
Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
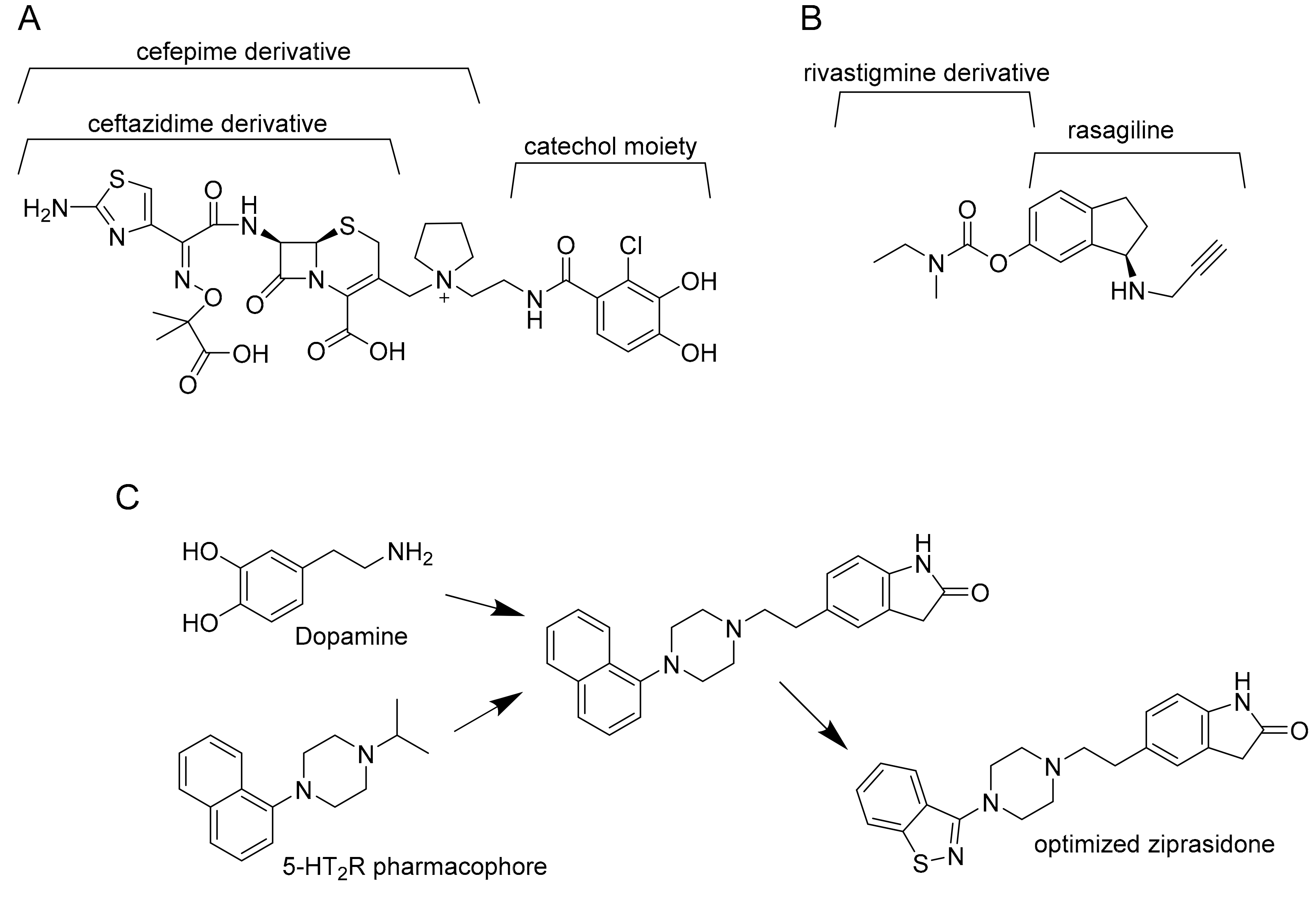
2. How Polypharmacology Can Help in Fighting MDR-TB?
3. Multitargeting Compounds Against M. tuberculosis
3.1. MmpL3 Inhibitors
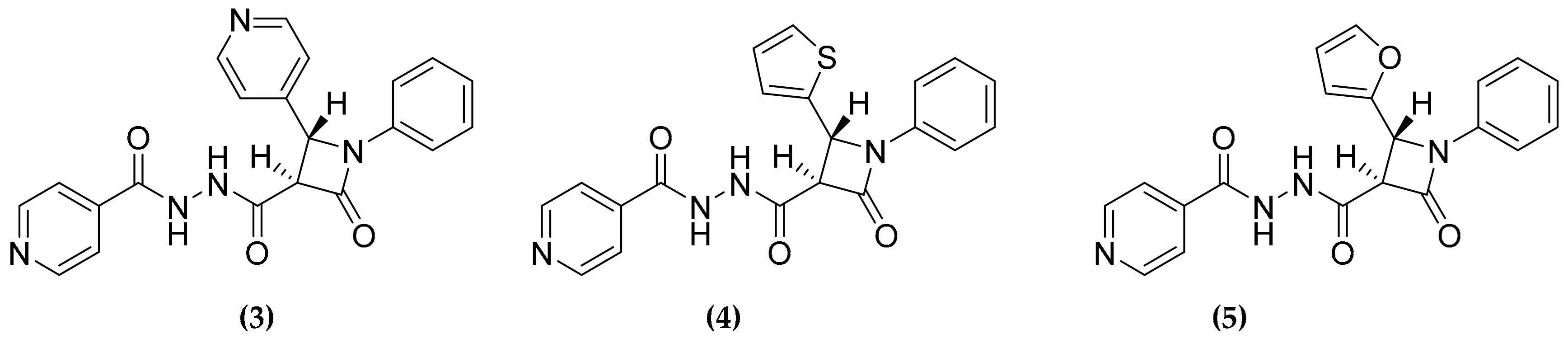
3.2. Designed Dual Inhibitors Against Fatty Acids Bypass Biosynthetic Pathways
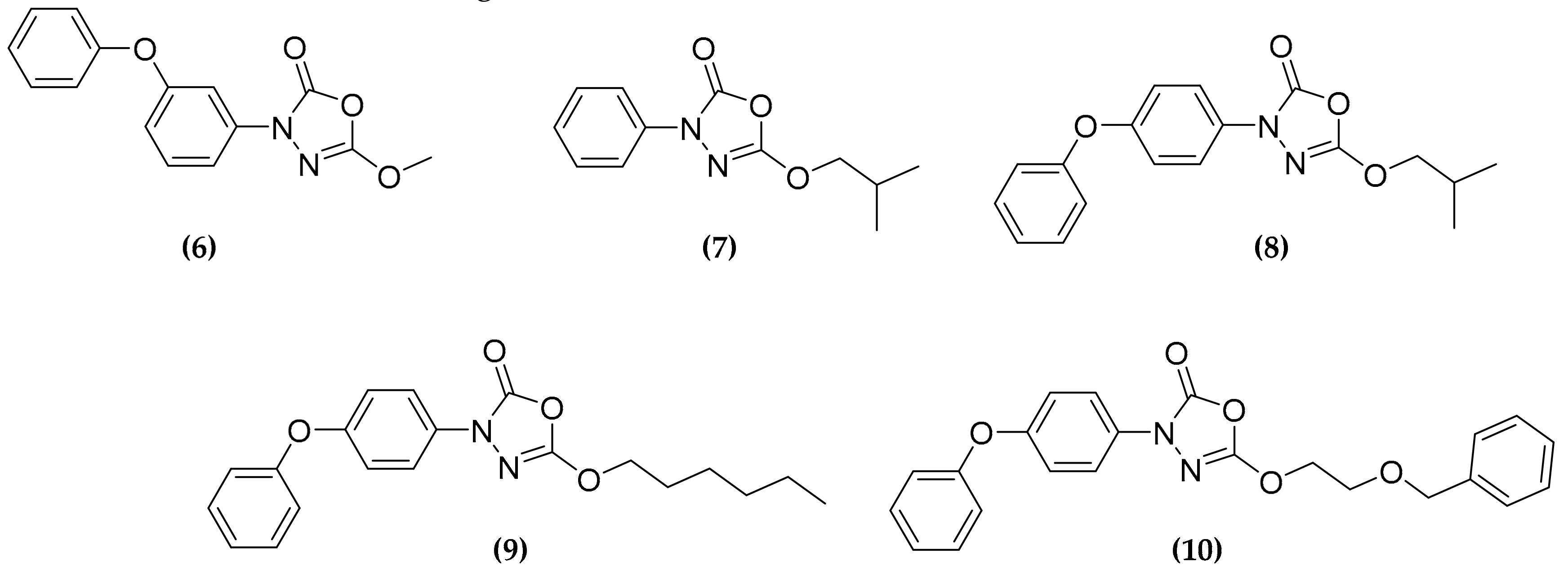
3.3. Oxadiazolone Derivatives Targeting (Ser/Cys)-Enzymes
3.4. CTP and CoA Biosynthesis Inhibition
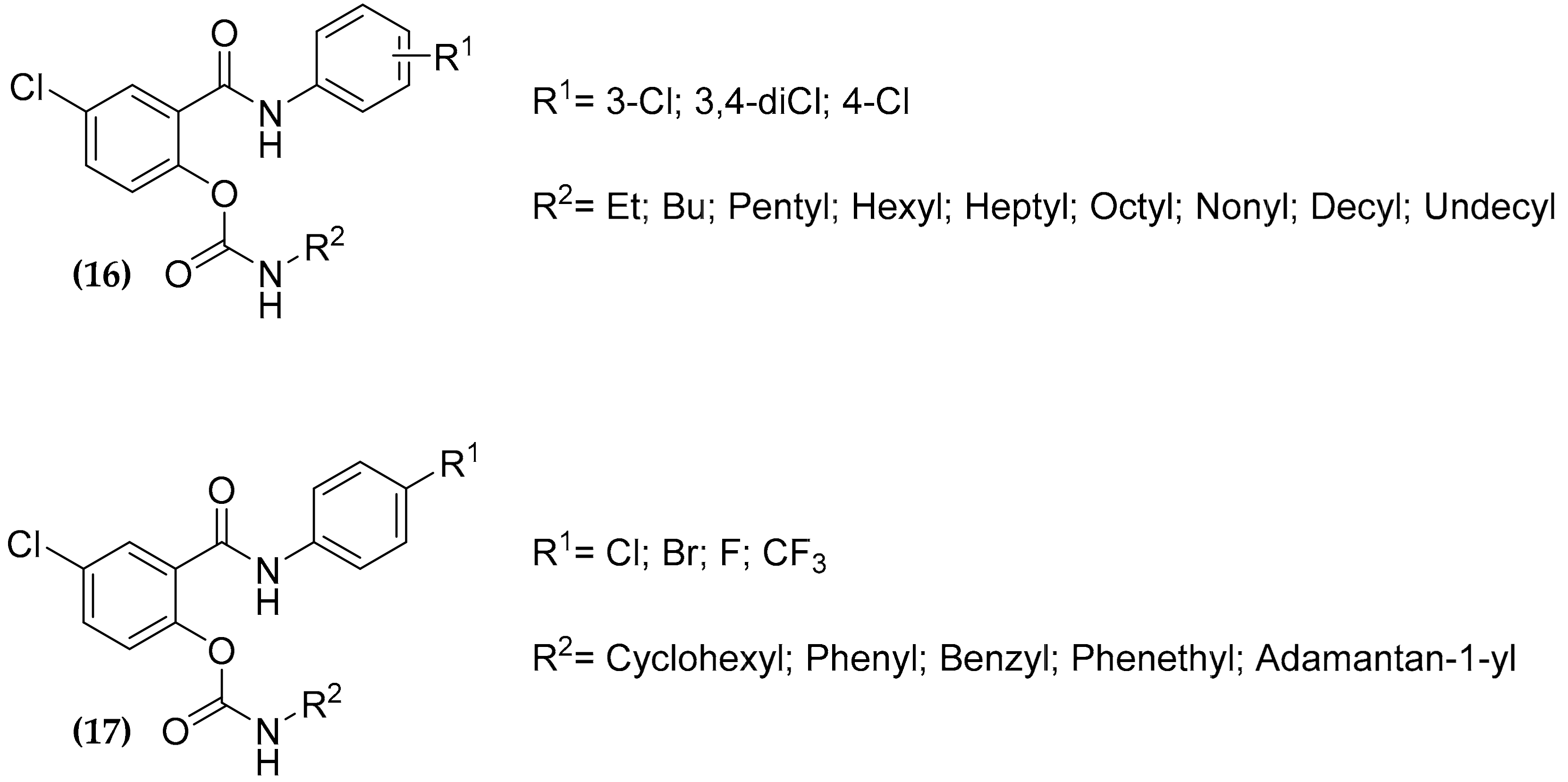
3.5. Salicylanilide Carbamate Compounds
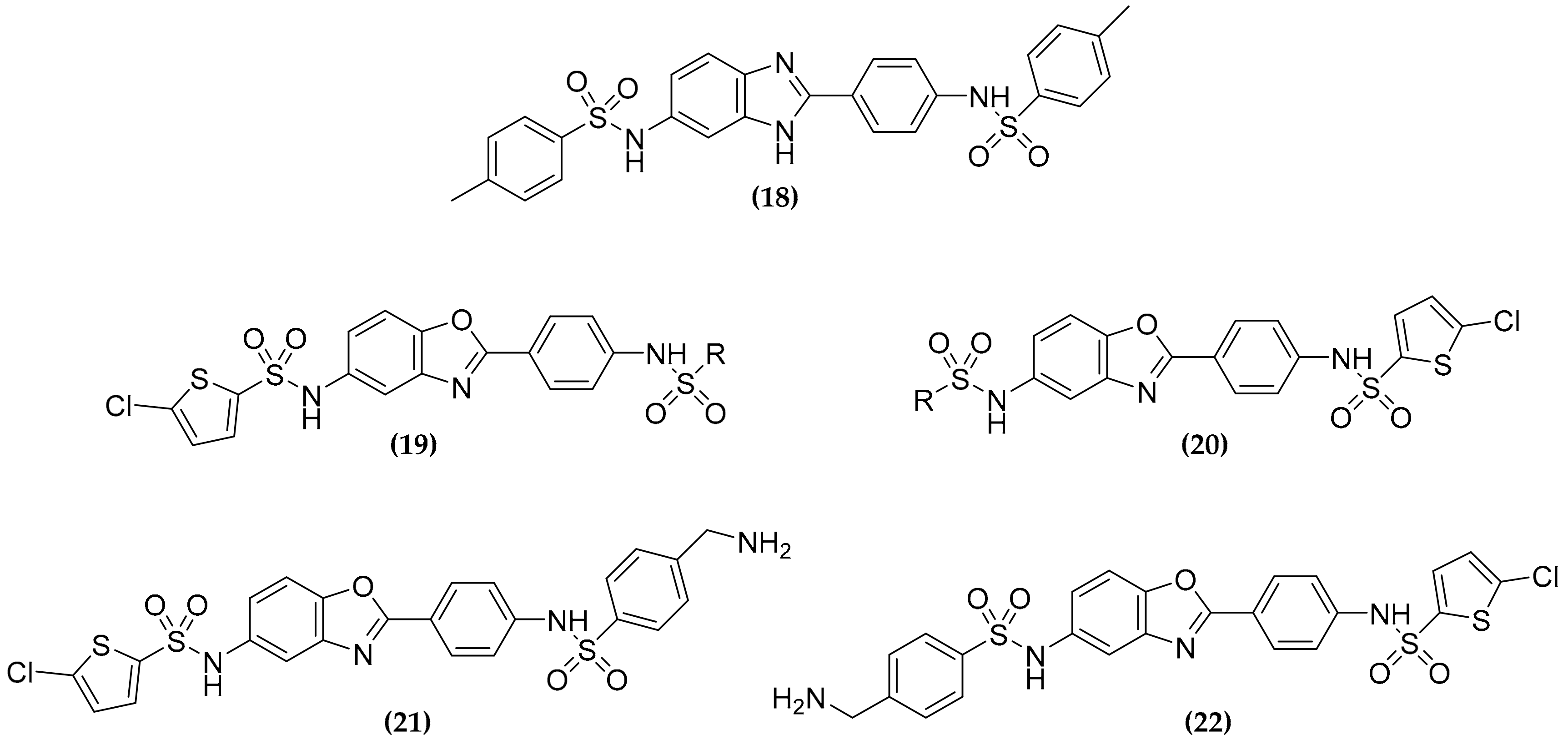
3.6. Dualtargeting GroEL/ES Chaperonin and Protein Tyrosine Phosphatase B
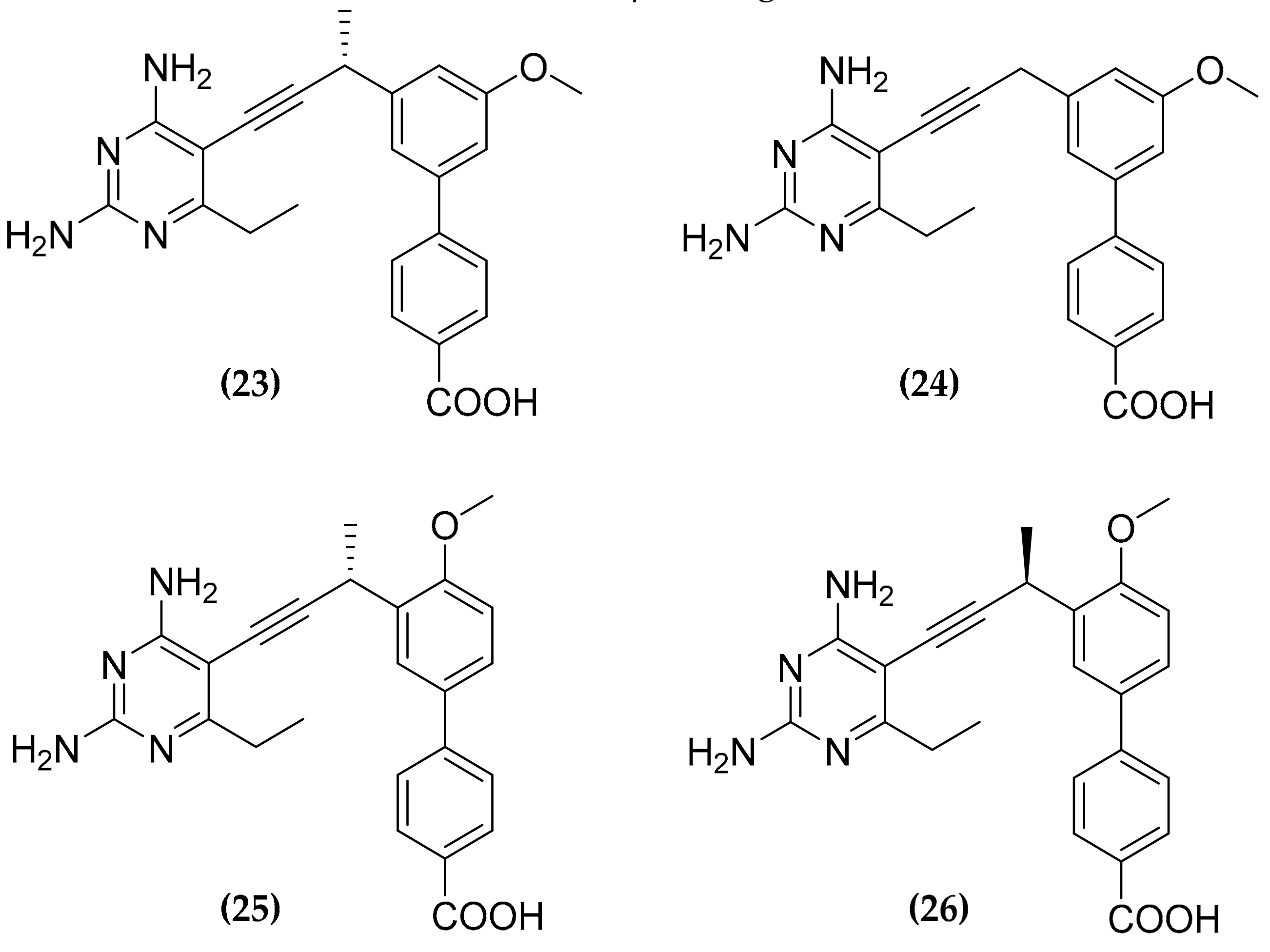
3.7. Multitargeting of the Folate Pathway
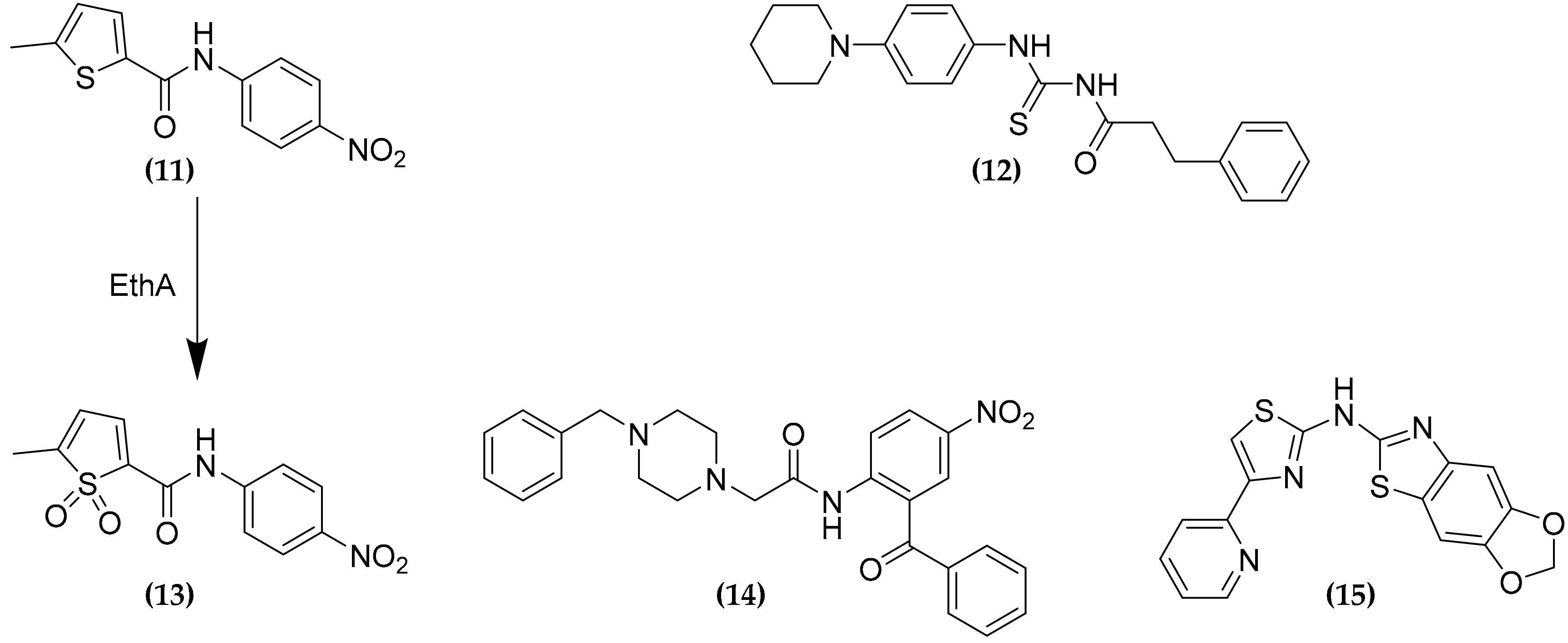
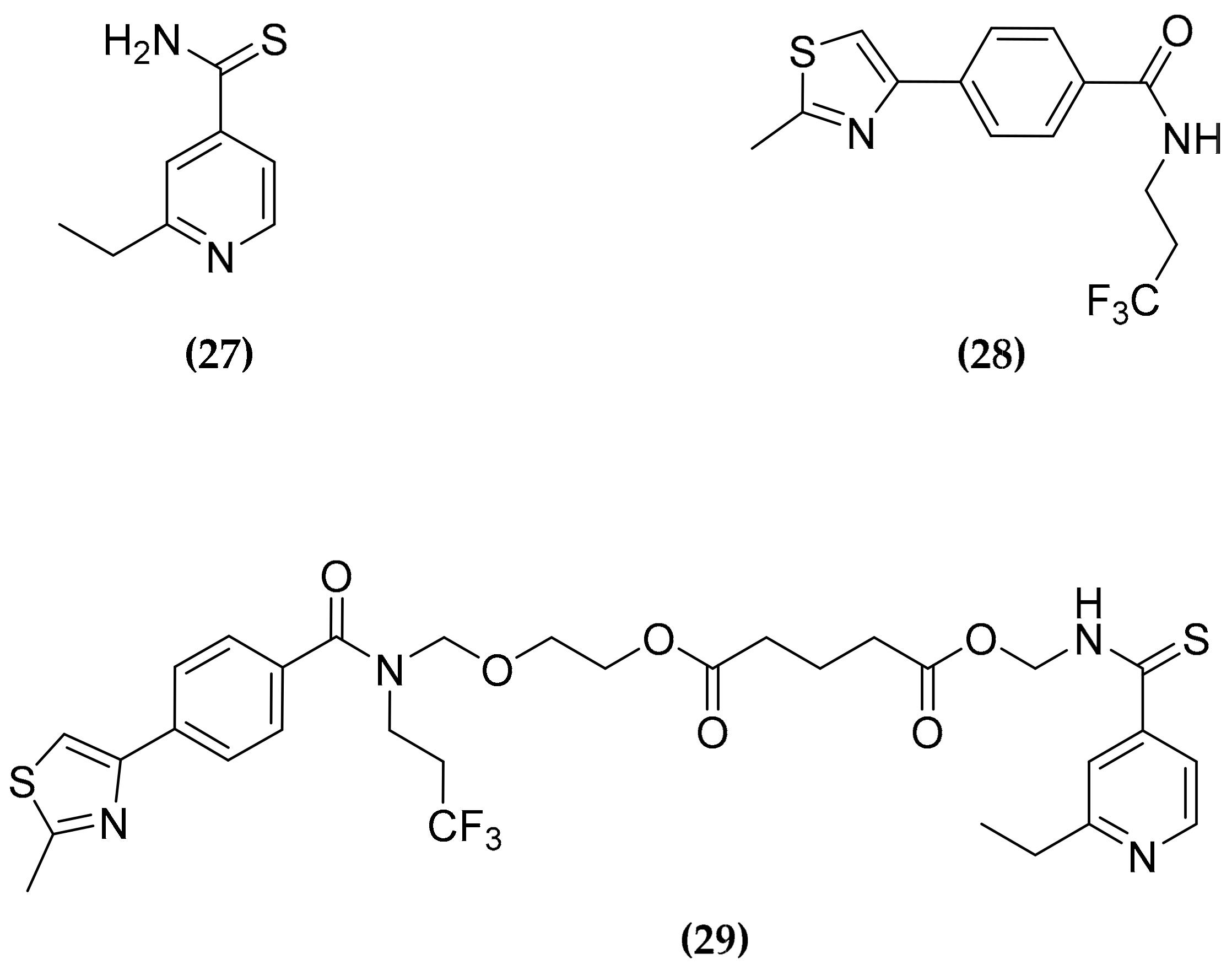
3.8. Ethionamide and Ethionamide Booster Co-Administration
3.9. In Silico Approaches for Multitargeting Compounds Development
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global tuberculosis report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 13 February 2020).
- Merker, M.; Kohl, T.A.; Niemann, S.; Supply, P. The evolution of strain typing in the Mycobacterium tuberculosis complex. Adv Exp Med. Biol. 2017, 1019, 43–78. [Google Scholar]
- Beites, T.; O’Brien, K.; Tiwari, D.; Engelhart, C.A.; Walters, S.; Andrews, J.; Yang, H.J.; Sutphen, M.L.; Weiner, D.M.; Dayao, E.K.; et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat. Commun. 2019, 10, 4970. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, J.P. Screening migrants for tuberculosis and latent TB infection: The reward will come later. Eur. Respir. J. 2019, 54, 1901719. [Google Scholar] [CrossRef] [PubMed]
- Huaman, M.A.; Sterling, T.R. Treatment of latent tuberculosis infection-an update. Clin Chest Med. 2019, 40, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Cegielski, J.P.; Kurbatova, E.; van der Walt, M.; Brand, J.; Ershova, J.; Tupasi, T.; Caoili, J.C.; Dalton, T.; Contreras, C.; Yagui, M.; et al. Multidrug-resistant tuberculosis treatment outcomes in relation to treatment and initial versus acquired second-line drug resistance. Clin. Infect. Dis. 2016, 62, 418–430. [Google Scholar] [CrossRef]
- World Health Organization. Consolidated guidelines on drug-resistant tuberculosis treatment of 2019. Available online: https://www.who.int/tb/publications/2019/consolidated-guidelines-drug-resistant-TB-treatment/en/ (accessed on 25 February 2020).
- World Health Organization. Treatment guidelines for rifampicin and multidrug-resistant tuberculosis, 2018 update. Available online: https://www.who.int/tb/areas-of-work/drug-resistant-tb/treatment/gdg-meeting-mdr-rr-tb-treatment-2018-update/en/ (accessed on 25 February 2020).
