, F.D.; Pasello, E.; Montana, M.; Baiocchi, C. Horse metabolism and the photocatalytic process as a tool to identify metabolic products formed from dopant substances: The case of sildenafil. Drug Test. Anal. 2011, 3, 724–734. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37. [Google Scholar] [CrossRef]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 Photocatalysis: A Historical Overview and Future Prospects. Jpn. J. Appl. Phys. 2005, 44, 8269. [Google Scholar] [CrossRef]
- Gawlik, M.; Trawiński, J.; Skibiński, R. Simulation of phase I metabolism reactions of selected calcium channel blockers by human liver microsomes and photochemical methods with the use of Q-TOF LC/MS. J. Pharm. Biomed. Anal. 2019, 175, 112776. [Google Scholar] [CrossRef]
- Gawlik, M.; Trawiński, J.; Skibiński, R. Photocatalysis as a Tool for in Vitro Drug Metabolism Simulation: Multivariate Comparison of Twelve Metal Oxides on a Set of Twenty Model Drugs. Catalysts 2020, 10, 26. [Google Scholar] [CrossRef]
- Gul, T.; Bischoff, R.; Permentier, H.P. Electrosynthesis methods and approaches for the preparative production of metabolites from parent drugs. TrAC Trends Anal. Chem. 2015, 70, 58–66. [Google Scholar] [CrossRef]
- Valentová, K.; Káňová, K.; Di Meo, F.; Pelantová, H.; Chambers, C.S.; Rydlová, L.; Petrásková, L.; Křenková, A.; Cvačka, J.; Trouillas, P.; et al. Chemoenzymatic Preparation and Biophysical Properties of Sulfated Quercetin Metabolites. Int. J. Mol. Sci. 2017, 18, 2231. [Google Scholar]
- Mukai, K.; de Sant’Ana, D.P.; Hirooka, Y.; Mercado-Marin, E.V.; Stephens, D.E.; Kou, K.G.M.; Richter, S.C.; Kelley, N.; Sarpong, R. Bioinspired chemical synthesis of monomeric and dimeric stephacidin A congeners. Nat. Chem. 2018, 10, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Bioreactor Systems in Drug Metabolism: Synthesis of Cytochrome P450-Generated Metabolites|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1096717600901472?token=7F42B63F45A09779997B4069F89536C73627361C2FAEDB42F9CF7608F19CDB6C98140D2A81789F3DBE34BAE0ED793480 (accessed on 5 February 2020).
- Johansson, T.; Weidolf, L.; Jurva, U. Mimicry of phase I drug metabolism—Novel methods for metabolite characterization and synthesis. Rapid Commun. Mass Spectrom. 2007, 21, 2323–2331. [Google Scholar] [CrossRef]
- Zhao, J.; He, X.; Yang, N.; Sun, L.; Li, G. Study of Drug Metabolism by Xanthine Oxidase. Int. J. Mol. Sci. 2012, 13, 4873–4879. [Google Scholar] [CrossRef]
- Wen, B. Mass Spectrometry in Drug Metabolism: Principles and Common Practice. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D.W., Eds.; Academic Press: Oxford, UK, 2017; pp. 728–734. ISBN 978-0-12-803224-4. [Google Scholar]
- Li, F.; Gonzalez, F.J.; Ma, X. LC–MS-based Metabolomics in Profiling of Drug Metabolism and Bioactivation. Acta Pharm. Sin. B 2012, 2, 118. [Google Scholar] [CrossRef]
- Bell, J.D.; Gadian, D.G.; Preece, N.E. NMR studies of drug metabolism and disposition. Eur. J. Drug Metab. Pharmacokinet. 1990, 15, 127–133. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, W.-G.; Lee, S.; Lee, H.S. Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics. Int. J. Mol. Sci. 2017, 18, 621. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Reaction Type | Retention Time [min] | Measured Mass [m/z] | Theoretical Mass [m/z] | Mass Error [ppm] | Molecular Formula [M + H]+ | MS/MS Fragmentation [m/z] | Fragmentation Ion Formula [M + H]+ |
|---|---|---|---|---|---|---|---|---|
| MO | – | 6.53 | 277.1910 | 277.1910 | 0.00 | C16H24N2O2 | 190.1206 176.1057 116.0708 100.0761 82.0657 70.0658 | C12H16NO C11H14NO C5H10NO2 C5H10NO C5H8N C4H8N |
| M1 | Al-OH | 2.50 | 293.1860 | 293.1859 | 0.34 | C16H25N2O3 | 275.1726 221.1169 160.0605 100.0761 70.0660 | C16H23N2O2 C12H17N2O2 C9H8N2O C5H10NO C4H8N |
| M2 | Dealk | 5.24 | 251.1762 | 251.1754 | 3.18 | C14H23N2O2 | 178.1220 160.1132 122.0929 74.0610 | C11H16NO C11H14NO C8H12N C3H8NO |
| M3 | Al-OH | 4.90 | 293.2012 | 293.1859 | 52.18 | C16H25N2O3 | 206.1131 194.1113 188.1022 100.0760 74.0586 | C12H16NO2 C11H16NO2 C12H14NO C5H10NO C3H8NO |
| M4 | Al-2OH Dehydrog | 3.32 | 307.1654 | 307.1652 | 0.65 | C13H23N2O4 | 289.1458 188.1040 100.0759 84.0793 | C16H21N2O3 C12H14NO C5H10NO C5H10N |
| M5 | Al-OH Dehydrog | 3.70 | 291.1694 | 291.1703 | −3.09 | C16H23N2O3 | 204.1039 176.1069 162.0896 136.0735 100.0760 | C12H14NO2 C11H14NO C10H12NO C8H10NO C5H10NO |
| M6 | Dehydrog | 4.30 | 275.1754 | 275.1754 | 0.00 | C16H23N2O2 | 188.1036 100.0769 | C12H14NO C5H10NO |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawlik, M.; Savic, V.; Jovanovic, M.; Skibiński, R. Mimicking of Phase I Metabolism Reactions of Molindone by HLM and Photocatalytic Methods with the Use of UHPLC-MS/MS. Molecules 2020, 25, 1367. https://doi.org/10.3390/molecules25061367
Gawlik M, Savic V, Jovanovic M, Skibiński R. Mimicking of Phase I Metabolism Reactions of Molindone by HLM and Photocatalytic Methods with the Use of UHPLC-MS/MS. Molecules. 2020; 25(6):1367. https://doi.org/10.3390/molecules25061367
Chicago/Turabian StyleGawlik, Maciej, Vladimir Savic, Milos Jovanovic, and Robert Skibiński. 2020. "Mimicking of Phase I Metabolism Reactions of Molindone by HLM and Photocatalytic Methods with the Use of UHPLC-MS/MS" Molecules 25, no. 6: 1367. https://doi.org/10.3390/molecules25061367
APA StyleGawlik, M., Savic, V., Jovanovic, M., & Skibiński, R. (2020). Mimicking of Phase I Metabolism Reactions of Molindone by HLM and Photocatalytic Methods with the Use of UHPLC-MS/MS. Molecules, 25(6), 1367. https://doi.org/10.3390/molecules25061367