
Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
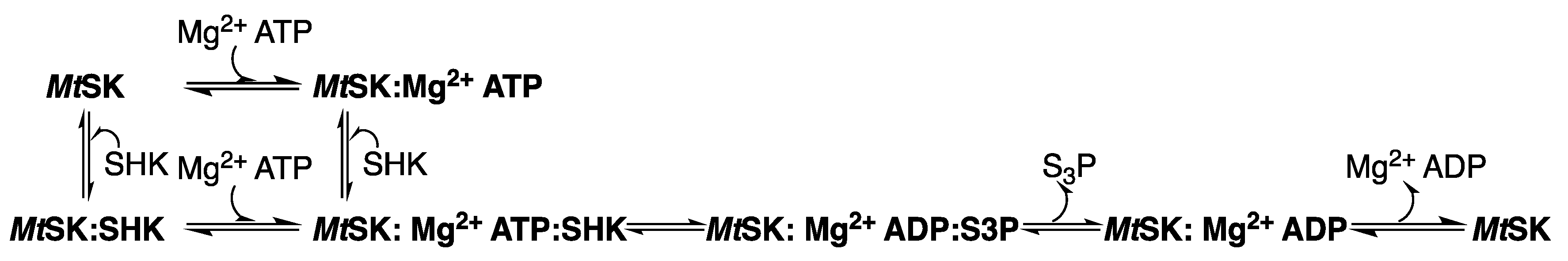{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Tuberculosis
1.2. Drug Screening
1.3. Preferable Characteristics of Targets and Enzyme Inhibitors/Anti-TB Agents
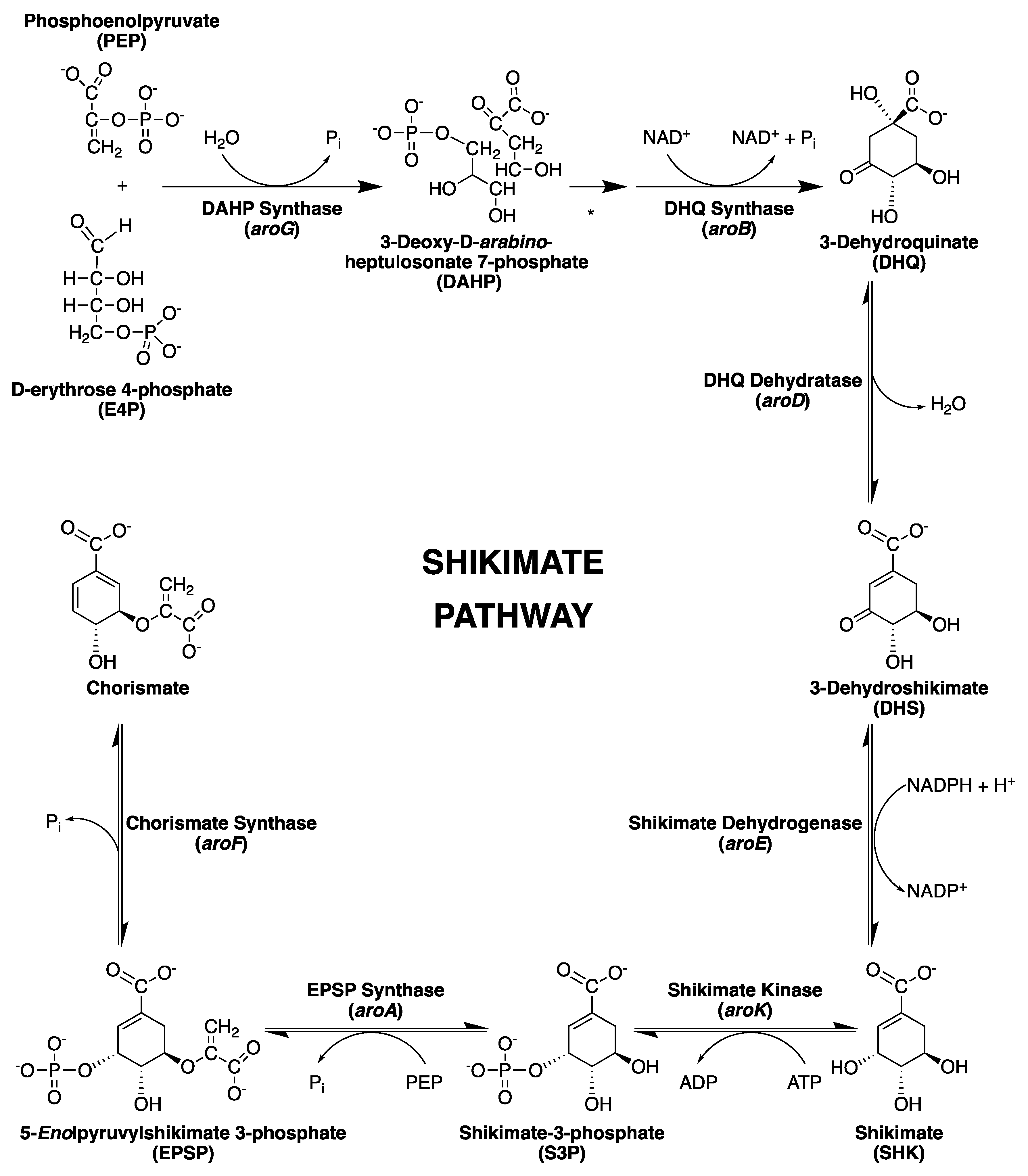
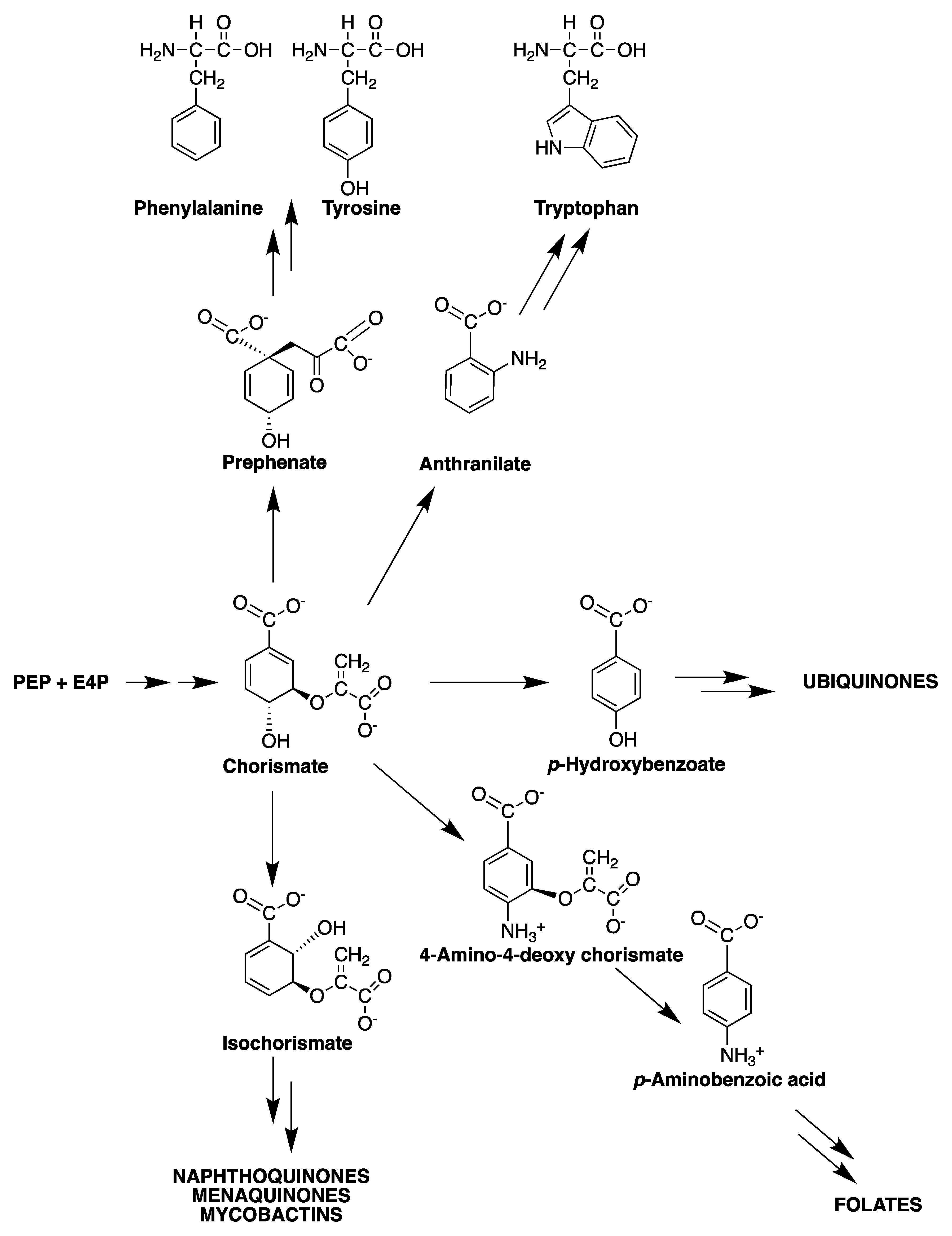
2. The Shikimate Pathway
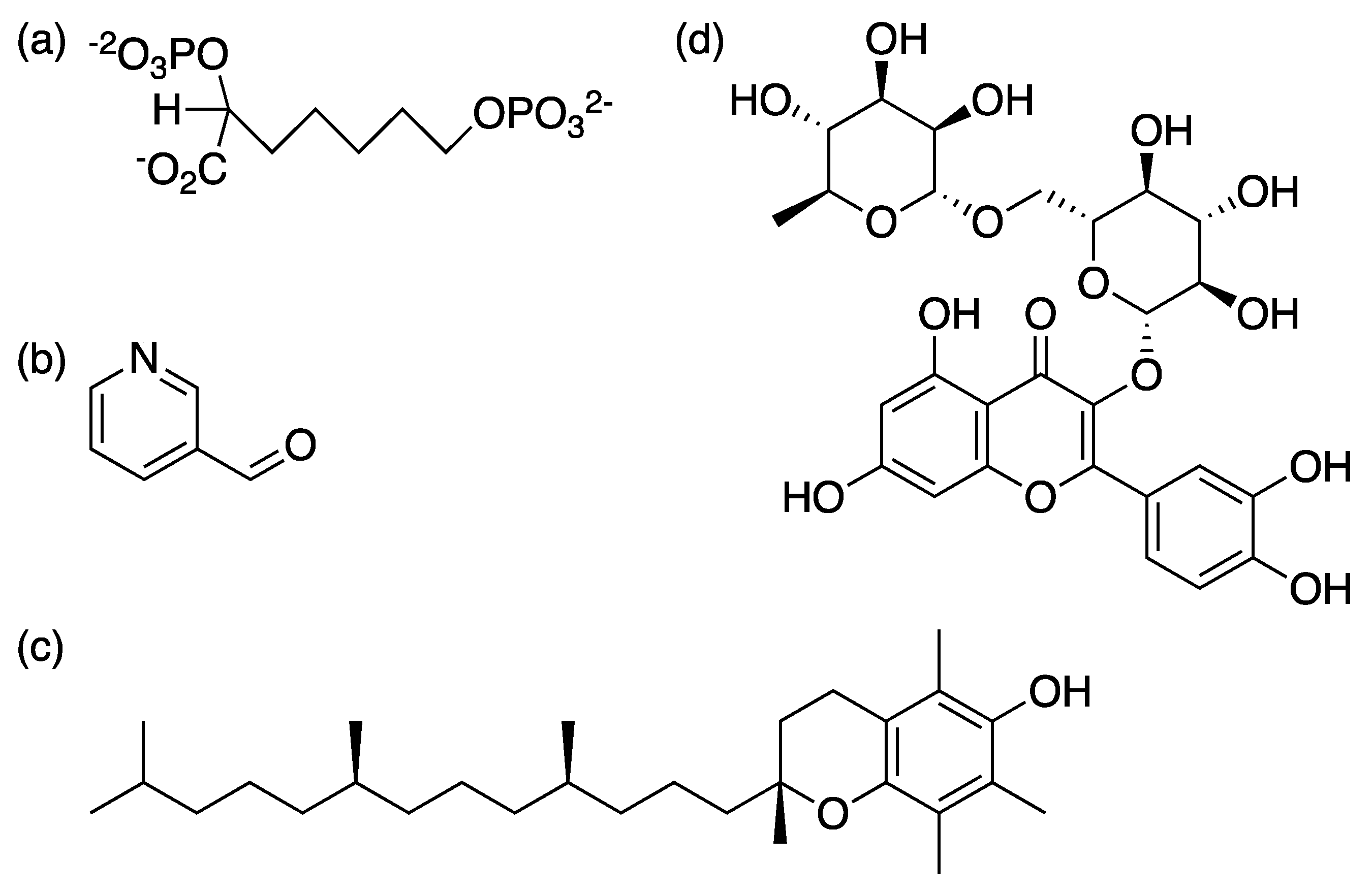