- Adejumo, O.A.; Olusola-Faleye, B.; Adepoju, V.; Bowale, A.; Adesola, S.; Falana, A.; Owuna, H.; Otemuyiwa, K.; Oladega, S.; Adegboye, O. Prevalence of rifampicin resistant tuberculosis and associated factors among presumptive tuberculosis patients in a secondary referral hospital in Lagos Nigeria. Afr. Health Sci. 2018, 18, 472–478. [Google Scholar] [CrossRef]
- Nguyen, H.B.; Nguyen, N.V.; Tran, H.T.; Nguyen, H.V.; Bui, Q.T. Prevalence of resistance to second-line tuberculosis drug among multidrug-resistant tuberculosis patients in VietNam, 2011. Western Pac. Surveill Resp. J. 2016, 7, 35–40. [Google Scholar] [CrossRef]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef]
- Anand, P.; Chandra, N. Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection. Sci. Rep. 2014, 4, 6356. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the "skeleton key approach" from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- East, S.P.; Silver, L.L. Multitarget ligands in antibacterial research: Progress and opportunities. Expert. Opin. Drug Discov. 2013, 8, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Petrelli, A.; Valabrega, G. Multitarget drugs: The present and the future of cancer therapy. Exp. Opin. Pharmaco. 2009, 10, 589–600. [Google Scholar] [CrossRef]
- Shahbazian, D.; Sznol, J.; Kluger, H.M. Vertical pathway targeting in cancer therapy. Adv. Pharmacol. 2012, 65, 1–26. [Google Scholar]
- Merk, D.; Schubert-Zsilavecz, M. The Linker Approach. In Drug Selectivity: An Evolving Concept in Medicinal Chemistry; Handler, N., Buschmann, H., Mannhold, R., Holenz, J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2017. [Google Scholar]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef]
- Jean, S.S.; Hsueh, S.C.; Lee, W.S.; Hsueh, P.R. Cefiderocol: A promising antibiotic against multidrug-resistant Gram-negative bacteria. Exp. Rev. Anti-Infect Ther. 2019, 17, 307–309. [Google Scholar] [CrossRef]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; et al. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Viana, J.; Ishiki, H.M.; Scotti, M.T.; Scotti, L. Multi-Target Antitubercular Drugs. Curr. Top. Med. Chem. 2018, 18, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Khambete, M.; Kundaikar, H.; Raju, A.; Lonkar, S.; Degani, M.; Ray, M.K. Design and synthesis of 5-(5-nitrothiophen-2-yl)-3-phenyl-4,5-dihydro-1H-pyrazole derivatives with improved solubility and potential antituberculosis activity. Chem. Biol. Drug Des. 2019, 93, 84–88. [Google Scholar] [CrossRef]
