
Glycosaminoglycan-Inspired Biomaterials for the Development of Bioactive Hydrogel Networks

 ,
,  and
and 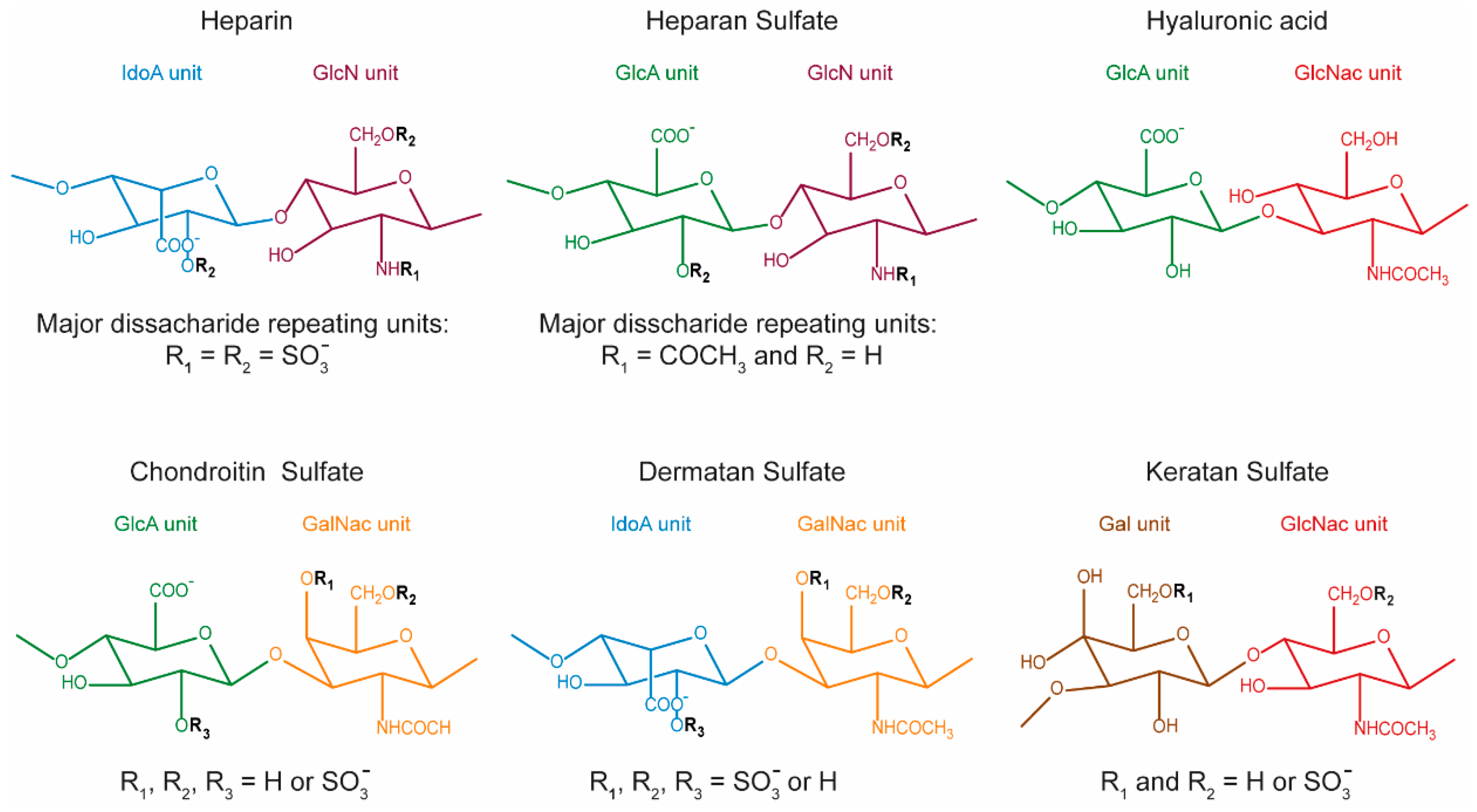{kind=link}
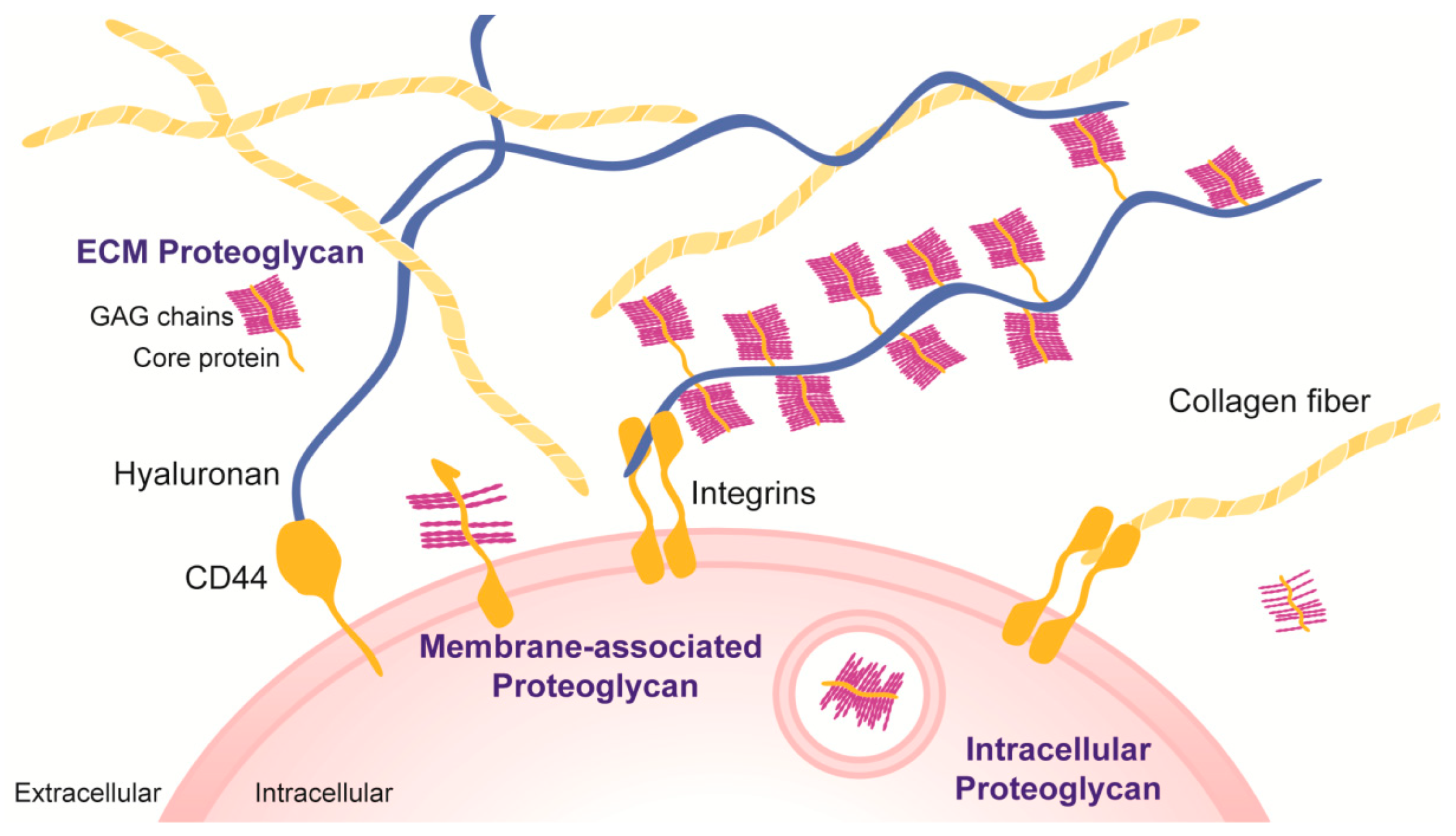{kind=link}
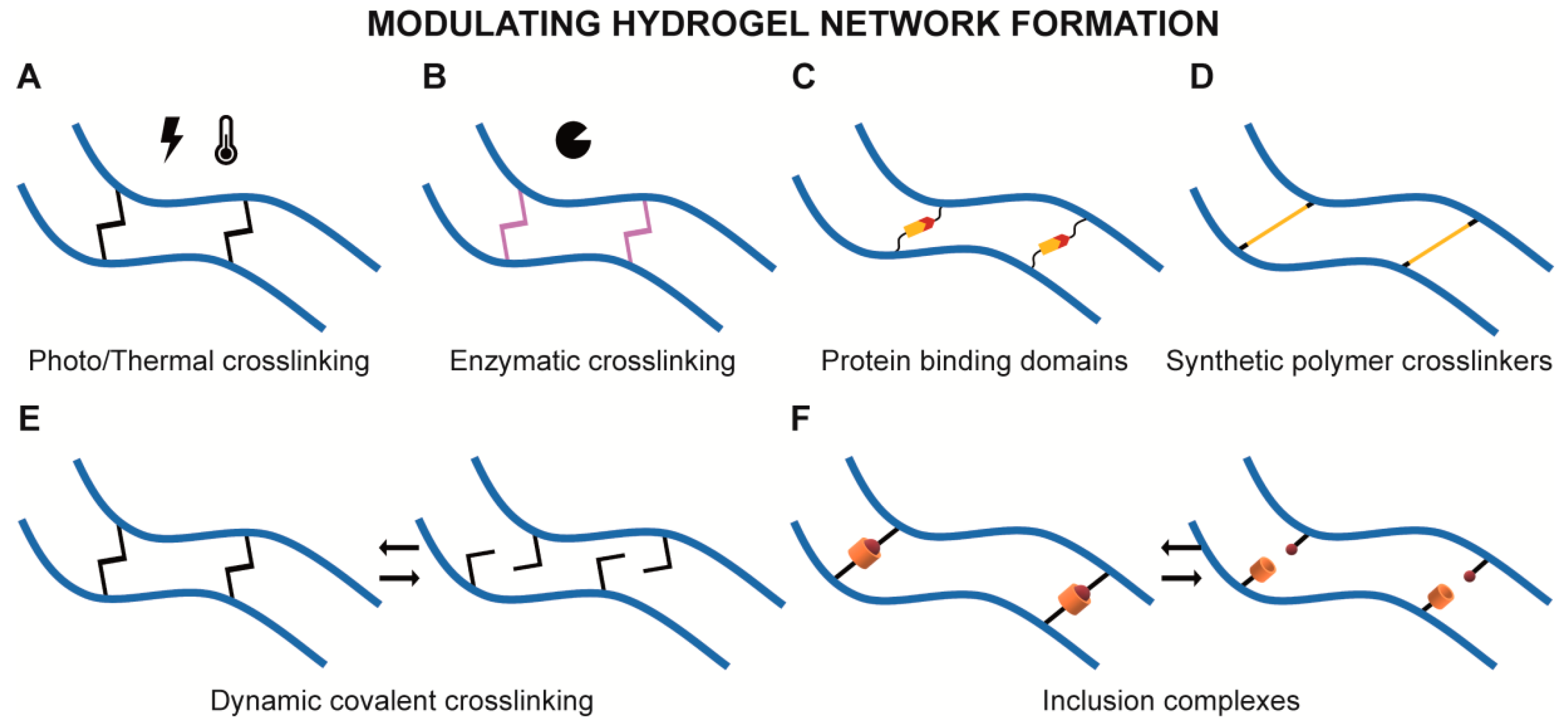{kind=link}
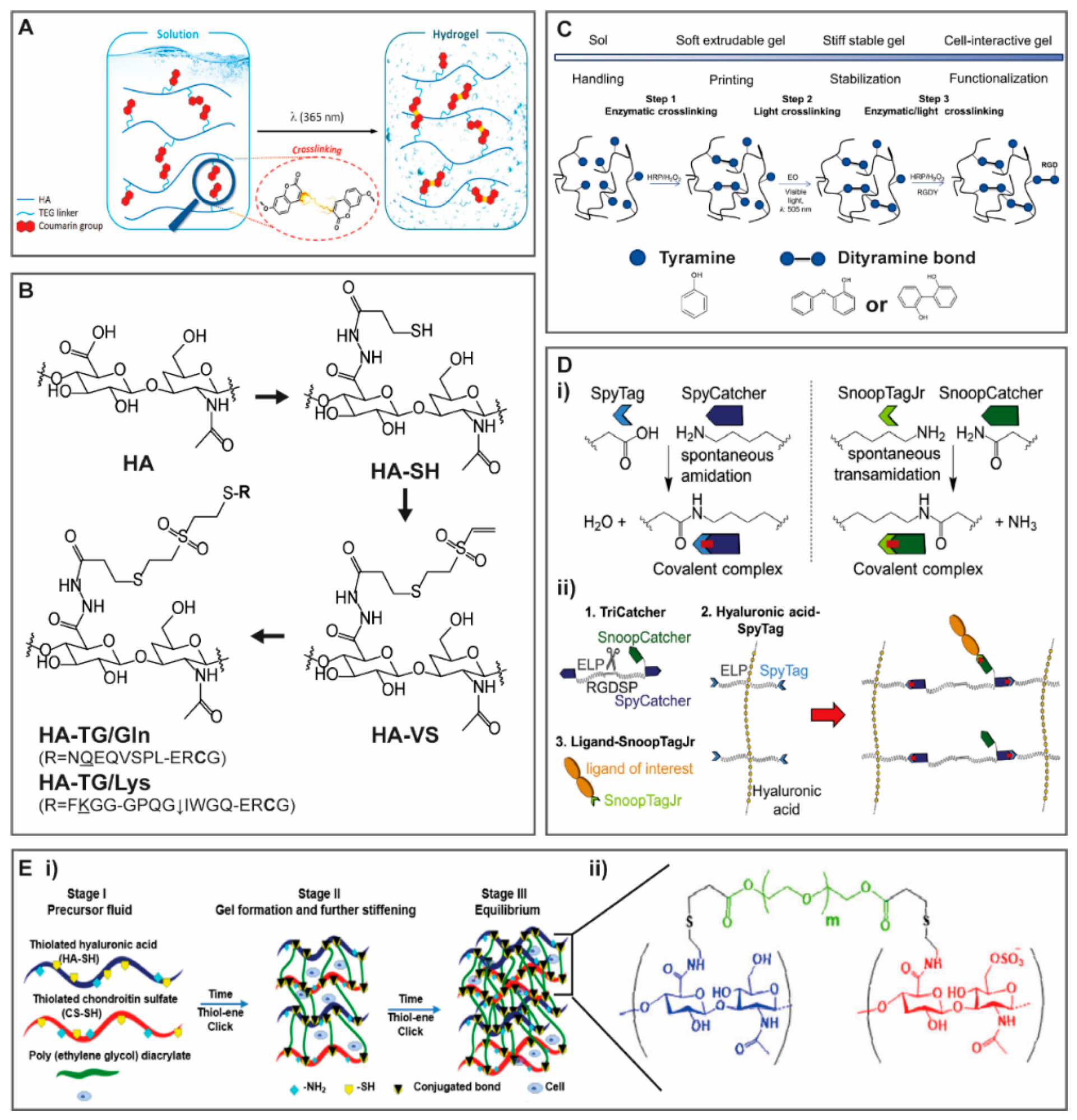{kind=link}
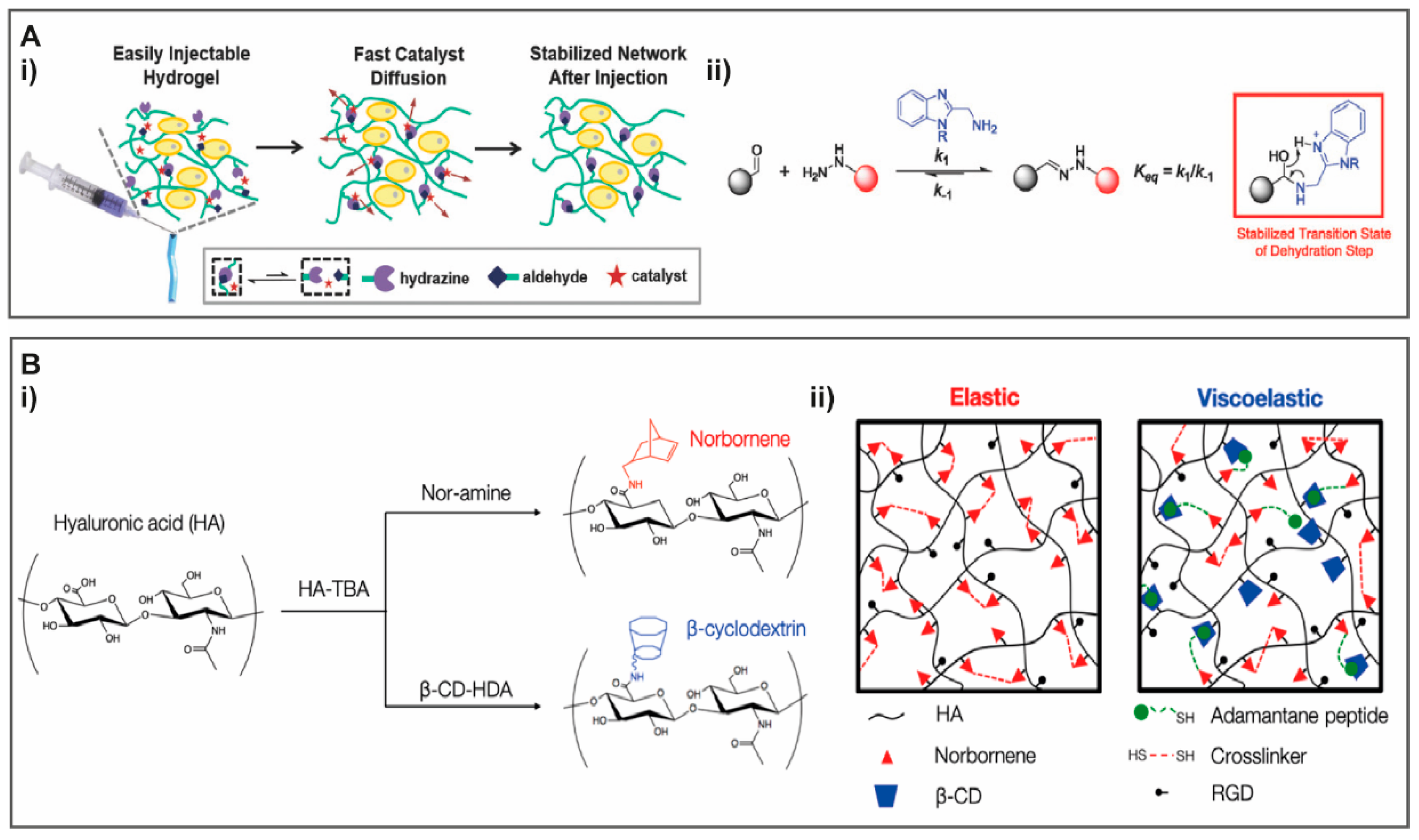{kind=link}
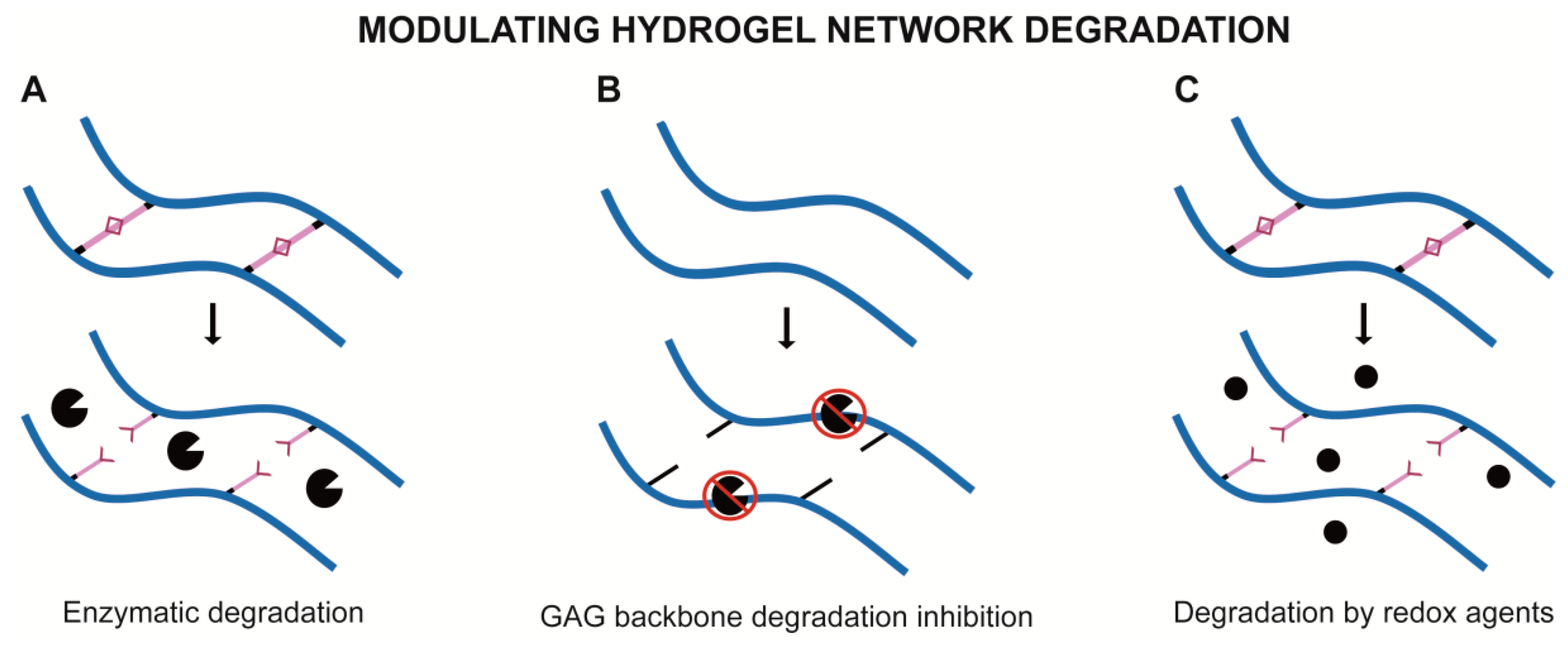{kind=link}
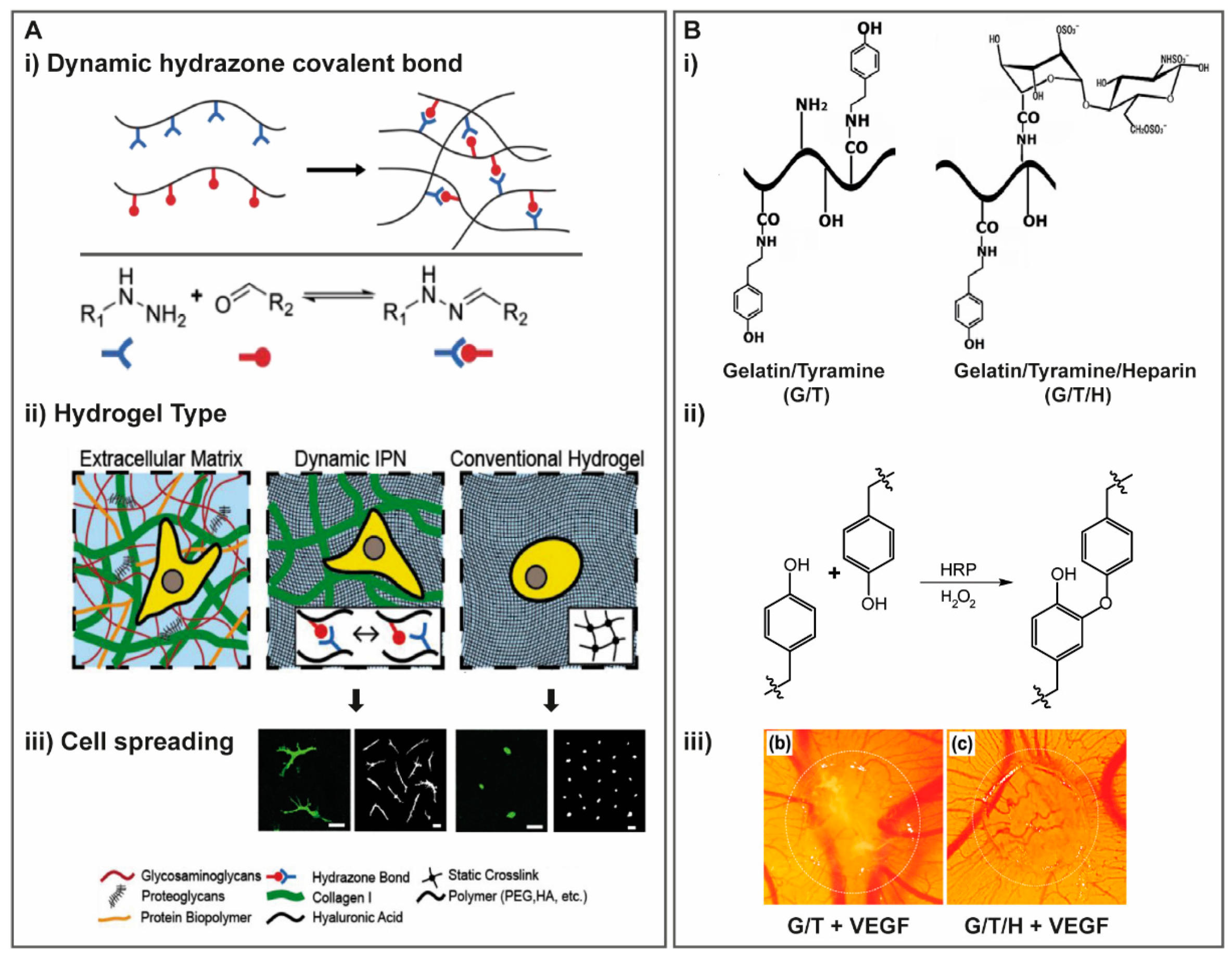{kind=link}
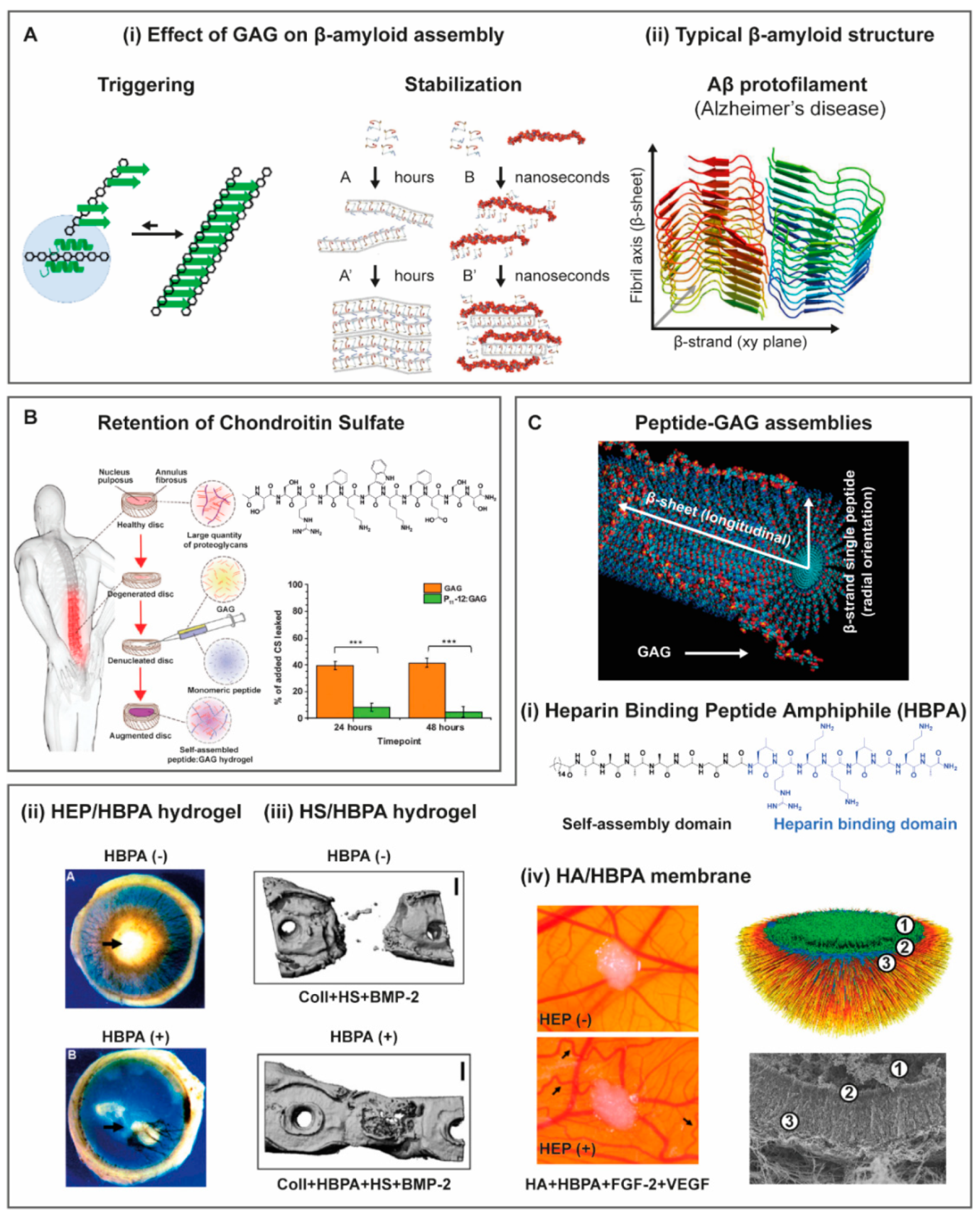{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. GAG-Based Hydrogels
2.1. Modulating Hydrogel Network Formation
2.1.1. Light and Temperature Induced Crosslinking
2.1.2. Enzymatically Driven Crosslinking
2.1.3. Crosslinking with Protein Binding Domains
2.1.4. Crosslinking with Synthetic Polymers
2.1.5. Reversible Crosslinking
Dynamic Covalent Crosslinking
Inclusion Complexes
2.2. Modulating Hydrogel Network Degradation
3. Hybrid Hydrogels Containing GAG-Based Modules
3.1. Proteins and Peptides
3.1.1. Collagen
3.1.2. Gelatin
3.1.3. Silk Fibroin
3.1.4. Self-Assembly Peptides
3.2. Non-Mammalian Non-GAG Polysaccharides
3.2.1. Chitosan
3.2.2. Alginate
3.2.3. Pectin
3.2.4. Dextran
3.2.5. Cellulose
3.2.6. Pullulan
3.3. Decellularized Extracellular Matrix (dECM)
4. GAG-Inspired Biomaterials
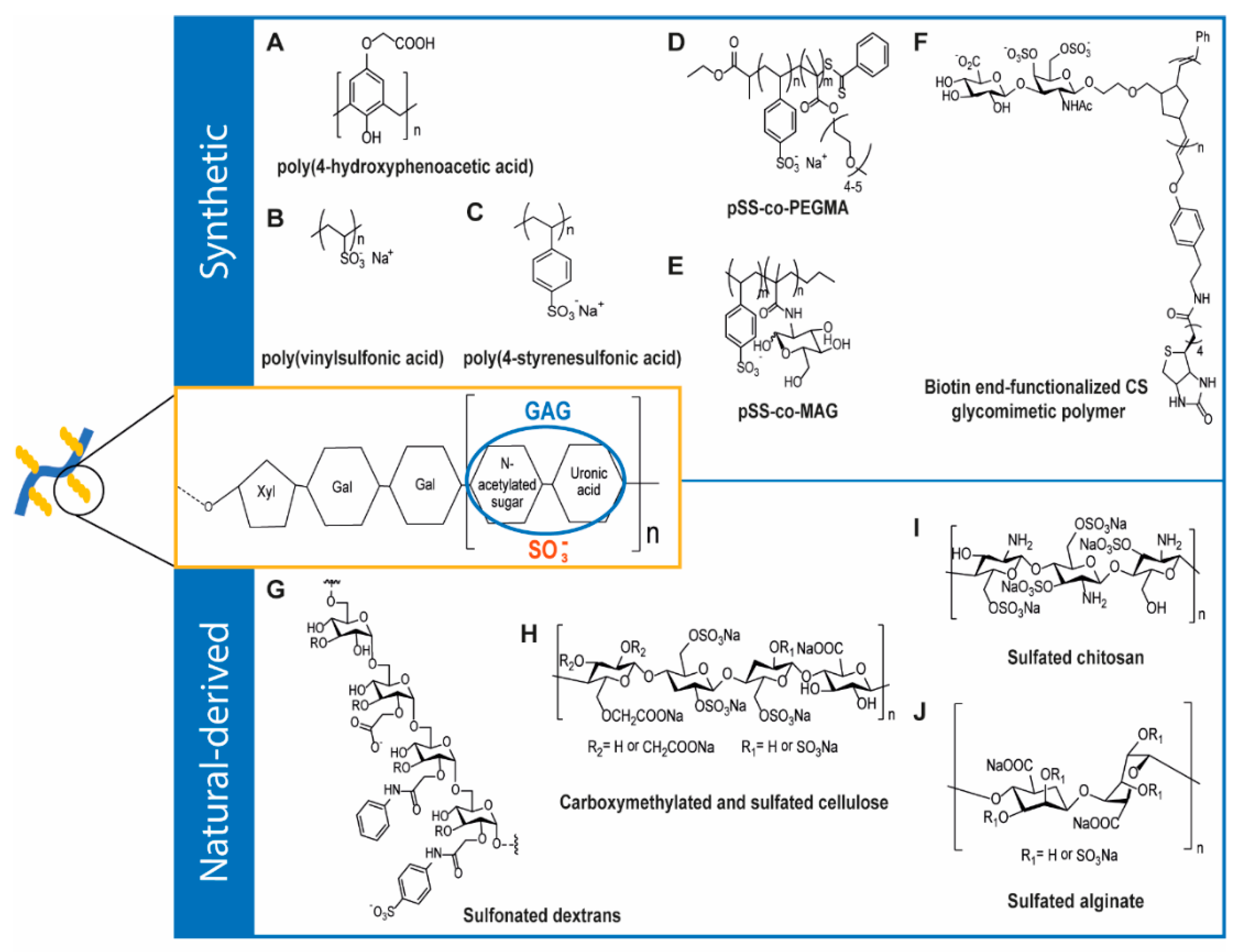
4.1. Synthetic GAG-Like Polymers
4.2. Natural-Derived GAG-Like Polymers
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and Proteoglycans. Pharmaceuticals (Basel) 2018, 11, 27. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Hogwood, J.; Naggi, A.; Torri, G.; Page, C.; Rigsby, P.; Mulloy, B.; Gray, E. The effect of increasing the sulfation level of chondroitin sulfate on anticoagulant specific activity and activation of the kinin system. PLoS ONE 2018, 13, e0193482. [Google Scholar] [CrossRef] [PubMed]
- Corsuto, L.; Rother, S.; Koehler, L.; Bedini, E.; Moeller, S.; Schnabelrauch, M.; Hintze, V.; Schiraldi, C.; Scharnweber, D. Sulfation degree not origin of chondroitin sulfate derivatives modulates keratinocyte response. Carbohydr. Polym. 2018, 191, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, Y.; Zhou, X.; Lu, H.; Ramacciotti, E.; Zhang, L. Chemically oversulfated glycosaminoglycans are potent modulators of contact system activation and different cell signaling pathways. J. Biol. Chem. 2010, 285, 22966–22975. [Google Scholar] [CrossRef] [PubMed]