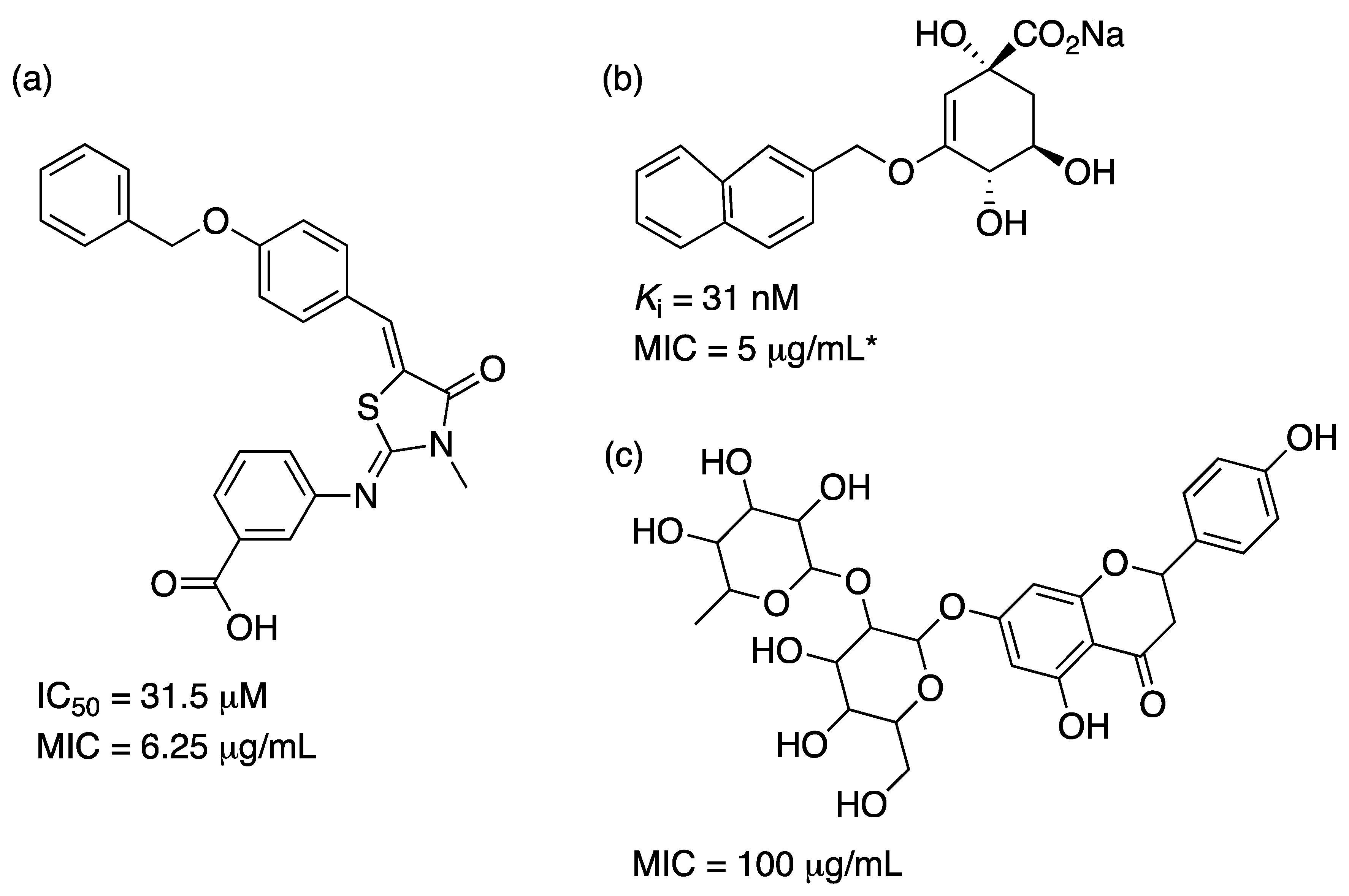
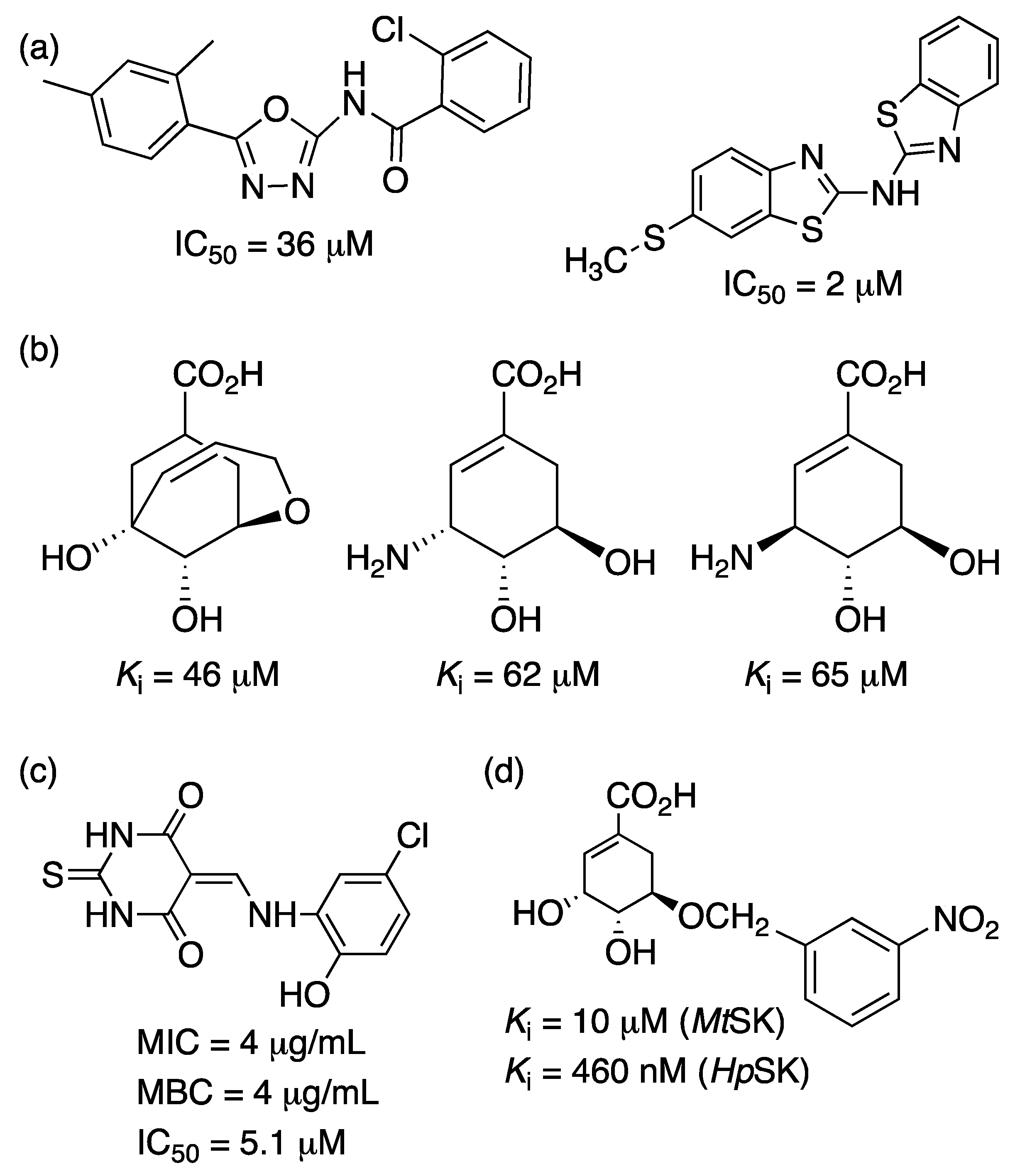
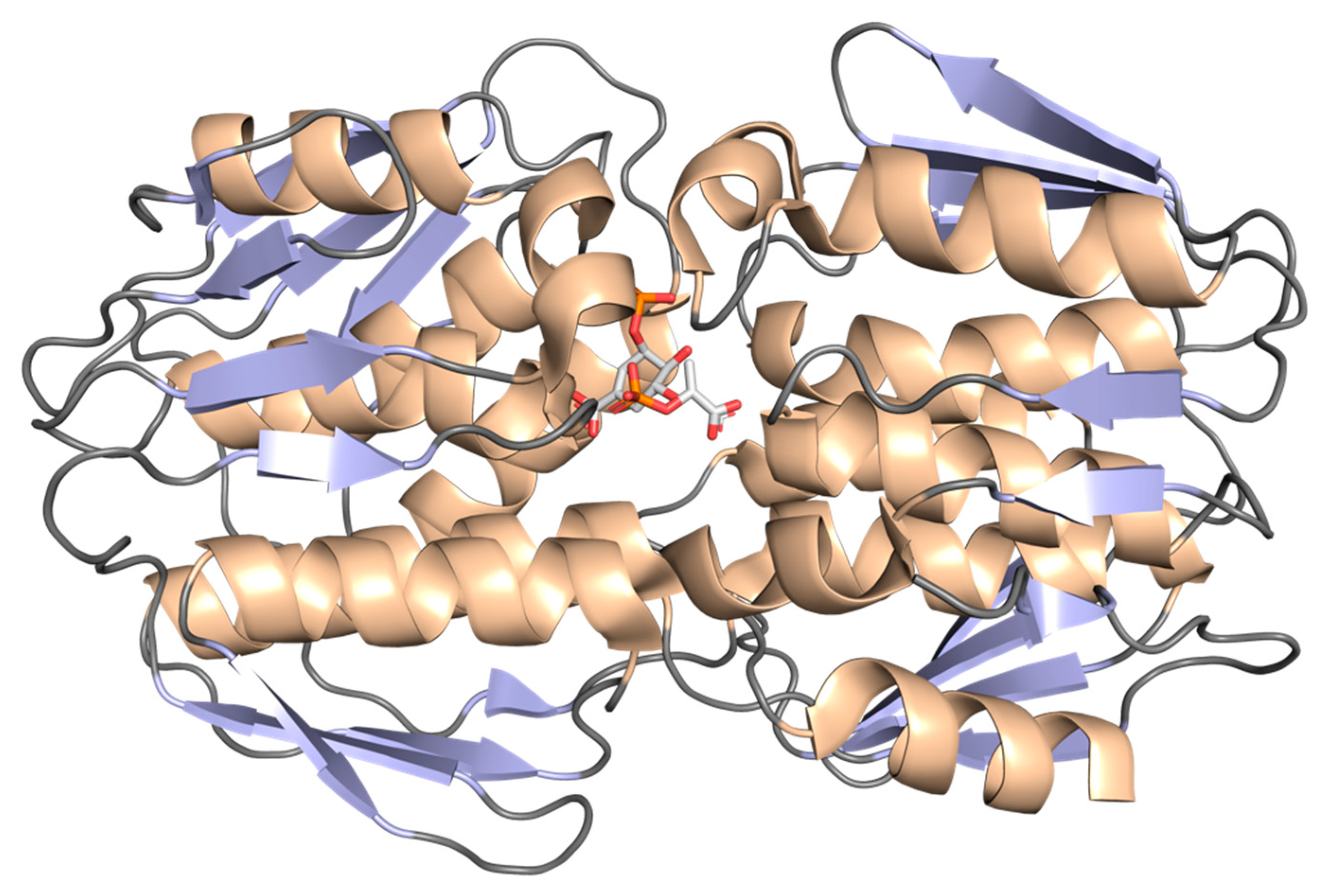
2.1. 3-Deoxy-D-Arabino-Heptulosonate-7-Phosphate Synthase (aroG Coding Sequence; EC 2.5.1.54)
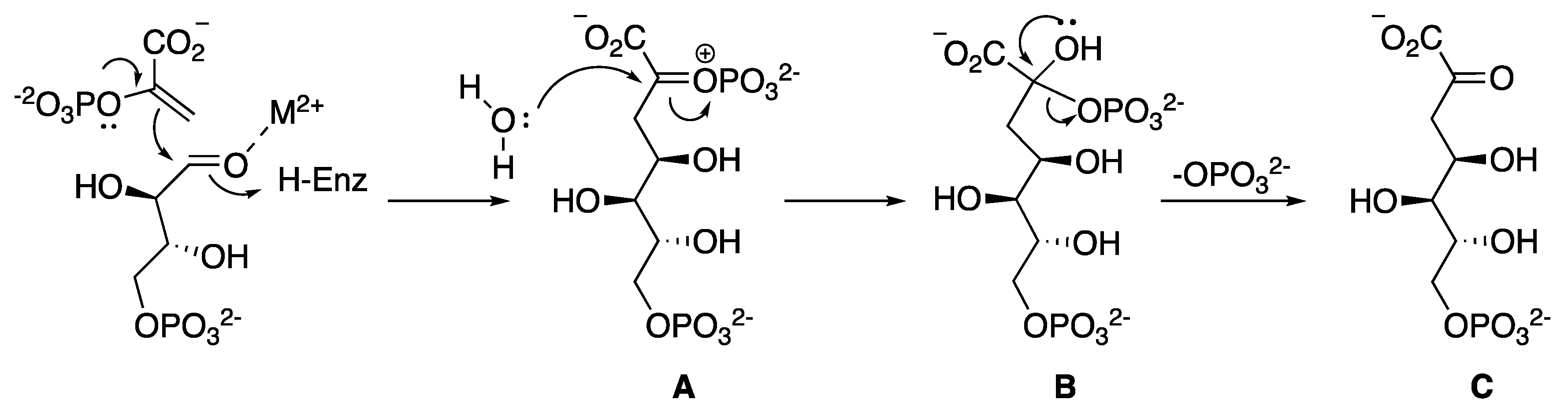
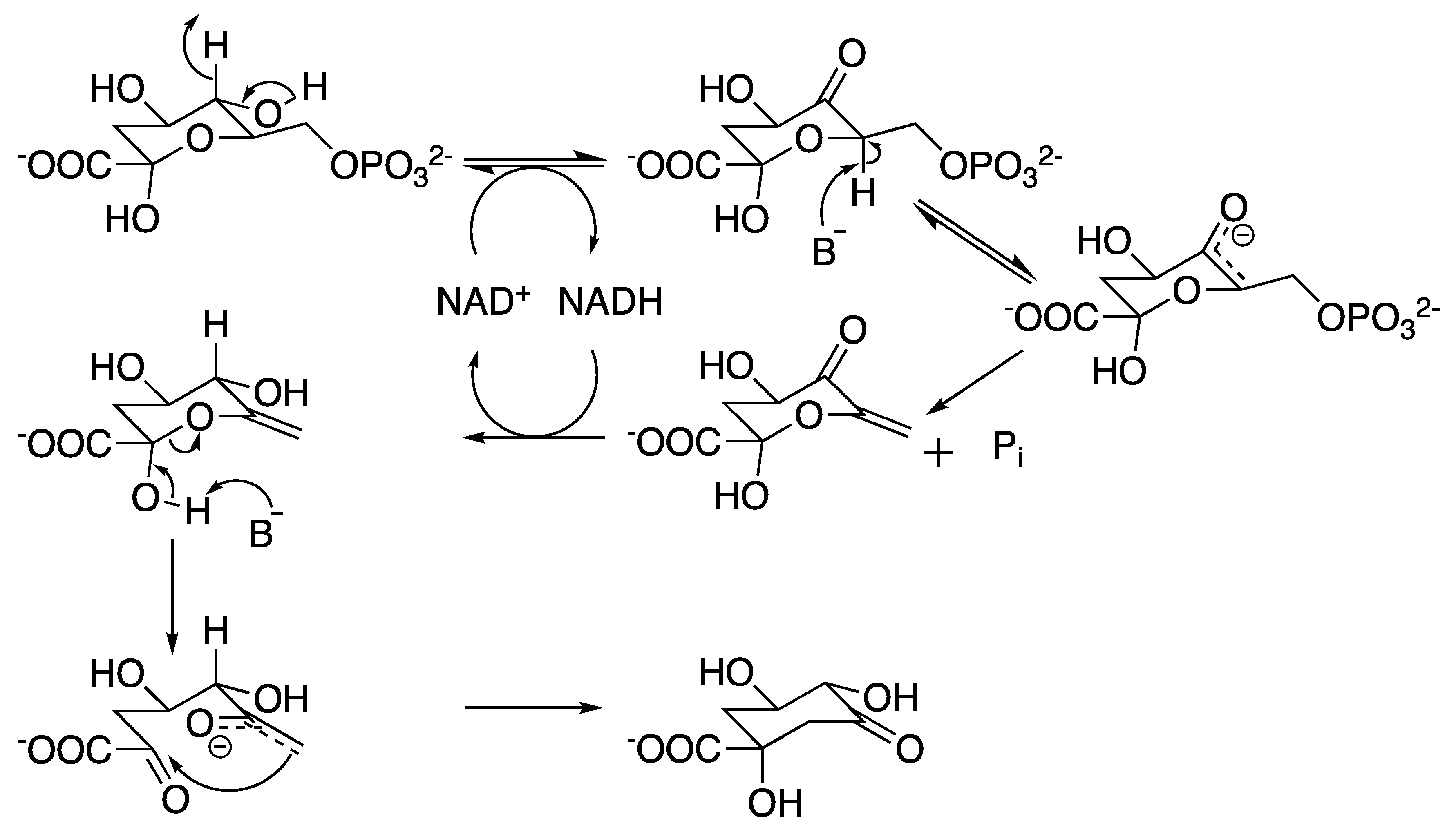
2.2. 3-Dehydroquinate Synthase (aroB Coding Sequence; EC 4.2.3.4)
2.3. 3-Dehydroquinate Dehydratase (aroD Coding Sequence; EC 4.2.1.10)