- Agre, N.; Khambete, M.; Maitra, A.; Gupta, A.; Munshi, T.; Bhakta, S.; Degani, M. Exploration of 5-(5-nitrothiophen-2-yl)-4,5- dihydro-1H-pyrazoles as selective, multitargeted antimycobacterial agents. Chem. Biol. Drug Des. 2020, 95, 192–199. [Google Scholar] [CrossRef]
- Guzman, J.D.; Pesnot, T.; Barrera, D.A.; Davies, H.M.; McMahon, E.; Evangelopoulos, D.; Mortazavi, P.N.; Munshi, T.; Maitra, A.; Lamming, E.D.; et al. Tetrahydroisoquinolines affect the whole-cell phenotype of Mycobacterium tuberculosis by inhibiting the ATP-dependent MurE ligase. J. Antimicrob. Chemother. 2015, 70, 1691–1703. [Google Scholar] [CrossRef]
- Washburn, A.; Abdeen, S.; Ovechkina, Y.; Ray, A.M.; Stevens, M.; Chitre, S.; Sivinski, J.; Park, Y.; Johnson, J.; Hoang, Q.Q.; et al. Dual-targeting GroEL/ES chaperonin and protein tyrosine phosphatase B (PtpB) inhibitors: A polypharmacology strategy for treating Mycobacterium tuberculosis infections. Bioorg. Med. Chem. Lett. 2019, 29, 1665–1672. [Google Scholar] [CrossRef]
- Banerjee, D.R.; Biswas, R.; Das, A.K.; Basak, A. Design, synthesis and characterization of dual inhibitors against new targets FabG4 and HtdX of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2015, 100, 223–234. [Google Scholar] [CrossRef]
- Degiacomi, G.; Belardinelli, J.M.; Pasca, M.R.; De Rossi, E.; Riccardi, G.; Chiarelli, L.R. Promiscuous targets for antitubercular drug discovery: The paradigm of DprE1 and MmpL3. Appl. Sci. 2020, 10, 623. [Google Scholar] [CrossRef]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef]
- Working Group on New TB Drugs. Available online: www.newtbdrugs.org/pipeline/compound/sq109 (accessed on 13 February 2020).
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Fut. Microbio. 2012, 7, 823–837. [Google Scholar] [CrossRef]
- Li, W.; Upadhyay, A.; Fontes, F.L.; North, E.J.; Wang, Y.; Crans, D.C.; Grzegorzewicz, A.E.; Jones, V.; Franzblau, S.G.; Lee, R.E.; et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Stevens, C.M.; Pandya, A.N.; Darzynkiewicz, Z.; Bhattarai, P.; Tong, W.; Gonzalez-Juarrero, M.; North, E.J.; Zgurskaya, H.I.; Jackson, M. Direct inhibition of MmpL3 by novel antitubercular compounds. ACS Infect. Dis. 2019, 5, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Cheng, X.; Liu, Z.; et al. Crystal structures of membrane transporter MmpL3, an anti-TB drug target. Cell 2019, 176, 636–648. [Google Scholar] [CrossRef]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbio. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Myers, T.G.; Copp, B.R.; McNeil, M.R.; Wilson, M.A.; Barry, C.E. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: Novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 279, 40174–40184. [Google Scholar] [CrossRef]