- Ialenti, A.; Di Rosa, M. Hyaluronic acid modulates acute and chronic inflammation. Agents Actions 1994, 43, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yamaguchi, T. Effects of hyaluronic acid on macrophage phagocytosis and active oxygen release. Agents Actions 1993, 38, 32–37. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, Y.; Yang, H.; Qiu, P.; Cong, Z.; Zou, Y.; Song, L.; Guo, J.; Anastassiades, T.P. A Low Molecular Weight Hyaluronic Acid Derivative Accelerates Excisional Wound Healing by Modulating Pro-Inflammation, Promoting Epithelialization and Neovascularization, and Remodeling Collagen. Int. J. Mol. Sci. 2019, 20, 3722. [Google Scholar] [CrossRef]
- Rayahin, J.E.; Buhrman, J.S.; Zhang, Y.; Koh, T.J.; Gemeinhart, R.A. High and Low Molecular Weight Hyaluronic Acid Differentially Influence Macrophage Activation. ACS Biomater. Sci. Eng. 2015, 1, 481–493. [Google Scholar] [CrossRef]
- Shute, J. Glycosaminoglycan and chemokine/growth factor interactions. Handb. Exp. Pharmacol. 2012, 307–324. [Google Scholar]
- Canning, D.R.; Brelsford, N.R.; Lovett, N.W. Chondroitin sulfate effects on neural stem cell differentiation. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lin, X. Functions of heparan sulfate proteoglycans in cell signaling during development. Dev. Camb. Engl. 2004, 131, 6009–6021. [Google Scholar] [CrossRef] [PubMed]
- Aquino, R.S.; Park, P.W. Glycosaminoglycans and infection. Front. Biosci. (Landmark Ed.) 2016, 21, 1260–1277. [Google Scholar] [PubMed]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Kimata, K.; Itano, N. Key Roles of Hyaluronan and Its CD44 Receptor in the Stemness and Survival of Cancer Stem Cells. Front. Oncol. 2015, 5, 180. [Google Scholar] [CrossRef]
- Gatto, F.; Volpi, N.; Nilsson, H.; Nookaew, I.; Maruzzo, M.; Roma, A.; Johansson, M.E.; Stierner, U.; Lundstam, S.; Basso, U.; et al. Glycosaminoglycan Profiling in Patients’ Plasma and Urine Predicts the Occurrence of Metastatic Clear Cell Renal Cell Carcinoma. Cell Rep. 2016, 15, 1822–1836. [Google Scholar] [CrossRef]
- Holmes, M.W.; Bayliss, M.T.; Muir, H. Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem. J. 1988, 250, 435–441. [Google Scholar] [CrossRef]
- Pacella, E.; Pacella, F.; De Paolis, G.; Parisella, F.R.; Turchetti, P.; Anello, G.; Cavallotti, C. Glycosaminoglycans in the human cornea: Age-related changes. Ophthalmol. Eye Dis. 2015, 7, 1–5. [Google Scholar] [CrossRef]
- Plaas, A.H.; West, L.A.; Wong-Palms, S.; Nelson, F.R. Glycosaminoglycan sulfation in human osteoarthritis. Disease-related alterations at the non-reducing termini of chondroitin and dermatan sulfate. J. Biol. Chem. 1998, 273, 12642–12649. [Google Scholar] [CrossRef]