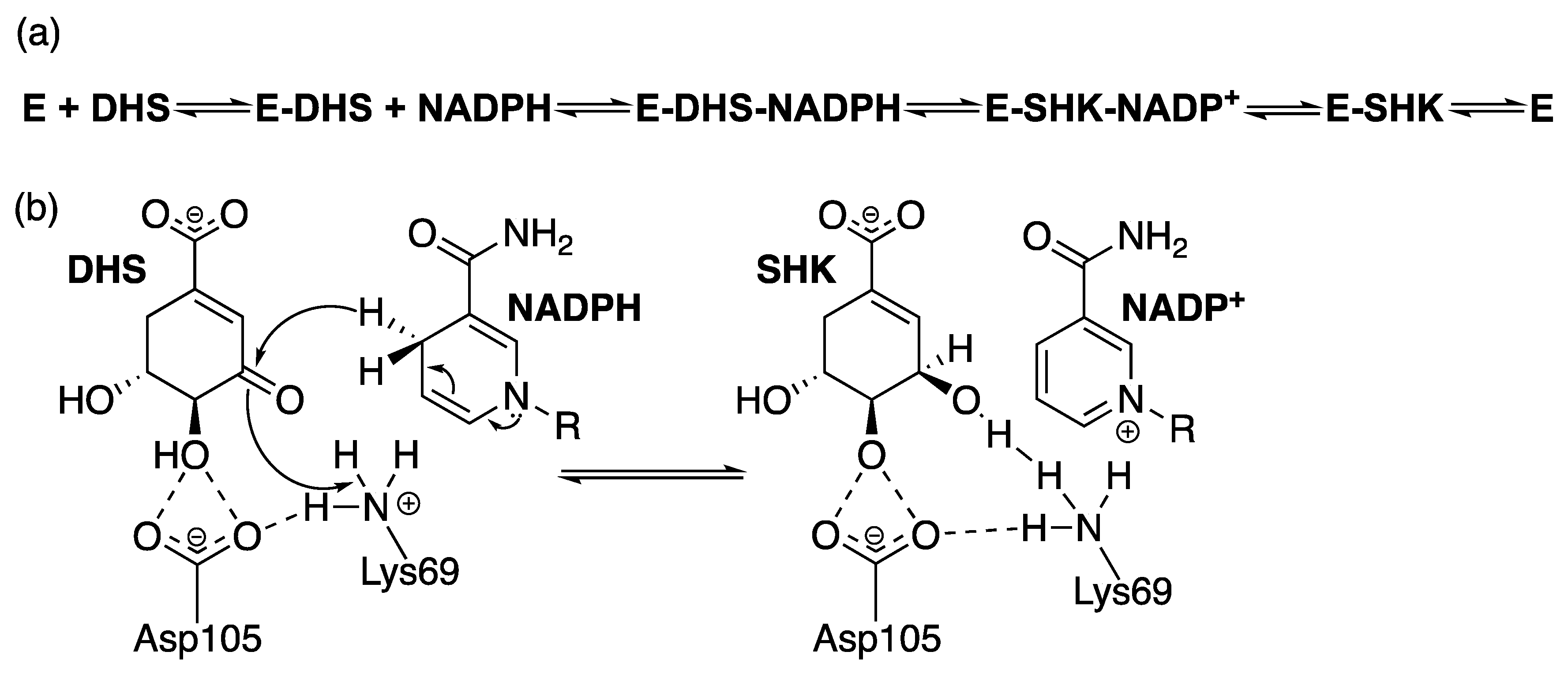
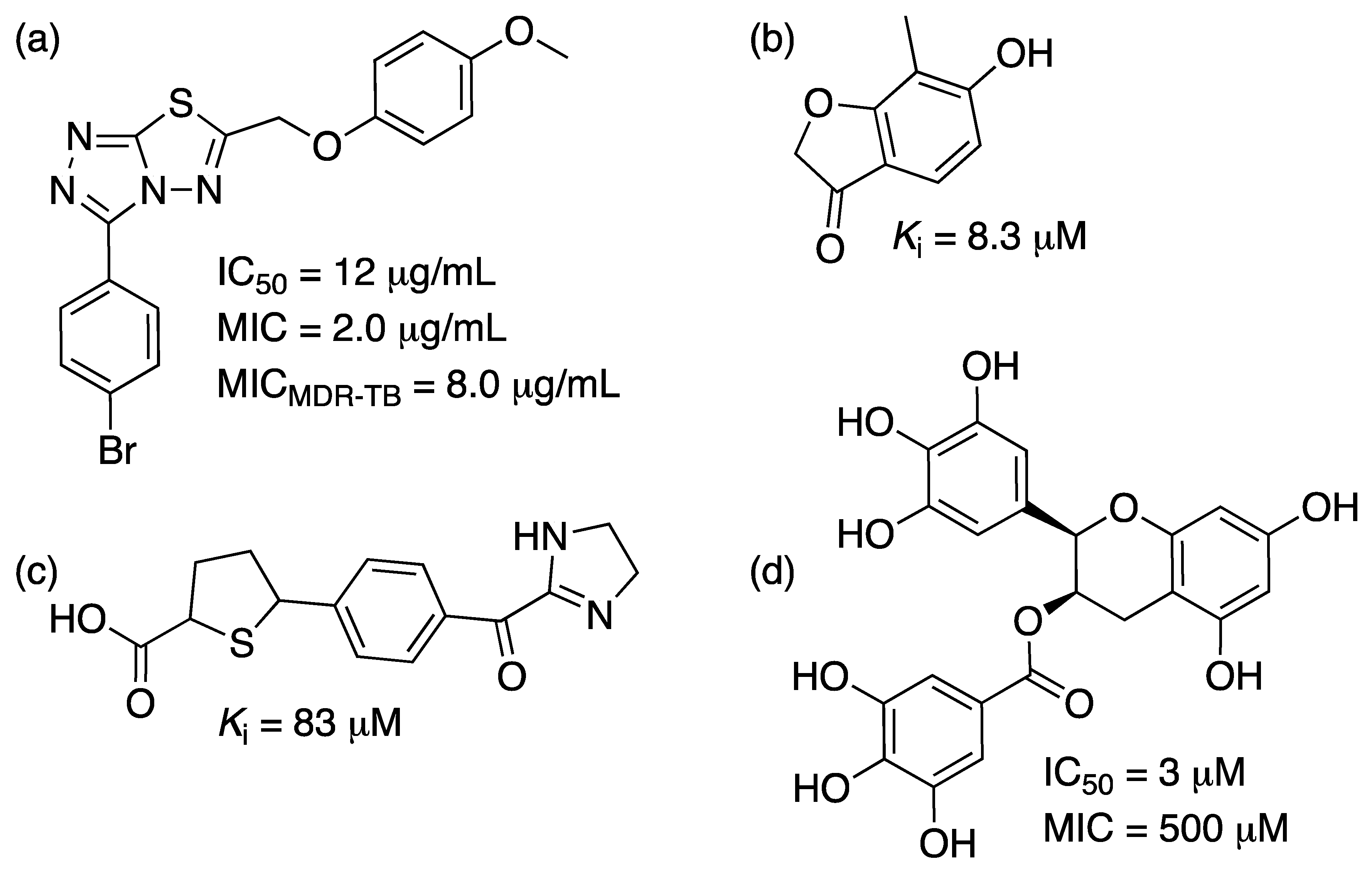
2.4. Shikimate 5-Dehydrogenase (aroE Coding Sequence; EC 1.1.1.25)
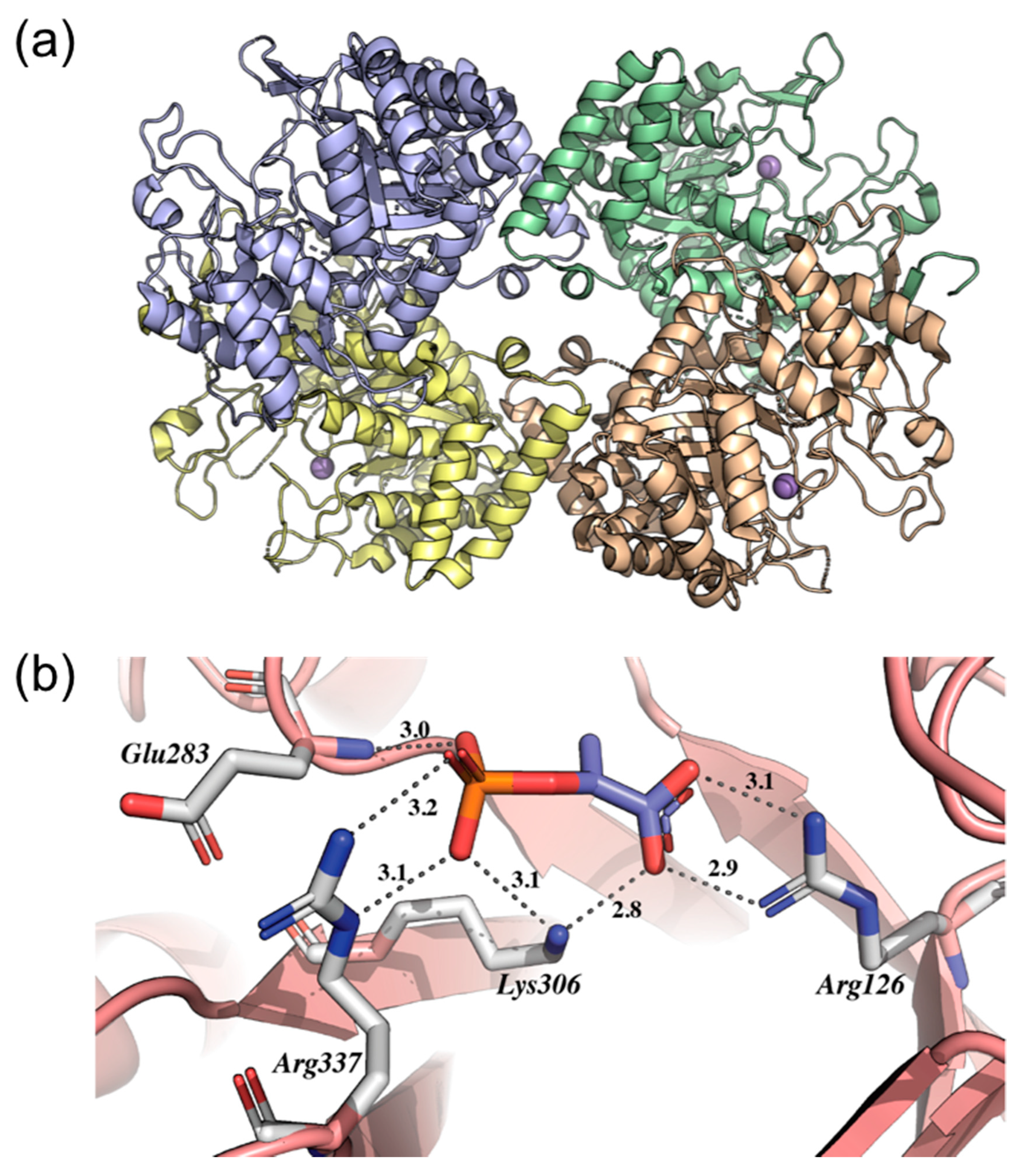
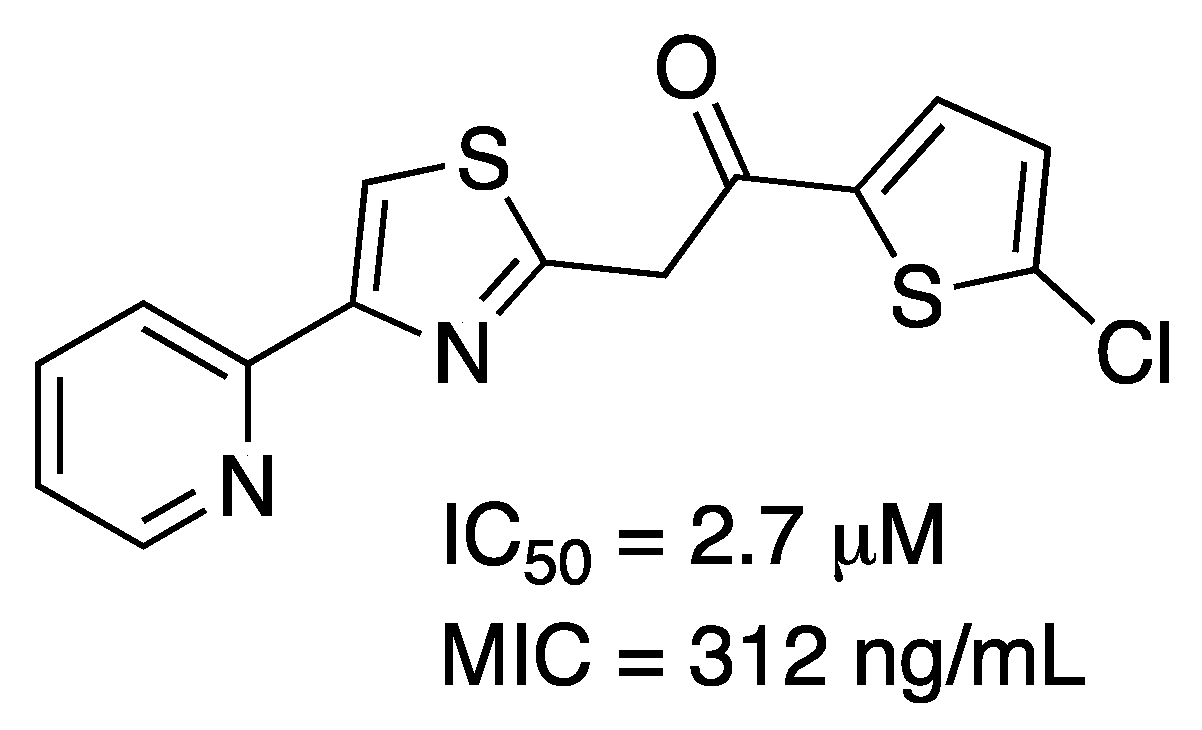
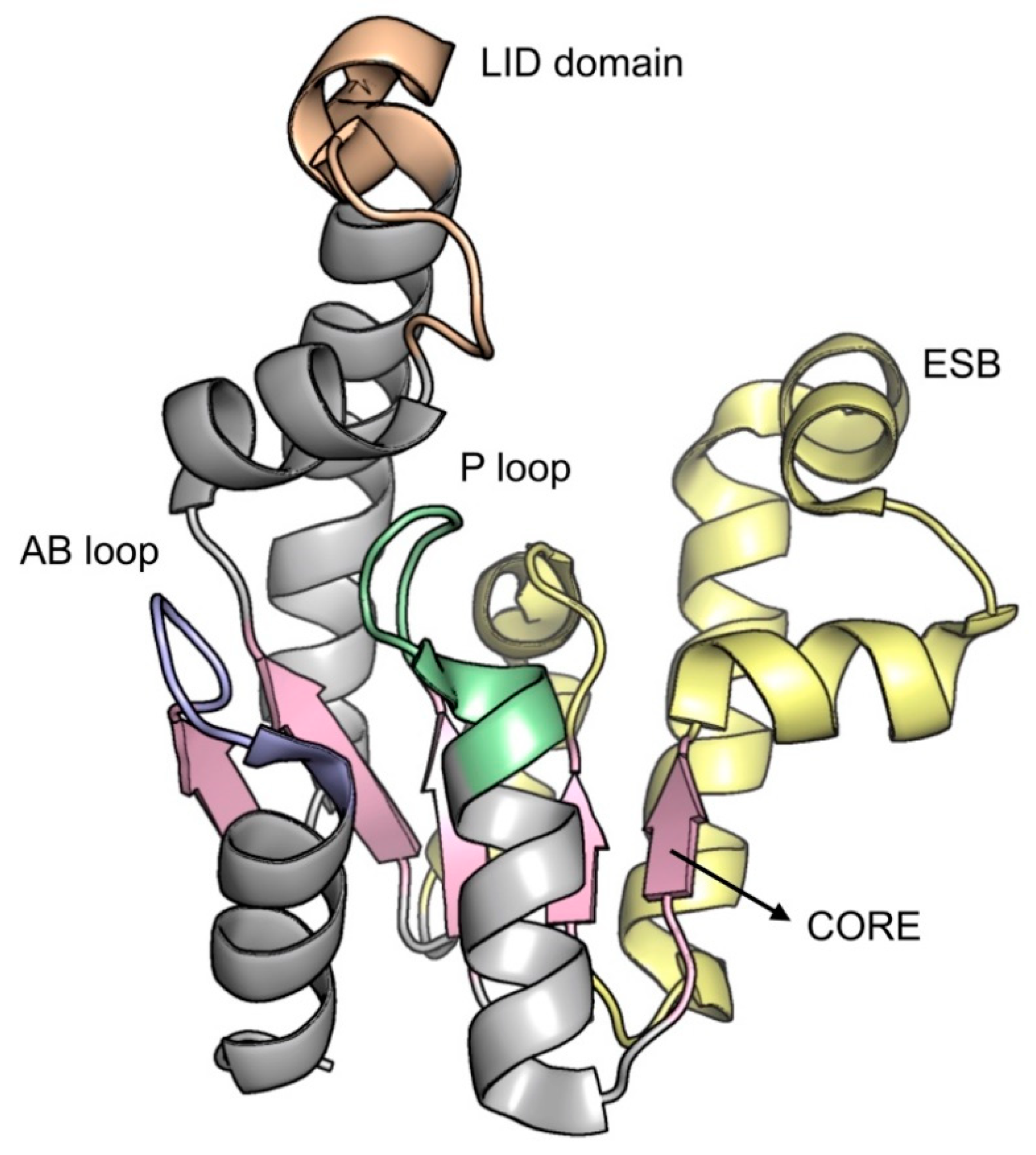
2.5. Shikimate Kinase (aroK Coding Sequence; EC 2.7.1.71)
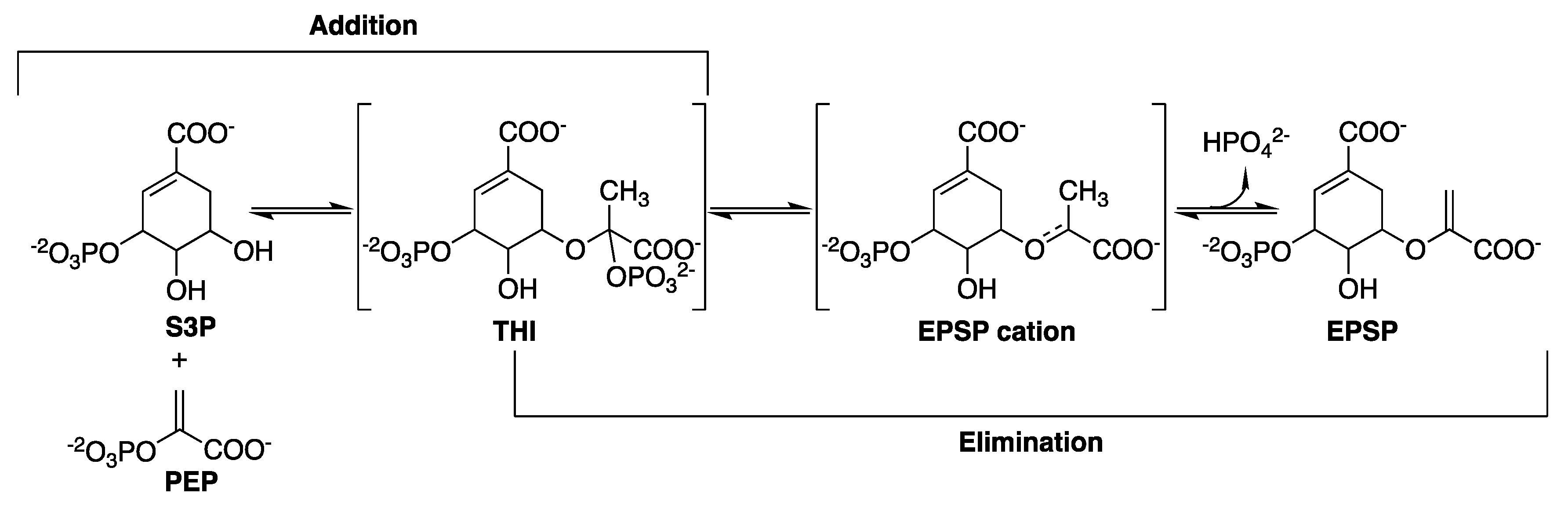
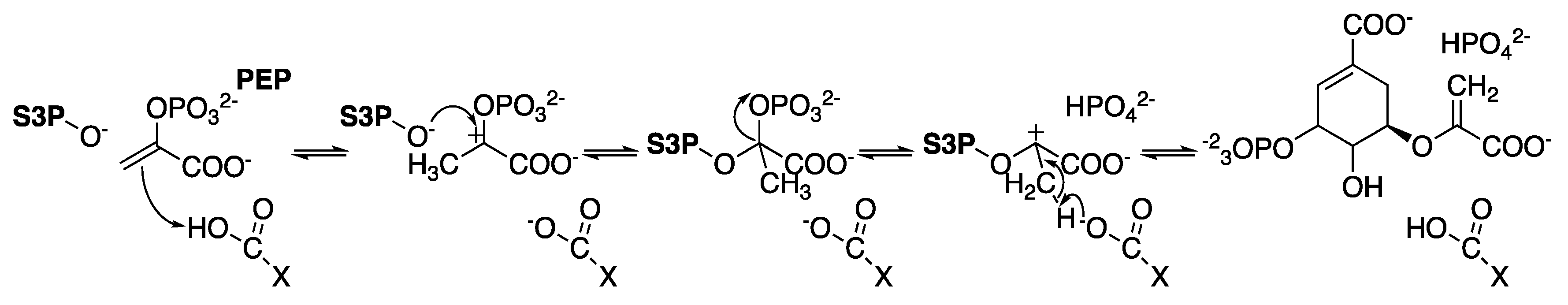
2.6. 5-Enolpyruvylshikimate-3-Phosphate Synthase (aroA Coding Sequence; EC 2.5.1.19)
2.7. Chorismate Synthase (aroF Coding Sequence; EC 4.2.3.5)
3. Future Prospects
Author Contributions
Acknowledgments
Conflicts of Interest
References
- WHO. Global tuberculosis report 2019. 2019. Licence: CC BY-NC-SA 3.0 IGO. Available online: www.who.int/tb/publications/global_report/en (accessed on 27 December 2019).