- Kikuchi, S.; Kusaka, T. New malonyl-CoA-dependent fatty acid elongation system in Mycobacterium smegmatis. J. Biochem. 1982, 92, 839–844. [Google Scholar] [CrossRef]
- Zampieri, D.; Mamolo, M.G.; Laurini, E.; Fermeglia, M.; Posocco, P.; Pricl, S.; Banfi, E.; Scialino, G.; Vio, L. Antimycobacterial activity of new 3,5-disubstituted 1,3,4-oxadiazol-2(3H)-one derivatives. Molecular modeling investigations. Bioorg. Med. Chem. 2009, 17, 4693–4707. [Google Scholar] [CrossRef]
- Mamolo, M.G.; Zampieri, D.; Vio, L.; Fermeglia, M.; Ferrone, M.; Pricl, S.; Scialino, G.; Banfi, E. Antimycobacterial activity of new 3-substituted 5-(pyridin-4-yl)-3H-1,3,4-oxadiazol-2-one and 2-thione derivatives. Preliminary molecular modeling investigations. Bioorg. Med. Chem. 2005, 13, 3797–3809. [Google Scholar] [CrossRef]
- Bénarouche, A.; Point, V.; Carrière, F.; Cavalier, J.F. Using the reversible inhibition of gastric lipase by Orlistat for investigating simultaneously lipase adsorption and substrate hydrolysis at the lipid-water interface. Biochimie 2014, 101, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Dedieu, L.; Serveau-Avesque, C.; Kremer, L.; Canaan, S. Mycobacterial lipolytic enzymes: A gold mine for tuberculosis research. Biochimie 2013, 95, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.C.; Delorme, V.; Bénarouche, A.; Guy, A.; Landry, V.; Audebert, S.; Pophillat, M.; Camoin, L.; Crauste, C.; Galano, J.M.; et al. Oxadiazolone derivatives, new promising multi-target inhibitors against M. tuberculosis. Bioorg. Chem. 2018, 81, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.; Chiarelli, L.R.; Esposito, M.; Makarov, V.; Bellinzoni, M.; Hartkoorn, R.C.; Degiacomi, G.; Boldrin, F.; Ekins, S.; De jesus lopes ribeiro, A.L.; et al. Thiophenecarboxamidederivatives activated by EthA kill Mycobacterium tuberculosis by inhibiting the CTP synthetase PyrG. Chem. Biol. 2015, 22, 917–927. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, G.; Orena, B.S.; Esposito, M.; Lane, T.; de Jesus Lopes Ribeiro, A.L.; Degiacomi, G.; Zemanova, J.; Szadocka, S.; Huszar, S.; et al. A multitarget approach to drug discovery inhibiting Mycobacterium tuberculosis PyrG and PanK. Sci. Rep. 2018, 8, 3187. [Google Scholar] [CrossRef]
- Esposito, M.; Szadocka, S.; Degiacomi, G.; Orena, B.S.; Mori, G.; Piano, V.; Boldrin, F.; Zemanová, J.; Huszár, S.; Barros, D.; et al. A phenotypic based target screening approach delivers new antitubercular CTP synthetase inhibitors. ACS Infect. Dis. 2017, 3, 428–437. [Google Scholar] [CrossRef]