- Ucakturk, E.; Akman, O.; Sun, X.; Baydar, D.E.; Dolgun, A.; Zhang, F.; Linhardt, R.J. Changes in composition and sulfation patterns of glycoaminoglycans in renal cell carcinoma. Glycoconj. J. 2016, 33, 103–112. [Google Scholar] [CrossRef]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Fongmoon, D.; Sugahara, K. Interaction of chondroitin sulfate and dermatan sulfate from various biological sources with heparin-binding growth factors and cytokines. Glycoconj. J. 2013, 30, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Rifkin, D.B. Interaction of heparin with human basic fibroblast growth factor: Protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J. Cell. Physiol. 1989, 138, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Eldor, A.; Vlodavsky, I. Binding of thrombin to subendothelial extracellular matrix. Protection and expression of functional properties. J. Clin. Investig. 1989, 84, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Munakata, H.; Takagaki, K.; Majima, M.; Endo, M. Interaction between collagens and glycosaminoglycans investigated using a surface plasmon resonance biosensor. Glycobiology 1999, 9, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Raspanti, M.; Viola, M.; Forlino, A.; Tenni, R.; Gruppi, C.; Tira, M.E. Glycosaminoglycans show a specific periodic interaction with type I collagen fibrils. J. Struct. Biol. 2008, 164, 134–139. [Google Scholar] [CrossRef]
- Rowley, J.A.; Madlambayan, G.; Mooney, D.J. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 1999, 20, 45–53. [Google Scholar] [CrossRef]
- Rowley, J.A.; Mooney, D.J. Alginate type and RGD density control myoblast phenotype. J. Biomed. Mater. Res. 2002, 60, 217–223. [Google Scholar] [CrossRef]
- Bidarra, S.J.; Barrias, C.C.; Fonseca, K.B.; Barbosa, M.A.; Soares, R.A.; Granja, P.L. Injectable in situ crosslinkable RGD-modified alginate matrix for endothelial cells delivery. Biomaterials 2011, 32, 7897–7904. [Google Scholar] [CrossRef]
- Fonseca, K.B.; Gomes, D.B.; Lee, K.; Santos, S.G.; Sousa, A.; Silva, E.A.; Mooney, D.J.; Granja, P.L.; Barrias, C.C. Injectable MMP-Sensitive Alginate Hydrogels as hMSC Delivery Systems. Biomacromolecules 2014, 15, 380–390. [Google Scholar] [CrossRef]
- Fonseca, K.B.; Bidarra, S.J.; Oliveira, M.J.; Granja, P.L.; Barrias, C.C. Molecularly designed alginate hydrogels susceptible to local proteolysis as three-dimensional cellular microenvironments. Acta Biomater. 2011, 7, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- You, J.-O.; Rafat, M.; Almeda, D.; Maldonado, N.; Guo, P.; Nabzdyk, C.S.; Chun, M.; LoGerfo, F.W.; Hutchinson, J.W.; Pradhan-Nabzdyk, L.K.; et al. pH-responsive scaffolds generate a pro-healing response. Biomaterials 2015, 57, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, C.; Li, Y.; Wang, K.; Wang, X.; Wei, Y.; Tao, L. Synthesis of an injectable, self-healable and dual responsive hydrogel for drug delivery and 3D cell cultivation. Polym. Chem. 2017, 8, 537–544. [Google Scholar] [CrossRef]