- Mandal, S.; Njikan, S.; Kumar, A.; Early, J.V.; Parish, T. The relevance of persisters in tuberculosis drug discovery. Microbiology 2019, 165, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Ducati, R.G.; Ruffino-Netto, A.; Basso, L.A.; Santos, D.S. The resumption of consumption—A review on tuberculosis. Mem. Inst. Oswaldo Cruz. 2006, 101, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Zhao, A.; Cohen, C.; Kang, W.; Lu, J.; Wang, G.; Zhao, Y.; Zheng, S. Current status of new tuberculosis vaccine in children. Hum. Vaccin. Immunother. 2016, 12, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Yuan, T.; Sampson, N.S. Hit generation in TB drug discovery: From genome to granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef]
- Wellington, S.; Hung, D.T. The expanding diversity of Mycobacterium tuberculosis drug targets. ACS Infect. Dis. 2018, 4, 696–714. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Comess, K.M.; McLoughlin, S.M.; Oyer, J.A.; Richardson, P.L.; Stöckmann, H.; Vasudevan, A.; Warder, S.E. Emerging approaches for the identification of protein targets of mall molecules—a practitioners’ perspective. J. Med. Chem. 2018, 61, 8504–8535. [Google Scholar] [CrossRef]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, E892. [Google Scholar] [CrossRef]
- McNeil, M.B.; Cook, G.M. Utilization of CRISPR interference to validate MmpL3 as a drug target in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00629. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Gashaw, I.; Ellinghaus, P.; Sommer, A.; Asadullah, K. What makes a good drug target? Drug Discov. Today. 2011, 16, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Holdgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132. [Google Scholar] [CrossRef]
- Greenwood, D.J.; Santos, M.S.; Huang, S.; Russell, M.R.G.; Collinson, L.M.; McRae, J.I.; West, A.; Jiang, H.; Gutierrez, M.G. Subcellular antibiotic visualization reveals a dynamic drug reservoir in infected macrophages. Science 2019, 364, 1279–1282. [Google Scholar] [CrossRef]
- Bentley, R. The shikimate pathway a metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 307–384. [Google Scholar] [CrossRef]
- Herrmann, K.M.; Weaver, L.M. The shikimate pathway. Annu. Rev. Plant. Physiol. Mol. Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef]
- Coggins, J.R.; Abell, C.; Evans, L.B.; Frederickson, M.; Robinson, D.A.; Roszak, A.W.; Lapthorn, A.P. Experiences with the shikimate-pathway enzymes as targets for rational drug design. Biochem. Soc. Trans. 2003, 31, 548–551. [Google Scholar] [CrossRef]
- Roberts, F.; Roberts, C.W.; Johnson, J.J.; Kyle, D.E.; Krell, T.; Coggins, J.R.; Coombs, G.H.; Milhous, W.K.; Tzipori, S.; Fergunson, D.J.P.; et al. Evidence for shikimate pathway in apicomplexan parasites. Nature 1998, 393, 801–805. [Google Scholar] [CrossRef]
- Ducati, R.G.; Basso, L.A.; Santos, D.S. Mycobacterial shikimate pathway enzymes as targets for drug design. Curr. Drug Targets. 2007, 8, 423–435. [Google Scholar] [CrossRef]
- Mir, R.; Jallu, S.; Singh, T.P. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit. Rev. Microbiol. 2015, 41, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Reddy, M.C.; Ioerger, T.R.; Rothchild, A.C.; Dartois, V.; Schuster, B.M.; Trauner, A.; Wallis, D.; Galaviz, S.; Huttenhower, C.; et al. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 2013, 155, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.M.; Weaver, L.M. 3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase. Purification, properties and kinetics of the tyrosine-sensitive isoenzyme from Escherichia coli. J. Biol. Chem. 1976, 251, 5440–5447. [Google Scholar]
- Tzin, V.; Galili, G.; Aharoni, A. Shikimate Pathway and Aromatic Amino Acid Biosynthesis. eLS. John Wiley & Sons 2012. [Google Scholar]
- Jiao, W.; Blackmore, N.J.; Nazmi, A.R.; Parker, E.J. Quaternary structure is an essential component that contributes to the sophisticated allosteric regulation mechanism in a key enzyme from Mycobacterium tuberculosis. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef]
- Jensen, R.A.; Xie, G..; Calhoun, D.H.; Bonner, C.A. The correct phylogenetic relationship of KdsA (3-deoxy-d-manno-octulosonate 8-phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-d-arabino-heptulosonate 7-phosphate synthase). J. Mol. Evol. 2002, 54, 416–423. [Google Scholar] [CrossRef]
- Webby, C.J.; Baker, H.M.; Lott, J.S.; Baker, E.N.; Parker, E.J. The structure of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J. Mol. Biol. 2005, 354, 927–939. [Google Scholar] [CrossRef]
- Shumilin, I.A.; Kretsinger, R.H.; Bauerle, R.H. Crystal structure of phenylalanine-regulated 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Escherichia coli. Structure 1999, 7, 865–875. [Google Scholar] [CrossRef]
- Shumilin, I.A.; Zhao, C.; Bauerle, R.; Kretsinger, R.H. Allosteric inhibition of 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase alters the coordination of both substrates. J. Mol. Biol. 2002, 320, 1147–1156. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 2002, 148, 3069–3077. [Google Scholar] [CrossRef]
- Furdui, C.; Zhou, L.; Woodard, R.W.; Anderson, K.S. Insights into the mechanism of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase (Phe) from Escherichia coli using a transient kinetic analysis. J. Biol. Chem. 2004, 279, 45618–45625. [Google Scholar] [CrossRef] [PubMed]
- Sterritt, O.W.; Lang, E.J.M.; Kessans, S.A.; Ryan, T.M.; Demeler, B.; Jameson, G.B.; Parker, E.J. Structural and functional characterisation of the entry point to pyocyanin biosynthesis in Pseudomonas aeruginosa defines a new 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase subclass. Bio. Sci. Rep. 2018, 38, BSR20181605. [Google Scholar] [CrossRef] [PubMed]
- Webby, C.J.; Patchett, M.L.; Parker, E.J. Characterization of a recombinant type II 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Helicobacter pylori. Bio. Chem. J. 2005, 390, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Reichau, S.; Jiao, W.; Walker, S.R.; Hutton, R.H.; Baker, E.N.; Parker, E.J. Potent inhibitors of a shikimate pathway enzyme from Mycobacterium tuberculosis: Combining mechanism- and modeling-based design. J. Bio. Chem. 2011, 286, 16197–16207. [Google Scholar] [CrossRef]
- Webby, C.J.; Jiao, W.; Hutton, R.D.; Blackmore, N.J.; Baker, H.M.; Baker, E.N.; Jameson, G.B.; Parker, E.J. Synergistic allostery: A sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2010, 285, 30567–30576. [Google Scholar] [CrossRef]
- Blackmore, N.J.; Reichau, S.; Jiao, W.; Hutton, R.D.; Baker, E.N.; Jameson, G.B.; Parker, E.J. Three sites and you are out: Ternary synergistic allostery controls aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Mol. Biol. 2013, 425, 1582–1592. [Google Scholar] [CrossRef]
- Munack, S.; Roderer, K.; Okvist, M.; Kamarauskaite, J.; Sasso, S.; van Eerde, A.; Kast, P.; Krengel, U. Remote control by inter-enzyme allostery: A novel paradigm for regulation of the shikimate pathway. J. Mol. Biol. 2016, 428, 1237–1255. [Google Scholar] [CrossRef]
- Sterritt, O.W.; Kessans, S.A.; Jameson, G.B.; Parker, E.J. A Pseudoisostructural Type II DAH7PS Enzyme from Pseudomonas aeruginosa: Alternative Evolutionary Strategies to Control Shikimate Pathway Flux. Biochemistry 2018, 57, 2667–2678. [Google Scholar] [CrossRef]
- Blackmore, N.J.; Nazmi, A.R.; Hutton, R.D.; Webby, M.N.; Baker, E.N.; Jameson, G.B.; Parker, E.J. Complex formation between two biosynthetic enzymes modifies the allosteric regulatory properties of both: An example of molecular symbiosis. J. Biol. Chem. 2015, 290, 18187–18198. [Google Scholar] [CrossRef]
- Nirmal, C.R.; Rao, R.; Hopper, W. Inhibition of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis: In silico screening and in vitro validation. Eur. J. Med. Chem. 2015, 105, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.W.; Bender, J.L.; Kadonaga, J.T.; Knowles, J.R. Dehydroquinate Synthase from Escherichia coli: Purification, Cloning and Construction of Overproducers of the Enzyme. Biochemistry 1984, 23, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, P.R.; Rothschild, J.; Sprinson, D.B. The Enzymic Conversion of 3-Deox-D-arabino-heptulosonic Acid 7-Phosphate to 5- Dehydroquinate. J. Biol. Chem. 1963, 238, 3176–3182. [Google Scholar] [PubMed]
- Coggins, J.R.; Duncan, K.; Anton, I.A.; Boocock, M.R.; Chaudhuri, S.; Lambert, J.M.; Lewendon, A.; Millar, G.; Mousdale, D.M.; Smith, D.D.S. The anatomy of a multifunctional enzyme. Bio. Chem. Soc. Trans. 1987, 15, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, A.R.; Lamb, H.K. The Molecular Biology of Multidomain Proteins Selected Examples. Eur. J. Biochem. 1995, 232, 7–18. [Google Scholar] [CrossRef]
- Carpenter, E.P.; Hawkins, A.R.; Frost, J.W.; Brown, K.A. Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 1998, 394, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.L.; Mehdi, S.; Knowles, J.R. Dehydroquinate Synthase: The Role of Divalent Metal Cations and of Nicotinamide Adenine Dinucleotide in Catalysis. Biochemistry 1989, 28, 7555–7560. [Google Scholar] [CrossRef]
- Widlanski, T.; Bender, S.L.; Knowles, J.R. Dehydroquinate Synthase: The Use of Substrate Analogues To Probe the Late Steps of the Catalyzed Reaction. Biochemistry 1989, 28, 7572–7582. [Google Scholar] [CrossRef]
- De Mendonça, J.D.; Ely, F.; Palma, M.S.; Frazzon, J.; Basso, L.A.; Santos, D.S. Functional characterization by genetic complementation of aroB-encoded dehydroquinate synthase from Mycobacterium tuberculosis H37Rv and its heterologous expression and purification. J. Bacteriol. 2007, 189, 6246–6252. [Google Scholar] [CrossRef]
- De Mendonça, J.D.; Adachi, O.; Rosado, L.A.; Ducati, R.G.; Santos, D.S.; Basso, L.A. Kinetic mechanism determination and analysis of metal requirement of dehydroquinate synthase from Mycobacterium tuberculosis H37Rv: An essential step in the function-based rational design of anti-TB drugs. Mol. Biosyst. 2011, 7, 119–128. [Google Scholar] [CrossRef]
- Moore, J.D.; Coggins, J.R.; Virden, R.; Hawkins, A.R. Efficient independent activity of a monomeric, monofunctional dehydroquinate synthase derived from the N-terminus of the pentafunctional AROM protein of Aspergillus nidulans. Biochem. J. 1994, 301, 297–304. [Google Scholar] [CrossRef]