- Reddy, B.K.; Landge, S.; Ravishankar, S.; Patil, V.; Shinde, V.; Tantry, S.; Kale, M.; Raichurkar, A.; Menasinakai, S.; Mudugal, N.V.; et al. Assessment of Mycobacterium tuberculosis pantothenate kinase vulnerability through target knockdown and mechanistically diverse inhibitors. Antimicrob. Agents Chemother. 2014, 58, 3312–3326. [Google Scholar] [CrossRef]
- Triola, G.; Wetzel, S.; Ellinger, B.; Koch, M.A.; Hübel, K.; Rauh, D.; Waldmann, H. ATP competitive inhibitors of D-alanine-D-alanine ligase based on protein kinase inhibitor scaffolds. Bioorg. Med. Chem. 2009, 17, 1079–1087. [Google Scholar] [CrossRef]
- Cheng, T.J.; Wu, Y.T.; Yang, S.T.; Lo, K.H.; Chen, S.K.; Chen, Y.H.; Huang, W.I.; Yuan, C.H.; Guo, C.W.; Huang, L.Y.; et al. High-throughput identification of antibacterials against methicillin-resistant Staphylococcus aureus (MRSA) and the transglycosylase. Bioorg. Med. Chem. 2010, 18, 8512–8529. [Google Scholar] [CrossRef]
- Wu, W.S.; Cheng, W.C.; Cheng, T.R.; Wong, C.H. Affinity-Based Screen for Inhibitors of Bacterial Transglycosylase. J. Am. Chem. Soc. 2018, 140, 2752–2755. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Wsól, V.; Trejtnar, F.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; et al. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis (Edinb) 2012, 92, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Novotná, E.; Saxena, S.; Yogeeswari, P.; Sriram, D.; Švarcová, M.; Vinšová, J. Salicylanilide diethyl phosphates as potential inhibitors of some mycobacterial enzymes. Sci. World J. 2014, 2014, 703053. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.Y.; Gruber, T.D.; Samuels, A.; Yun, M.; Nam, B.; Kang, M.; Crowley, K.; Winterroth, B.; Boshoff, H.I.; Barry, C.E. Structure-activity relationships of antitubercular salicylanilides consistent with disruption of the proton gradient via proton shuttling. Bioorg. Med. Chem. 2013, 21, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Janďourek, O.; Baranyai, Z.; Novotná, E.; Stolaříková, J.; Bősze, S.; Vinšová, J. Phenolic N-monosubstituted carbamates: Antitubercular and toxicity evaluation of multi-targeting compounds. Eur. J. Med. Chem. 2019, 181, 111578. [Google Scholar] [CrossRef]
- Thorberg, S.O.; Berg, S.; Lundström, J.; Pettersson, B.; Wijkström, A.; Sanchez, D.; Lindberg, P.; Nilsson, J.L. Carbamate ester derivatives as potential prodrugs of the presynaptic dopamine autoreceptor agonist (-)-3-(3-hydroxyphenyl)-N-propylpiperidine. J. Med. Chem. 1987, 30, 2008–2012. [Google Scholar] [CrossRef]