- Therien-Aubin, H.; Wang, Y.; Nothdurft, K.; Prince, E.; Cho, S.; Kumacheva, E. Temperature-Responsive Nanofibrillar Hydrogels for Cell Encapsulation. Biomacromolecules 2016, 17, 3244–3251. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, Y.; Yan, J.; Li, W.; Zhang, A. Thermo- and redox-responsive dendronized polymer hydrogels. Polym. Chem. 2018, 9, 712–721. [Google Scholar] [CrossRef]
- Zhou, M.L.; Qian, Z.G.; Chen, L.; Kaplan, D.L.; Xia, X.X. Rationally Designed Redox-Sensitive Protein Hydrogels with Tunable Mechanical Properties. Biomacromolecules 2016, 17, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Williams, C.G.; Sun, D.D.N.; Wang, J.; Leong, K.; Elisseeff, J.H. Photocrosslinkable polysaccharides based on chondroitin sulfate. J. Biomed. Mater. Res. Part. A 2004, 68, 28–33. [Google Scholar] [CrossRef]
- Donnelly, P.E.; Chen, T.; Finch, A.; Brial, C.; Maher, S.A.; Torzilli, P.A. Photocrosslinked tyramine-substituted hyaluronate hydrogels with tunable mechanical properties improve immediate tissue-hydrogel interfacial strength in articular cartilage. J. Biomater. Sci. Polym. Ed. 2017, 28, 582–600. [Google Scholar] [CrossRef]
- Broguiere, N.; Isenmann, L.; Zenobi-Wong, M. Novel enzymatically cross-linked hyaluronan hydrogels support the formation of 3D neuronal networks. Biomaterials 2016, 99, 47–55. [Google Scholar] [CrossRef]
- Karvinen, J.; Joki, T.; Ylä-Outinen, L.; Koivisto, J.T.; Narkilahti, S.; Kellomäki, M. Soft hydrazone crosslinked hyaluronan- and alginate-based hydrogels as 3D supportive matrices for human pluripotent stem cell-derived neuronal cells. React. Funct. Polym. 2018, 124, 29–39. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Palomares, T.; Castro, B.; Herrero, J.; Alonso-Varona, A. Generation of tunable glycosaminoglycan hydrogels to mimic extracellular matrices. J. Tissue Eng. Regen. Med. 2016, 10, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martos, S.; Calvo-Sanchez, M.; Garcia-Alonso, K.; Castro, B.; Hashtroody, B.; Espada, J. Sustained Human Hair Follicle Growth Ex Vivo in a Glycosaminoglycan Hydrogel Matrix. Int. J. Mol. Sci. 2019, 20, 1741. [Google Scholar] [CrossRef] [PubMed]
- Karumbaiah, L.; Enam, S.F.; Brown, A.C.; Saxena, T.; Betancur, M.I.; Barker, T.H.; Bellamkonda, R.V. Chondroitin Sulfate Glycosaminoglycan Hydrogels Create Endogenous Niches for Neural Stem Cells. Bioconj. Chem. 2015, 26, 2336–2349. [Google Scholar] [CrossRef] [PubMed]
- Ornell, K.J.; Lozada, D.; Phan, N.V.; Coburn, J.M. Controlling methacryloyl substitution of chondroitin sulfate: Injectable hydrogels with tunable long-term drug release profiles. J. Mater. Chem. B 2019, 7, 2151–2161. [Google Scholar] [CrossRef]