- Skinner, M.A.; Günel-Ozcan, A.; Moore, J.; Hawkins, A.R.; Brown, K.A. Dehydroquinate Synthase Binds Divalent and Trivalent Cations: Role of Metal Binding in Catalysis. Biochem. Soc. Trans. 1997, 25, S609. [Google Scholar] [CrossRef] [PubMed]
- Le Marechal, P.; Froussios, C.; Level, M.; Azerad, R. The Interaction of Phosphonate and Homophosphonate Analogues of 3-Deoxy-D-Arabino Heptulosonate 7-Phospate With 3-Dehydroquinate Synthase from Escherichia coli. Biochem. Biophys. Res. Commun. 1980, 92, 1104–1109. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, X.; Li, D.; Lin, Y.; You, X.; Jiang, J.; Xu, Y.; Jiang, W.; Si, S. IMB-T130 targets 3-dehydroquinate synthase and inhibits Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Lin, Y.; Li, D.; Gao, N.; Liu, C.; You, X.; Jiang, J.; Jiang, W.; Si, S. Identification of an anti-TB compound targeting the tyrosyl-tRNA synthetase. J. Antimicrob. Chemother. 2015, 70, 2287–2294. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Davis, B.D. Aromatic biosynthesis. XII. Conversion of 5-dehydroquinic acid to 5-dehydroshikimic acid dy 5-dehydroquinase. Biochim. Biophys. Acta. - Gen. Subj. 1954, 15, 54–61. [Google Scholar]
- Gourley, D.G.; Shrive, A.K.; Polikarpov, I.; Krell, T.; Coggins, J.R.; Hawkins, A.R.; Isaacs, N.W.; Sawyer, L. The two types of 3-dehydroquinase have distinct structures but catalyze the same overall reaction. Nat. Struct. Biol. 1999, 6, 521–525. [Google Scholar]
- Garbe, T.; Servos, S.; Hawkins, A.; Dimitriadis, G.; Young, D.; Dougan, G.; Charles, I. The Mycobacterium tuberculosis shikimate pathway genes: Evolutionary relationship between biosynthetic and catabolic 3-dehydroquinases. MGG Mol. Gen. Genet. 1991, 228, 385–392. [Google Scholar] [CrossRef]
- Kleanthous, C.; Deka, R.; Davis, K.; Kelly, S.M.; Cooper, A.; Harding, S.E.; Price, N.C.; Hawkins, A.R.; Coggins, J.R. A comparison of the enzymological and biophysical properties of two distinct classes of dehydroquinase enzymes. Biochem. J. 1992, 282, 687–695. [Google Scholar] [CrossRef]
- Harris, J.; Kleanthous, C.; Coggins, J.R.; Hawkins, A.R.; Abell, C. Different Mechanistic and Stereochemical Courses for the Reactions catalysed by Type I and Type II Dehydroquinases. J. Chem. Soc. Chem. Commun. 1993, 13, 1080–1081. [Google Scholar] [CrossRef]
- Roszak, A.W.; Robinson, D.A.; Krell, T.; Hunter, I.S.; Fredrickson, M.; Abell, C.; Coggins, J.R.; Lapthorn, A.J. The structure and mechanism of the type II dehydroquinase from Streptomyces coelicolor. Structure 2002, 10, 493–503. [Google Scholar] [CrossRef]
- Turner, M.J.; Smith, B.W.; Haslam, E. The shikimate pathway. Part IV. The stereochemistry of the 3-dehydroquinate dehydratase reaction and observations on 3-dehydroquinate synthetase. J. Chem. Soc. Perkin Trans. 1975, 1, 52–55. [Google Scholar] [CrossRef]
- Shneier, A.; Harris, J.; Kleanthous, C.; Coggins, J.R.; Hawkins, A.R.; Abell, C. Evidence for Opposite Stereochemical Courses for the Reactions Catalysed by Type I and Type II Dehydroquinases. Bioorganic Med. Chem. 1993, 3, 1399–1402. [Google Scholar] [CrossRef]
- Le Sann, C.; Gower, M.A.; Abell, A.D. Inhibitors of Types I and II Dehydroquinase. Mini Rev. Med. Chem. 2004, 4, 747–756. [Google Scholar] [PubMed]
- González-Bello, C.; Tizón, L.; Lence, E.; Otero, J.M.; Van Raaij, M.J.; Martinez-Guitian, M.; Beceiro, A.; Thompson, P.; Hawkins, A.R. Chemical Modification of a Dehydratase Enzyme Involved in Bacterial Virulence by an Ammonium Derivative: Evidence of its Active Site Covalent Adduct. J. Am. Chem. Soc. 2015, 137, 9333–9343. [Google Scholar] [CrossRef]
- Harris, J.M.; Gonzalez-bello, C.; Kleanthous, C.; Hawkins, A.R.; Coggins, J.R.; Abell, C. Evidence from kinetic isotope studies for an enolate intermediate in the mechanism of type II dehydroquinases. Biochem. J. 1996, 319, 333–336. [Google Scholar] [CrossRef]
- Devi, A.S.; Ebihara, A.; Kuramitsu, S.; Yokoyama, S.; Kumarevel, T.; Ponnuraj, K. Crystal structure of type I 3-dehydroquinate dehydratase of Aquifex aeolicus suggests closing of active site flap is not essential for enzyme action. Biochem. Biophys. Res. Commun. 2013, 432, 350–354. [Google Scholar] [CrossRef]
- Cheung, V.W.N.; Xue, B.; Hernandez-Valladares, M.; Go, M.K.; Tung, A.; Aguda, A.H.; Robinson, R.C.; Yew, W.S. Identification of polyketide inhibitors targeting 3-dehydroquinate dehydratase in the shikimate pathway of Enterococcus faecalis. PLoS ONE 2014, 9, 1–9. [Google Scholar] [CrossRef]
- Petersen, G.O.; Saxena, S.; Renuka, J.; Soni, V.; Yogeeswari, P.; Santos, D.S.; Bizarro, C.V.; Sriram, D. Structure-based virtual screening as a tool for the identification of novel inhibitors against Mycobacterium tuberculosis 3-dehydroquinate dehydratase. J. Mol. Graph. Model. 2015, 60, 124–131. [Google Scholar] [CrossRef]
- Lee, B.I.; Kwak, J.E.; Suh, S.W. Crystal structure of the type II 3-dehydroquinase from Helicobacter pylori. Proteins Struct. Funct. Genet. 2003, 51, 616–617. [Google Scholar] [CrossRef]
- Iqbal, N.; Kumar, M.; Sharma, P.; Yadav, S.P.; Kaur, P.; Sharma, S.; Singh, T.P. Binding studies and structure determination of the recombinantly produced type-II 3-dehydroquinate dehydratase from Acinetobacter baumannii. Int. J. Biol. Macromol. 2017, 94, 459–465. [Google Scholar] [CrossRef]
- Reiling, S.; Kelleher, A.; Matsumoto, M.M.; Robinson, G.; Asojo, O.A. Structure of type II dehydroquinase from Pseudomonas aeruginosa. Acta Crystallogr. Sect. F Structural Biol. Commun. 2014, 70, 1485–1491. [Google Scholar] [CrossRef]
- Dias, M.V.B.; Blundell, T.L.; Snee, W.C.; Palaninathan, S.K.; Sacchettini, J.C.; Bromfield, K.M.; Payne, R.J.; Ciulli, A.; Howard, N.I.; Abell, C. Structural investigation of inhibitor designs targeting 3-dehydroquinate dehydratase from the shikimate pathway of Mycobacterium tuberculosis. Biochem. J. 2011, 436, 729–739. [Google Scholar] [CrossRef]
- Krell, T.; Horsburght, M.J.; Cooper, A.; Kelly, S.M.; Coggins, J.R. Localization of the active site of type II dehydroquinases. Identification of a common arginine-containing motif in the two classes of dehydroquinases. J. Biol. Chem. 1996, 271, 24492–24497. [Google Scholar] [CrossRef] [PubMed]
- Peón, A.; Otero, J.M.; Tizón, L.; Prazeres, V.F.V.; Llamas-Saiz, A.L.; Fox, G.C.; Van Raaij, M.J.; Lamb, H.; Hawkins, A.R.; Gago, F.; et al. Understanding the key factors that control the inhibition of type II dehydroquinase by (2R)-2-benzyl-3-dehydroquinic acids. ChemMedChem 2010, 5, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.J.; Peyrot, F.; Kerbarh, O.; Abell, A.D.; Abell, C. Rational design, synthesis and evaluation of nanomolar type II dehydroquinase inhibitors. ChemMedChem 2007, 2, 1015–1029. [Google Scholar] [CrossRef]
- Prazeres, V.F.V.; Sánchez-Sixto, C.; Castedo, L.; Lamb, H.; Hawkins, A.R.; Riboldi-Tunnicliffe, A.; Coggins, J.R.; Lapthorn, A.J.; González-Bello, C. Nanomolar competitive inhibitors of Mycobacterium tuberculosis and Streptomyces coelicolor type II dehydroquinase. ChemMedChem 2007, 2, 194–207. [Google Scholar] [CrossRef]
- Prazeres, V.F.V.; Castedo, L.; Lamb, H.; Hawkins, A.R.; González-Bello, C. 2-Substituted-3-dehydroquinic acids as potent competitive inhibitors of type II dehydroquinase. ChemMedChem 2009, 4, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Tizón, L.; Otero, J.M.; Prazeres, V.F.V.; Llamas-Saiz, A.L.; Fox, G.C.; Van Raaij, M.J.; Lamb, H.; Hawkins, A.R.; Ainsa, J.A.; Castedo, L.; et al. A prodrug approach for improving antituberculosis activity of potent Mycobacterium tuberculosis type II dehydroquinase inhibitors. J. Med. Chem. 2011, 54, 6063–6084. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ze-Sheng, L. Structure-and-Mechanism-Based Design and Discovery of Type II Mycobacterium tuberculosis Dehydroquinate Dehydratase Inhibitors. Curr. Top. Med. Chem. 2014, 14, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lone, M.Y.; Athar, M.; Gupta, V.K.; Jha, P.C. Prioritization of natural compounds against Mycobacterium tuberculosis 3-dehydroquinate dehydratase: A combined in-silico and in-vitro study. Biochem. Biophys. Res. Commun. 2017, 491, 1105–1111. [Google Scholar] [CrossRef]
- Yaniv, H.; Gilvarg, C. Aromatic Biosynthesis XIV. 5-Dehydroshikimic Reductase. J. Chem. Inf. Model. 1955, 213, 787–795. [Google Scholar]
- Michel, G.; Roszak, A.W.; Sauvé, V.; Maclean, J.; Matte, A.; Coggins, J.R.; Cygler, M.; Lapthorn, A.J. Structures of shikimate dehydrogenase AroE and its paralog YdiB: A common structural framework for different activities. J. Biol. Chem. 2003, 278, 19463–19472. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Hao, F.; Lai, X.; Yu, H.; Huang, Y.; Wang, H. Expression, purification and properties of shikimate dehydrogenase from Mycobacterium tuberculosis. J. Biochem. Mol. Biol. 2005, 38, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, I.O.; Magalhães, M.L.B.; Oliveira, J.S.; Silva, R.G.; Mendes, M.A.; Palma, M.S.; Santos, D.S.; Basso, L.A. Functional shikimate dehydrogenase from Mycobacterium tuberculosis H37Rv: Purification and characterization. Protein Expr. Purif. 2006, 46, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Padyana, A.K.; Burley, S.K. Crystal structure of shikimate 5-dehydrogenase (SDH) bound to NADP: Insights into function and evolution. Structure 2003, 11, 1005–1013. [Google Scholar] [CrossRef]
- Kubota, T.; Tanaka, Y.; Hiraga, K.; Inui, M.; Yukawa, H. Characterization of shikimate dehydrogenase homologues of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2013, 97, 8139–8149. [Google Scholar] [CrossRef] [PubMed]
- Giles, N.H.; Case, M.E.; Baum, J.; Geever, R.; Huiet, L.; Patel, V.; Tyler, B. Gene organization and regulation in the qa (quinic acid) gene cluster of Neurospora crassa. Microbiol. Rev. 1985, 49, 338–358. [Google Scholar] [CrossRef]
- Hawkins, A.R.; Lamb, H.K.; Smith, M.; Keyte, J.W.; Roberts, C.F. Molecular organisation of the quinic acid utilization (QUT) gene cluster in Aspergillus nidulans. MGG Mol. Gen. Genet. 1988, 214, 224–231. [Google Scholar] [CrossRef]
- Peek, J.; Christendat, D. The shikimate dehydrogenase family: Functional diversity within a conserved structural and mechanistic framework. Arch. Biochem. Biophys. 2015, 566, 85–99. [Google Scholar] [CrossRef]
- Guo, J.; Frost, J.W. Synthesis of aminoshikimic acid. Org. Lett. 2004, 6, 1585–1588. [Google Scholar] [CrossRef]