- Férriz, J.M.; Vávrová, K.; Kunc, F.; Imramovský, A.; Stolaríková, J.; Vavríková, E.; Vinsová, J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010, 18, 1054–1061. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Salicylanilide N-monosubstituted carbamates: Synthesis and in vitro antimicrobial activity. Bioorg. Med. Chem. 2016, 24, 1322–1330. [Google Scholar] [CrossRef]
- Baranyai, Z.; Krátký, M.; Vinšová, J.; Szabó, N.; Senoner, Z.; Horváti, K.; Stolaříková, J.; Dávid, S.; Bősze, S. Combating highly resistant emerging pathogen Mycobacterium abscessus and Mycobacterium tuberculosis with novel salicylanilide esters and carbamates. Eur. J. Med. Chem. 2015, 101, 692–704. [Google Scholar] [CrossRef]
- Krátký, M.; Volková, M.; Novotná, E.; Trejtnar, F.; Stolaříková, J.; Vinšová, J. Synthesis and biological activity of new salicylanilide N,N-disubstituted carbamates and thiocarbamates. Bioorg. Med. Chem. 2014, 22, 4073–4082. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012, 19, 6126–6137. [Google Scholar] [CrossRef]
- Lupoli, T.J.; Vaubourgeix, J.; Burns-Huang, K.; Gold, B. Targeting the proteostasis network for mycobacterial drug discovery. ACS Infect. Dis. 2018, 4, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Henderson, B.; Lund, P.A.; Tormay, P.; Ahmed, M.T.; Gurcha, S.S.; Besra, G.S.; Coates, A.R. A Mycobacterium tuberculosis mutant lacking the groEL homologue cpn60.1 is viable but fails to induce an inflammatory response in animal models of infection. Infect. Immun. 2008, 76, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Sharif, O.; Mak, P.A.; Wang, H.T.; Engels, I.H.; Brinker, A.; Schultz, P.G.; Horwich, A.L.; Chapman, E. A biochemical screen for GroEL/GroES inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 786–789. [Google Scholar] [CrossRef]
- Abdeen, S.; Salim, N.; Mammadova, N.; Summers, C.M.; Frankson, R.; Ambrose, A.J.; Anderson, G.G.; Schultz, P.G.; Horwich, A.L.; Chapman, E.; et al. GroEL/ES inhibitors as potential antibiotics. Bioorg. Med. Chem. Lett. 2016, 26, 3127–3134. [Google Scholar] [CrossRef]
- Abdeen, S.; Salim, N.; Mammadova, N.; Summers, C.M.; Goldsmith-Pestana, K.; McMahon-Pratt, D.; Schultz, P.G.; Horwich, A.L.; Chapman, E.; Johnson, S.M. Targeting the HSP60/10 chaperonin systems of Trypanosoma brucei as a strategy for treating African sleeping sickness. Bioorg. Med. Chem. Lett. 2016, 26, 5247–5253. [Google Scholar] [CrossRef]
- Abdeen, S.; Kunkle, T.; Salim, N.; Ray, A.M.; Mammadova, N.; Summers, C.; Stevens, M.; Ambrose, A.J.; Park, Y.; Schultz, P.G.; et al. Sulfonamido-2-arylbenzoxazole GroEL/ES inhibitors as potent antibacterials against Methicillin-Resistant Staphylococcus aureus (MRSA). J. Med. Chem. 2018, 61, 7345–7357. [Google Scholar] [CrossRef]