- Beninatto, R.; Barbera, C.; De Lucchi, O.; Borsato, G.; Serena, E.; Guarise, C.; Pavan, M.; Luni, C.; Martewicz, S.; Galesso, D.; et al. Photocrosslinked hydrogels from coumarin derivatives of hyaluronic acid for tissue engineering applications. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 96, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Jachak, S.M. Coumarins as privileged scaffold for anti-inflammatory drug development. RSC Adv. 2015, 5, 38892–38905. [Google Scholar] [CrossRef]
- El-Haggar, R.; Al-Wabli, R.I. Anti-inflammatory screening and molecular modeling of some novel coumarin derivatives. Molecules 2015, 20, 5374–5391. [Google Scholar] [CrossRef]
- Al-Majedy, Y.; Al-Amiery, A.; Kadhum, A.A.; BakarMohamad, A. Antioxidant Activity of Coumarins. Syst. Rev. Pharm. 2016, 8, 24–30. [Google Scholar] [CrossRef]
- Thomas, V.; Giles, D.; Basavarajaswamy, G.P.M.; Das, A.K.; Patel, A. Coumarin Derivatives as Anti-inflammatory and Anticancer Agents. Anti-Cancer Agents Med. Chem. 2017, 17, 415–423. [Google Scholar] [CrossRef]
- Lim, J.J.; Temenoff, J.S. The effect of desulfation of chondroitin sulfate on interactions with positively charged growth factors and upregulation of cartilaginous markers in encapsulated MSCs. Biomaterials 2013, 34, 5007–5018. [Google Scholar] [CrossRef]
- Toh, W.S.; Lim, T.C.; Kurisawa, M.; Spector, M. Modulation of mesenchymal stem cell chondrogenesis in a tunable hyaluronic acid hydrogel microenvironment. Biomaterials 2012, 33, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Loebel, C.; Broguiere, N.; Alini, M.; Zenobi-Wong, M.; Eglin, D. Microfabrication of Photo-Cross-Linked Hyaluronan Hydrogels by Single- and Two-Photon Tyramine Oxidation. Biomacromolecules 2015, 16, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Petta, D.; Grijpma, D.W.; Alini, M.; Eglin, D.; D’Este, M. Three-Dimensional Printing of a Tyramine Hyaluronan Derivative with Double Gelation Mechanism for Independent Tuning of Shear Thinning and Postprinting Curing. ACS Biomater. Sci. Eng. 2018, 4, 3088–3098. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Nakamura, T.; Brenner, M.D.; Gayet, R.V.; Yan, J.; Robinson, C.V.; Howarth, M. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [Google Scholar] [CrossRef]
- Wieduwild, R.; Howarth, M. Assembling and decorating hyaluronan hydrogels with twin protein superglues to mimic cell-cell interactions. Biomaterials 2018, 180, 253–264. [Google Scholar] [CrossRef]
- Hiraga, T.; Ito, S.; Nakamura, H. EpCAM expression in breast cancer cells is associated with enhanced bone metastasis formation. Int. J. Cancer 2016, 138, 1698–1708. [Google Scholar] [CrossRef]
- Kuang, L.; Damayanti, N.P.; Jiang, C.; Fei, X.; Liu, W.; Narayanan, N.; Irudayaraj, J.; Campanella, O.; Deng, M. Bioinspired glycosaminoglycan hydrogels via click chemistry for 3D dynamic cell encapsulation. J. Appl. Polym. Sci. 2019, 136. [Google Scholar] [CrossRef]