- Peek, J.; Garcia, C.; Lee, J.; Christendat, D. Insights into the function of RifI2: Structural and biochemical investigation of a new shikimate dehydrogenase family protein. Biochim. Biophys. Acta. - Proteins Proteomics. 2013, 1834, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Choorapoikayil, S.; Schoepe, J.; Buchinger, S.; Schomburg, D. Analysis of in vivo function of predicted isoenzymes-a metabolomic approach. Omi. A J. Integr. Biol. 2012, 16, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Peek, J.; Lee, J.; Hu, S.; Senisterra, G.; Christendat, D. Structural and mechanistic analysis of a novel class of shikimate dehydrogenases: Evidence for a conserved catalytic mechanism in the shikimate dehydrogenase family. Biochemistry 2011, 50, 8616–8627. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, I.O.; Silva, R.G.; Fernandes, C.L.; de Souza, O.N.; Basso, L.A.; Santos, D.S. Kinetic and chemical mechanisms of shikimate dehydrogenase from Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2007, 457, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, H.A.; Borges, J.C.; Fonseca, I.O.; Pereira, J.H.; Neto, J.R.; Basso, L.A.; Santos, D.S.; De Azevedo, W.F. Structural studies of shikimate 5-dehydrogenase from Mycobacterium tuberculosis. Proteins Struct. Funct. Genet. 2008, 72, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Bagautdinov, B.; Kunishima, N. Crystal Structures of Shikimate Dehydrogenase AroE from Thermus thermophilus HB8 and its Cofactor and Substrate Complexes: Insights into the Enzymatic Mechanism. J. Mol. Biol. 2007, 373, 424–438. [Google Scholar] [CrossRef]
- Singh, S.A.; Christendat, D. Structure of Arabidopsis dehydroquinate dehydratase-shikimate dehydrogenase and implications for metabolic channeling in the shikimate pathway. Biochemistry 2006, 45, 7787–7796. [Google Scholar] [CrossRef]
- Han, C.; Hu, T.; Wu, D.; Qu, S.; Zhou, J.; Ding, J.; Shen, X.; Qu, D.; Jiang, H. X-ray crystallographic and enzymatic analyses of shikimate dehydrogenase from Staphylococcus epidermidis: Implications for substrate binding and conformational change. FEBS J. 2009, 276, 1125–1139. [Google Scholar] [CrossRef]
- Balinsky, D.; Dennis, A.W.; Cleland, W.W. Kinetic and Isotope-Exchange Studies on Shikimate Dehydrogenase from Pisum sativum. Biochemistry 1971, 10, 1947–1952. [Google Scholar]
- Avitia-Domínguez, C.; Sierra-Campos, E.; Salas-Pacheco, J.M.; Nájera, H.; Rojo-Domínguez, A.; Cisneros-Martínez, J.; Téllez-Valencia, A. Inhibition and biochemical characterization of methicillin-resistant Staphylococcus aureus shikimate dehydrogenase: An in silico and kinetic study. Molecules 2014, 19, 4491–4509. [Google Scholar] [CrossRef]
- Lim, S.; Schröder, I.; Monbouquette, H.G. A thermostable shikimate 5-dehydrogenase from the archaeon Archaeoglobus fulgidus. FEMS Microbiol. Lett. 2004, 238, 101–106. [Google Scholar] [PubMed]
- Park, A.; Lamb, H.K.; Nichols, C.; Moore, J.D.; Brown, K.A.; Cooper, A.; Charles, I.G.; Stammers, D.K.; Hawkins, A.R. Biophysical and kinetic analysis of wild-type and site-directed mutants of the isolated and native dehydroquinate synthase domain of the AROM protein. Protein Sci. 2004, 13, 2108–2119. [Google Scholar] [CrossRef]
- Gan, J.; Wu, Y.; Prabakaran, P.; Gu, Y.; Li, Y.; Andrykovitch, M.; Liu, H.; Gong, Y.; Yan, H.; Ji, X. Structural and biochemical analyses of shikimate dehydrogenase AroE from Aquifex aeolicus: Implications for the catalytic mechanism. Biochemistry 2007, 46, 9513–9522. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Junior, V.; Breda, A.; Santos, D.; Basso, L. The conserved Lysine69 residue plays a catalytic role in Mycobacterium tuberculosis shikimate dehydrogenase. BMC Res. Notes. 2009, 2, 1–7. [Google Scholar]
- Dansette, P.; Azerad, R. The shikimate pathway: II. Stereospecificity of hydrogen transfer catalyzed by NADPH-dehydroshikimate reductase of E. coli. Biochimie. 1974, 56, 751–755. [Google Scholar] [CrossRef]
- Han, C.; Wang, L.; Yu, K.; Chen, L.; Hu, L.; Chen, K.; Jiang, H.; Shen, X. Biochemical characterization and inhibitor discovery of shikimate dehydrogenase from Helicobacter pylori. FEBS J. 2006, 273, 4682–4692. [Google Scholar] [CrossRef]
- Peek, J.; Shi, T.; Christendat, D. Identification of novel polyphenolic inhibitors of shikimate dehydrogenase (AroE). J. Biomol. Screen. 2014, 19, 1090–1098. [Google Scholar] [CrossRef]
- Díaz-Quiroz, D.C.; Cardona-Félix, C.S.; Viveros-Ceballos, J.L.; Reyes-González, M.A.; Bolívar, F.; Ordoñez, M.; Escalante, A. Synthesis, biological activity and molecular modelling studies of shikimic acid derivatives as inhibitors of the shikimate dehydrogenase enzyme of Escherichia coli. J. Enzyme Inhib. Med. Chem. 2018, 33, 397–404. [Google Scholar] [CrossRef]
- Enríquez-Mendiola, D.; Téllez-Valencia, A.; Sierra-Campos, E.; Campos-Almazán, M.; Valdez-Solana, M.; Gómez Palacio-Gastélum, M.; Avitia-Domínguez, C. Kinetic and molecular dynamic studies of inhibitors of shikimate dehydrogenase from methicillin-resistant Staphylococcus aureus. Chem. Biol. Drug Des. 2019, 94, 1504–1517. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Bai, X.; Deng, Q.; Wang, J.; Zhang, G.; Xiao, C.; Mei, Y.; Wang, Y. SAR studies on 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles as inhibitors of Mtb shikimate dehydrogenase for the development of novel antitubercular agents. RSC Adv. 2015, 5, 97089–97101. [Google Scholar] [CrossRef]
- Li, Z.; Bai, X.; Deng, Q.; Zhang, G.; Zhou, L.; Liu, Y.; Wang, J.; Wang, Y. Preliminary SAR and biological evaluation of antitubercular triazolothiadiazine derivatives against drug-susceptible and drug-resistant Mtb strains. Bioorganic Med. Chem. 2017, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Meng, J.; Liu, Y.; Guan, Y.; Xiao, C. IMB-SD62, a triazolothiadiazoles derivative with promising action against tuberculosis. Tuberculosis 2018, 112, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Isa, M.A.; Majumdar, R.S.; Haider, S. In silico identification of potential inhibitors against shikimate dehydrogenase through virtual screening and toxicity studies for the treatment of tuberculosis. Int. Microbiol. 2019, 22, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.D.; Bourenkov, G.P.; Oberschall, A.; Strizhov, N.; Bartunik, H.D. Mechanism of phosphoryl transfer catalyzed by shikimate kinase from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 364, 411–423. [Google Scholar] [CrossRef]
- Cheng, W.C.; Chen, Y.F.; Wang, H.J.; Hsu, K.C.; Lin, S.C.; Chen, T.J.; Yang, J.M.; Wang, W.C. Structures of Helicobacter pylori shikimate kinase reveal a selective inhibitor-induced-fit mechanism. PLoS ONE 2012, 7, e33481. [Google Scholar] [CrossRef]
- Blanco, B.; Prado, V.; Lence, E.; Otero, J.M.; García-Doval, C.; Van Raaij, M.J.; Llamas-Saiz, A.L.; Lamb, H.; Hawkins, A.R.; González-Bello, C. Mycobacterium tuberculosis shikimate kinase inhibitors: Design and simulation studies of the catalytic turnover. J. Am. Chem. Soc. 2013, 135, 12366–12376. [Google Scholar] [CrossRef]
- Whipp, M.J.; Pittard, A.J. A reassessment of the relationship between aroK-and aroL-encoded shikimate kinase enzymes of Escherichia coli. J. Bacteriol. 1995, 177, 1627–1629. [Google Scholar] [CrossRef]
- Gu, Y.; Reshetnikova, L.; Li, Y.; Wu, Y.; Yan, H.; Singh, S.; Ji, X. Crystal structure of shikimate kinase from Mycobacterium tuberculosis reveals the dynamic role of the LID domain in catalysis. J. Mol. Biol. 2002, 319, 779–789. [Google Scholar] [CrossRef]
- Vonrhein, C.; Schlauderer, G.J.; Schulz, G.E. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure 1995, 3, 483–490. [Google Scholar] [CrossRef]
- Favela-Candia, A.; Téllez-Valencia, A.; Campos-Almazán, M.; Sierra-Campos, E.; Valdez-Solana, M.; Oria-Hernández, J.; Castillo-Villanueva, A.; Nájera, H.; Avitia-Domínguez, C. Biochemical, Kinetic and Computational Structural Characterization of Shikimate Kinase from Methicillin-Resistant Staphylococcus aureus. Mol. Biotechnol. 2019, 61, 274–285. [Google Scholar] [CrossRef]
- Dhaliwal, B.; Nichols, C.E.; Ren, J.; Lockyer, M.; Charles, I.; Hawkins, A.R.; Stammers, D.K. Crystallographic studies of shikimate binding and induced conformational changes in Mycobacterium tuberculosis shikimate kinase. FEBS Lett. 2004, 574, 49–54. [Google Scholar] [CrossRef]
- Simithy, J.; Reeve, N.; Hobrath, J.V.; Reynolds, R.C.; Calderon, A.I. Identification of shikimate kinase inhibitors among anti-Mycobacterium tuberculosis compounds by LC-MS. Tuberculosis 2014, 94, 152–158. [Google Scholar] [CrossRef]
- Krell, T.; Maclean, J.; Boam, D.J.; Cooper, A.; Resmini, M.; Brocklehurst, K.; Kelly, S.M.; Price, N.C.; Lapthorn, A.J.; Coggins, J.R. Biochemical and X-ray crystallographic studies on shikimate kinase: The important structural role of the P-loop lysine. Protein Sci. 2001, 10, 1137–1149. [Google Scholar] [CrossRef]
- Rosado, L.A.; Vasconcelos, I.B.; Palma, M.S.; Frappier, V.; Najmanovich, R.J.; Santos, D.S.; Basso, L.A. The mode of action of recombinant Mycobacterium tuberculosis shikimate kinase: Kinetics and thermodynamics analyses. PLoS ONE 2013, 8, e61918. [Google Scholar] [CrossRef]
- Prado, V.; Lence, E.; Vallejo, J.A.; Beceiro, A.; Thompson, P.; Hawkins, A.R.; Gonzalez-Bello, C. Study of the Phosphoryl-Transfer Mechanism of Shikimate Kinase by NMR Spectroscopy. Chem. Eur. J. 2016, 22, 2758–2768. [Google Scholar] [CrossRef]
- Simithy, J.; Fuanta, N.R.; Alturki, M.; Hobrath, J.V.; Wahba, A.E.; Pina, I.; Rath, J.; Hamann, M.T.; DeRuiter, J.; Goodwin, D.C.; et al. Slow-Binding Inhibition of Mycobacterium tuberculosis Shikimate Kinase by Manzamine Alkaloids. Biochemistry 2018, 57, 4923–4933. [Google Scholar] [CrossRef] [PubMed]
- Alturki, M.S.; Fuanta, N.R.; Jarrard, M.A.; Hobrath, J.V.; Goodwin, D.C.; Rants’o, T.A.; Calderón, A.I. A multifaceted approach to identify non-specific enzyme inhibition: Application to Mycobacterium tuberculosis shikimate kinase. Bioorg. Med. Chem. Lett. 2018, 28, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Rajput, V.S.; Mehra, R.; Kumar, S.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of antitubercular compound library identifies novel shikimate kinase inhibitors of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2016, 100, 5415–5426. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.S.; Mendes, M.A.; Palma, M.S.; Basso, L.A.; Santos, D.S. One-step purification of 5-enolpyruvylshikimate-3-phosphate synthase enzyme from Mycobacterium tuberculosis. Protein Expr. Purif. 2003, 28, 287–292. [Google Scholar] [CrossRef]