- Fanzani, L.; Porta, F.; Meneghetti, F.; Villa, S.; Gelain, A.; Lucarelli, A.P.; Parisini, E. Mycobacterium tuberculosis low molecular weight phosphatases (MPtpA and MPtpB): From biological insight to inhibitors. Curr. Med. Chem. 2015, 22, 3110–3132. [Google Scholar] [CrossRef]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. North. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef]
- Nixon, M.R.; Saionz, K.W.; Koo, M.S.; Szymonifka, M.J.; Jung, H.; Roberts, J.P.; Nandakumar, M.; Kumar, A.; Liao, R.; Rustad, T.; et al. Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis. Chem. Biol. 2014, 21, 819–830. [Google Scholar] [CrossRef]
- Keshipeddy, S.; Reeve, S.M.; Anderson, A.C.; Wright, D.L. Nonracemic antifolates stereoselectively recruit alternate cofactors and overcome resistance in S. aureus. J. Am. Chem. Soc. 2015, 137, 8983–8990. [Google Scholar] [CrossRef]
- Hajian, B.; Scocchera, E.; Keshipeddy, S.; G-Dayanandan, N.; Shoen, C.; Krucinska, J.; Reeve, S.; Cynamon, M.; Anderson, A.C.; Wright, D.L. Propargyl-linked antifolates are potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0161740. [Google Scholar] [CrossRef] [PubMed]
- Hajian, B.; Scocchera, E.; Shoen, C.; Krucinska, J.; Viswanathan, K.; G-Dayanandan, N.; Erlandsen, H.; Estrada, A.; Mikušová, K.; Korduláková, J.; et al. Drugging the Folate Pathway in Mycobacterium tuberculosis: The Role of Multi-targeting Agents. Cell Chem. Biol. 2019, 26, 781–791. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Sacchettini, J.C. Structural insights into Mycobacterium tuberculosis Rv2671 Protein as a Dihydrofolate Reductase Functional Analogue Contributing to para-Aminosalicylic Acid Resistance. Biochemistry 2016, 55, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Yu, L.; Karunaratne, K.; Mondal, D.; Corcoran, J.M.; Choi, M.A.; Kohen, A. An unprecedented mechanism of nucleotide methylation in organisms containing thyX. Science 2016, 351, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.; Chiarelli, L.R.; Riccardi, G.; Pasca, M.R. New prodrugs against tuberculosis. Drug Discov. Today 2017, 22, 519–525. [Google Scholar] [CrossRef]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.G.; Barry, C.E. 3rd. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9682. [Google Scholar] [CrossRef]
- Willand, N.; Dirié, B.; Carette, X.; Bifani, P.; Singhal, A.; Desroses, M.; Leroux, F.; Willery, E.; Mathys, V.; Déprez-Poulain, R.; et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 2009, 15, 537–544. [Google Scholar] [CrossRef]
- Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Dirié, B.; Carette, X.; Leroux, F.; Piveteau, C.; Demirkaya, F.; Lens, Z.; Rucktooa, P.; et al. Ethionamide boosters: Synthesis, biological activity, and structure-activity relationships of a series of 1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 2011, 54, 2994–3010. [Google Scholar] [CrossRef]
- Villemagne, B.; Flipo, M.; Blondiaux, N.; Crauste, C.; Malaquin, S.; Leroux, F.; Piveteau, C.; Villeret, V.; Brodin, P.; Villoutreix, B.O.; et al. Ligand efficiency driven design of new inhibitors of Mycobacterium tuberculosis transcriptional repressor EthR using fragment growing, merging, and linking approaches. J. Med. Chem. 2014, 57, 4876–4888. [Google Scholar] [CrossRef]
- Costa-Gouveia, J.; Pancani, E.; Jouny, S.; Machelart, A.; Delorme, V.; Salzano, G.; Iantomasi, R.; Piveteau, C.; Queval, C.J.; Song, O.; et al. Combination therapy for tuberculosis treatment: Pulmonary administration of ethionamide and booster co-loaded nanoparticles. Sci. Rep. 2017, 7, 5390. [Google Scholar] [CrossRef]
- Pastor, A.; Machelart, A.; Li, X.; Willand, N.; Baulard, A.R.; Brodin, P.; Gref, R.; Desmaële, D. A novel codrug made of the combination of ethionamide and its potentiating booster: Synthesis, self-assembly into nanoparticles and antimycobacterial evaluation. Org. Biomol. Chem. 2019, 17, 5129–5137. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, S.; John, L.; Prasanthi, M.; Poroikov, V.; Narahari Sastry, G. A QSAR and molecular modelling study towards new lead finding: Polypharmacological approach to Mycobacterium tuberculosis. SAR QSAR Environ. Res. 2017, 28, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Volynets, G.P.; Starosyla, S.A.; Rybak, M.Y.; Bdzhola, V.G.; Kovalenko, O.P.; Vdovin, V.S.; Yarmoluk, S.M.; Tukalo, M.A. Dual-targeted hit identifcation using pharmacophore screening. J. Computer-Aided Mol. Des. 2019, 33, 955–964. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stelitano, G.; Sammartino, J.C.; Chiarelli, L.R. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules 2020, 25, 1239. https://doi.org/10.3390/molecules25051239
Stelitano G, Sammartino JC, Chiarelli LR. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules. 2020; 25(5):1239. https://doi.org/10.3390/molecules25051239
Chicago/Turabian StyleStelitano, Giovanni, José Camilla Sammartino, and Laurent Roberto Chiarelli. 2020. "Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis" Molecules 25, no. 5: 1239. https://doi.org/10.3390/molecules25051239
APA StyleStelitano, G., Sammartino, J. C., & Chiarelli, L. R. (2020). Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules, 25(5), 1239. https://doi.org/10.3390/molecules25051239