- Grab, A.L.; Seckinger, A.; Horn, P.; Hose, D.; Cavalcanti-Adam, E.A. Hyaluronan hydrogels delivering BMP-6 for local targeting of malignant plasma cells and osteogenic differentiation of mesenchymal stromal cells. Acta Biomater. 2019, 96, 258–270. [Google Scholar] [CrossRef]
- Tae, G.; Kim, Y.-J.; Choi, W.-I.; Kim, M.; Stayton, P.S.; Hoffman, A.S. Formation of a Novel Heparin-Based Hydrogel in the Presence of Heparin-Binding Biomolecules. Biomacromolecules 2007, 8, 1979–1986. [Google Scholar] [CrossRef]
- Kool, E.T.; Park, D.H.; Crisalli, P. Fast hydrazone reactants: Electronic and acid/base effects strongly influence rate at biological pH. J. Am. Chem. Soc. 2013, 135, 17663–17666. [Google Scholar] [CrossRef] [PubMed]
- Azagarsamy, M.A.; Marozas, I.A.; Spaans, S.; Anseth, K.S. Photoregulated Hydrazone-Based Hydrogel Formation for Biochemically Patterning 3D Cellular Microenvironments. ACS Macro Lett. 2015, 5, 19–23. [Google Scholar] [CrossRef]
- Lou, J.; Liu, F.; Lindsay, C.D.; Chaudhuri, O.; Heilshorn, S.C.; Xia, Y. Dynamic Hyaluronan Hydrogels with Temporally Modulated High Injectability and Stability Using a Biocompatible Catalyst. Adv Mater. 2018, 30, e1705215. [Google Scholar] [CrossRef] [PubMed]
- Stefanello, T.F.; Couturaud, B.; Szarpak-Jankowska, A.; Fournier, D.; Louage, B.; Garcia, F.P.; Nakamura, C.V.; De Geest, B.G.; Woisel, P.; van der Sanden, B.; et al. Coumarin-containing thermoresponsive hyaluronic acid-based nanogels as delivery systems for anticancer chemotherapy. Nanoscale 2017, 9, 12150–12162. [Google Scholar] [CrossRef]
- Payne, W.M.; Svechkarev, D.; Kyrychenko, A.; Mohs, A.M. The role of hydrophobic modification on hyaluronic acid dynamics and self-assembly. Carbohydr. Polym. 2018, 182, 132–141. [Google Scholar] [CrossRef]
- Manzi, G.; Zoratto, N.; Matano, S.; Sabia, R.; Villani, C.; Coviello, T.; Matricardi, P.; Di Meo, C. "Click" hyaluronan based nanohydrogels as multifunctionalizable carriers for hydrophobic drugs. Carbohydr. Polym. 2017, 174, 706–715. [Google Scholar] [CrossRef]
- Palumbo, F.S.; Puleio, R.; Fiorica, C.; Pitarresi, G.; Loria, G.R.; Cassata, G.; Giammona, G. Matrices of a hydrophobically functionalized hyaluronic acid derivative for the locoregional tumour treatment. Acta Biomater. 2015, 25, 205–215. [Google Scholar] [CrossRef]
- Palumbo, F.S.; Agnello, S.; Fiorica, C.; Pitarresi, G.; Puleio, R.; Tamburello, A.; Loria, R.; Giammona, G. Hyaluronic Acid Derivative with Improved Versatility for Processing and Biological Functionalization. Macromol. Biosci. 2016, 16, 1485–1496. [Google Scholar] [CrossRef]
- Rosales, A.M.; Rodell, C.B.; Chen, M.H.; Morrow, M.G.; Anseth, K.S.; Burdick, J.A. Reversible Control of Network Properties in Azobenzene-Containing Hyaluronic Acid-Based Hydrogels. Bioconj. Chem. 2018, 29, 905–913. [Google Scholar] [CrossRef]