- Oliveira, J.S.; Pinto, C.A.; Basso, L.A.; Santos, D.S. Cloning and overexpression in soluble form of functional shikimate kinase and 5-enolpyruvylshikimate 3-phosphate synthase enzymes from Mycobacterium tuberculosis. Protein Expr. Purif. 2001, 22, 430–435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vaso, A.; Santos, D.S.; Basso, L.A.; Palma, M.S. Hydrogen/deuterium exchange mass spectrometry for characterizing phosphoenolpyruvate-induced structural transitions in Mycobacterium tuberculosis 5-enolpyruvylshikimate-3-phosphate synthase (EC 2.5.1.1). Int. J. Mass Spectrom. 2011, 302, 12–18. [Google Scholar] [CrossRef]
- Marques, M.R.; Pereira, J.H.; Oliveira, J.S.; Basso, L.A.; de Azevedo, W.F., Jr.; Santos, D.S.; Palma, M.S. The inhibition of 5-enolpyruvylshikimate-3-phosphate synthase as a model for development of novel antimicrobials. Curr. Drug Targets. 2007, 8, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.R.; Vaso, A.; Neto, J.R.; Fossey, M.A.; Oliveira, J.S.; Basso, L.A.; Santos, D.S.; Azevedo, W.F., Jr.; Palma, M.S. Dynamics of glyphosate-induced conformational changes of Mycobacterium tuberculosis 5-enolpyruvylshikimate-3-phosphate synthase (EC 2.5.1.19) determined by hydrogen-deuterium exchange and electrospray mass spectrometry. Biochemistry 2008, 47, 7509–7522. [Google Scholar] [CrossRef]
- Stallings, W.C.; Abdel-Meguid, S.S.; Lim, L.W.; Shieh, H.S.; Dayringer, H.E.; Leimgruber, N.K.; Kishore, G.M. Structure and topological symmetry of the glyphosate target 5-enolpyruvylshikimate-3-phosphate synthase: A distinctive protein fold. Proc. Natl. Acad. Sci. USA 1991, 88, 5046–5050. [Google Scholar] [CrossRef]
- Funke, T.; Healy-Fried, M.L.; Han, H.; Alberg, D.G.; Bartlett, P.A.; Schönbrunn, E. Differential inhibition of class I and class II 5-enolpyruvylshikimate-3-phosphate synthases by tetrahedral reaction intermediate analogues. Biochemistry 2007, 46, 13344–13351. [Google Scholar] [CrossRef]
- Clark, M.E.; Berti, P.J. Enolpyruvyl activation by enolpyruvylshikimate-3-phosphate synthase. Biochemistry 2007, 46, 1933–1940. [Google Scholar] [CrossRef]
- Lou, M.; Burger, S.K.; Gilpin, M.E.; Gawuga, V.; Capretta, A.; Berti, P.J. Transition state analysis of enolpyruvylshikimate 3-phosphate (EPSP) synthase (aroA)-catalyzed EPSP hydrolysis. J. Am. Chem. Soc. 2012, 134, 12958–12969. [Google Scholar] [CrossRef]
- Dos Santos, A.M.; Lima, A.H.; Alves, C.N.; Lameira, J. Unraveling the Addition-Elimination Mechanism of EPSP Synthase through Computer Modeling. J. Phys. Chem. B 2017, 121, 8626–8637. [Google Scholar] [CrossRef] [PubMed]
- Steinrücken, H.C.; Amrhein, N. The herbicide glyphosate is a potent inhibitor of 5-enolpyruvylshikimic acid-3-phosphate synthase. Biochem. Biophys. Res. Commun. 1980, 94, 1207–1212. [Google Scholar] [CrossRef]
- Sergiev, I.G.; Alexieva, V.S.; Ivanov, S.V.; Moskova, I.I.; Karanov, E.N. The phenylurea cytokinin 4PU-30 protects maize plants against glyphosate action. Pestic Biochem. Physiol. 2006, 85, 139–146. [Google Scholar] [CrossRef]
- Haghani, K.; Salmanian, A.H.; Ranjbar, B.; Zakikhan-Alang, K.; Khajeh, K. Comparative studies of wild type Escherichia coli 5-enolpyruvylshikimate 3-phosphate synthase with three glyphosate-insensitive mutated forms: Activity, stability and structural characterization. Biochim. Biophys. Acta. 2008, 1784, 1167–1175. [Google Scholar] [CrossRef]
- Ramachandran, V.; Singh, R.; Yang, X.; Tunduguru, R.; Mohapatra, S.; Khandelwal, S.; Datta, S. Genetic and chemical knockdown: A complementary strategy for evaluating an anti-infective target. Adv. Appl. Bioinform. Chem. 2013, 6, 1–13. [Google Scholar] [CrossRef]
- Borges, J.C.; Pereira, J.H.; Vasconcelos, I.B.; Santos, G.C.; Olivieri, J.R.; Ramos, C.H.I.; Palma, M.S.; Basso, L.A.; Santos, D.S.; Azevedo, W.F., Jr. Phosphate closes the solution structure of the 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) from Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2006, 452, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Timmers, L.F.S.M.; Neto, A.M.; Montalvão, R.W.; Basso, L.A.; Santos, D.S.; de Souza, O.N. EPSP synthase flexibility is determinant to its function: Computational molecular dynamics and metadynamics studies. J. Mol. Model. 2017, 23, 197. [Google Scholar] [CrossRef]
- Priestman, M.A.; Healy, M.L.; Becker, A.; Alberg, D.G.; Bartlett, P.A.; Lushington, G.H.; Schönbrunn, E. Interaction of phosphonate analogues of the tetrahedral reaction intermediate with 5-enolpyruvylshikimate-3-phosphate synthase in atomic detail. Biochemistry. 2005, 44, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.S.; Xu, J.; Han, J.; Zhao, W.; Fu, X.Y.; Peng, R.H.; Yao, Q.H. Complementary screening, identification and application of a novel class II 5-enopyruvylshikimate-3-phosphate synthase from Bacillus cereus. World J. Microbiol. Biotechnol. 2013, 29, 549–557. [Google Scholar] [CrossRef]
- Dias, M.V.; Ely, F.; Canduri, F.; Pereira, J.H.; Frazzon, J.; Basso, L.A.; Santos, D.S. Crystallization and preliminary X-ray crystallographic analysis of chorismate synthase from Mycobacterium tuberculosis. Acta Crystallogr. D: Biol. Crystallogr. 2004, 60, 2003–2005. [Google Scholar] [CrossRef] [PubMed]
- Ely, F.; Nunes, J.E.; Schroeder, E.K.; Frazzon, J.; Palma, M.S.; Santos, D.S.; Basso, L.A. The Mycobacterium tuberculosis Rv2540c DNA sequence encodes a bifunctional chorismate synthase. BMC Biochem. 2008, 9, 13. [Google Scholar] [CrossRef]
- Dias, M.V.; Borges, J.C.; Ely, F.; Pereira, J.H.; Canduri, F.; Ramos, C.H.; de Azevedo, W.F., Jr. Structure of chorismate synthase from Mycobacterium tuberculosis. J. Struct. Biol. 2006, 154, 130–143. [Google Scholar] [CrossRef]
- Arcuri, H.; Palma, M. Understanding the structure, activity and inhibition of chorismate synthase from Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1311–1317. [Google Scholar] [CrossRef]
- Dias, M.V.; Ely, F.; Palma, M.S.; Azevedo, W.F., Jr.; Basso, L.A.; Santos, D.S. Chorismate synthase: An attractive target for drug development against orphan diseases. Curr. Drug Targets. 2007, 8, 437–444. [Google Scholar] [CrossRef]
- Khera, H.K.; Singh, S.K.; Mir, R.; Bharadwaj, V.; Singh, S. A HR-MS based method for the determination of Chorismate Synthase activity. Protein Pep. Lett. 2017, 24, 229–234. [Google Scholar] [CrossRef]
- Lawan, N. QM-MM investigation on chorismate synthase enzyme role. Chiang Mai, J. Sci. 2014, 41, 618–626. [Google Scholar]
- Lawan, N.; Chasing, P.; Santatiwongchai, J.; Muangpil, S. QM/MM molecular modelling on mutation effect of chorismate synthase enzyme catalysis. J. Mol. Graph. Model. 2019, 87, 250–256. [Google Scholar] [CrossRef]
- Lawan, N.; Ranaghan, K.E.; Manby, F.R.; Mulholland, A.J. Comparison of DFT and ab initio QM/MM methods for modelling reaction in chorismate synthase. Chem. Phys. Lett. 2014, 608, 380–385. [Google Scholar] [CrossRef]
- Thomas, M.G.; Lawson, C.; Allanson, N.M.; Leslie, B.W.; Bottomley, J.R.; McBride, A.; Olusanya, O.A. A series of 2 (Z)-2-benzylidene-6, 7- dihydroxybenzofuran-3 [2H]-ones as inhibitors of chorismate synthase. Bioorg. Med. Chem. Lett. 2003, 13, 423–426. [Google Scholar] [CrossRef]
- Pitchandi, P.; Hopper, W.; Rao, R. Comprehensive database of chorismate synthase enzyme from shikimate pathway in pathogenic bacteria. BMC Pharmacol. Toxicol. 2013, 14, 29. [Google Scholar] [CrossRef]
- Fernandes, C.L.; Breda, A.; Santos, D.S.; Basso, L.A.; Souza, O.N. A structural model for chorismate synthase from Mycobacterium tuberculosis in complex with coenzyme and substrate. Comput. Biol. Med. 2007, 37, 149–158. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Madder, A.; Singh, I. Teixobactins: A new class of 21st century antibiotics to combat multidrug-resistant bacterial pathogens. Future Microbiol. 2019, 14, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Basaraba, R.J.; Ojha, A.K. Mycobacterial Biofilms: Revisiting Tuberculosis Bacilli in Extracellular Necrotizing Lesions. Microbiol. Spectr. 2017, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Flentie, K.; Harrison, G.A.; Tükenmez, H.; Livny, J.; Good, J.A.D.; Sarkar, S.; Zhu, D.X.; Kinsella, R.L.; Weiss, L.A.; Solomon, S.D.; et al. Chemical disarming of isoniazid resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10510–10517. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.; Bizarro, C.V.; Basso, L.A. Resistance Reversed in KatG Mutants of Mycobacterium tuberculosis. Trends Microbiol. 2019, 27, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Baindara, P. Host-directed therapies to combat tuberculosis and associated non-communicable diseases. Microb. Pathog. 2019, 130, 156–168. [Google Scholar] [CrossRef]
- Frank, D.J.; Horne, D.J.; Dutta, N.K.; Shaku, M.T.; Madensein, R.; Hawn, T.R.; Steyn, A.J.C.; Karakousis, P.C.; Kana, B.D.; Meintjes, G.; et al. Remembering the host in tuberculosis drug development. J. Infect. Dis. 2019, 219, 1518–1524. [Google Scholar] [CrossRef]
- Johnson, E.O.; LaVerriere, E.; Office, E.; Stanley, M.; Meyer, E.; Kawate, T.; Gomez, J.E.; Audette, R.E.; Bandyopadhyay, N.; Betancourt, N.; et al. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature 2019, 571, 72–78. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, J.E.S.; Duque, M.A.; de Freitas, T.F.; Galina, L.; Timmers, L.F.S.M.; Bizarro, C.V.; Machado, P.; Basso, L.A.; Ducati, R.G. Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules 2020, 25, 1259. https://doi.org/10.3390/molecules25061259
Nunes JES, Duque MA, de Freitas TF, Galina L, Timmers LFSM, Bizarro CV, Machado P, Basso LA, Ducati RG. Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules. 2020; 25(6):1259. https://doi.org/10.3390/molecules25061259
Chicago/Turabian StyleNunes, José E. S., Mario A. Duque, Talita F. de Freitas, Luiza Galina, Luis F. S. M. Timmers, Cristiano V. Bizarro, Pablo Machado, Luiz A. Basso, and Rodrigo G. Ducati. 2020. "Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs" Molecules 25, no. 6: 1259. https://doi.org/10.3390/molecules25061259
APA StyleNunes, J. E. S., Duque, M. A., de Freitas, T. F., Galina, L., Timmers, L. F. S. M., Bizarro, C. V., Machado, P., Basso, L. A., & Ducati, R. G. (2020). Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules, 25(6), 1259. https://doi.org/10.3390/molecules25061259