- Hu, Q.D.; Tang, G.P.; Chu, P.K. Cyclodextrin-based host-guest supramolecular nanoparticles for delivery: From design to applications. Acc Chem. Res. 2014, 47, 2017–2025. [Google Scholar] [CrossRef]
- Cheirsilp, B.; Rakmai, J. Inclusion complex formation of cyclodextrin with its guest and their applications. Biol. Eng. Med. 2017, 2. [Google Scholar] [CrossRef]
- Hui, E.; Gimeno, K.I.; Guan, G.; Caliari, S.R. Spatiotemporal Control of Viscoelasticity in Phototunable Hyaluronic Acid Hydrogels. Biomacromolecules 2019, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Pavan, M.; Galesso, D.; Secchieri, C.; Guarise, C. Hyaluronic acid alkyl derivative: A novel inhibitor of metalloproteases and hyaluronidases. Int. J. Biol. Macromol. 2016, 84, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Caley, M.P.; Martins, V.L.C.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.Q.; Chen, A.B.; Li, W.; Song, J.H.; Gao, C.Y. High MMP-1, MMP-2, and MMP-9 protein levels in osteoarthritis. Genet. Mol. Res.: GMR 2015, 14, 14811–14822. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Golland, B.; Tronci, G.; Thornton, P. A Redox-Responsive Hyaluronic Acid-Based Hydrogel for Chronic Wound Management. J. Mater. Chem. B 2019. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Koh, R.H.; Jin, Y.; Kang, B.J.; Hwang, N.S. Chondrogenically primed tonsil-derived mesenchymal stem cells encapsulated in riboflavin-induced photocrosslinking collagen-hyaluronic acid hydrogel for meniscus tissue repairs. Acta Biomater. 2017, 53, 318–328. [Google Scholar] [CrossRef]
- Heo, J.; Koh, R.H.; Shim, W.; Kim, H.D.; Yim, H.G.; Hwang, N.S. Riboflavin-induced photo-crosslinking of collagen hydrogel and its application in meniscus tissue engineering. Drug Deliv. Transl. Res. 2016, 6, 148–158. [Google Scholar] [CrossRef]
- Zhang, L.; Li, K.; Xiao, W.; Zheng, L.; Xiao, Y.; Fan, H.; Zhang, X. Preparation of collagen–chondroitin sulfate–hyaluronic acid hybrid hydrogel scaffolds and cell compatibility in vitro. Carbohydr. Polym. 2011, 84, 118–125. [Google Scholar] [CrossRef]
- Walimbe, T.; Calve, S.; Panitch, A.; Sivasankar, M.P. Incorporation of types I and III collagen in tunable hyaluronan hydrogels for vocal fold tissue engineering. Acta Biomater. 2019, 87, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhou, J.; Wang, M.; Su, D.; Ma, Q.; Lv, G.; Chen, J. In situ formed collagen-hyaluronic acid hydrogel as biomimetic dressing for promoting spontaneous wound healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 101, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Harrington, S.; Williams, J.; Rawal, S.; Ramachandran, K.; Stehno-Bittel, L. Hyaluronic Acid/Collagen Hydrogel as an Alternative to Alginate for Long-Term Immunoprotected Islet Transplantation. Tissue Eng. Part A 2017, 23, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ying, H.; Wang, M.; Su, D.; Lu, G.; Chen, J. Dual layer collagen-GAG conduit that mimic vascular scaffold and promote blood vessel cells adhesion, proliferation and elongation. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 447–452. [Google Scholar] [CrossRef]
- Van der Smissen, A.; Samsonov, S.; Hintze, V.; Scharnweber, D.; Moeller, S.; Schnabelrauch, M.; Pisabarro, M.T.; Anderegg, U. Artificial extracellular matrix composed of collagen I and highly sulfated hyaluronan interferes with TGFbeta(1) signaling and prevents TGFbeta(1)-induced myofibroblast differentiation. Acta Biomater. 2013, 9, 7775–7786. [Google Scholar] [CrossRef]
- Rother, S.; Kronert, V.; Hauck, N.; Berg, A.; Moeller, S.; Schnabelrauch, M.; Thiele, J.; Scharnweber, D.; Hintze, V. Hyaluronan/collagen hydrogel matrices containing high-sulfated hyaluronan microgels for regulating transforming growth factor-beta1. J. Mater. Sci Mater. Med. 2019, 30, 65. [Google Scholar] [CrossRef]
- Hempel, U.; Hintze, V.; Moller, S.; Schnabelrauch, M.; Scharnweber, D.; Dieter, P. Artificial extracellular matrices composed of collagen I and sulfated hyaluronan with adsorbed transforming growth factor beta1 promote collagen synthesis of human mesenchymal stromal cells. Acta Biomater. 2012, 8, 659–666. [Google Scholar] [CrossRef]
- Brodsky, B.; Ramshaw, J.A.M. The collagen triple-helix structure. Matrix Biol. 1997, 15, 545–554. [Google Scholar] [CrossRef]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23 (8 Suppl 1), S20–S23. [Google Scholar] [CrossRef]
- Achilli, M.; Mantovani, D. Tailoring Mechanical Properties of Collagen-Based Scaffolds for Vascular Tissue Engineering: The Effects of pH, Temperature and Ionic Strength on Gelation. Polymers 2010, 2, 664–680. [Google Scholar] [CrossRef]
- Lou, J.; Stowers, R.; Nam, S.; Xia, Y.; Chaudhuri, O. Stress relaxing hyaluronic acid-collagen hydrogels promote cell spreading, fiber remodeling, and focal adhesion formation in 3D cell culture. Biomaterials 2018, 154, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Nochel, U.; Lowenberg, C.; Lendlein, A.; Neffe, A.T. Supramolecular hydrogel networks formed by molecular recognition of collagen and a peptide grafted to hyaluronic acid. Acta Biomater. 2016, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.T.; Harrison, R.W.; Iozzo, R.V. Model structure of decorin and implications for collagen fibrillogenesis. J. Biol. Chem. 1996, 271, 31767–31770. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Qian, G.; Chen, S.; Xu, D.; Zhao, X.; Du, C. A tracheal scaffold of gelatin-chondroitin sulfate-hyaluronan-polyvinyl alcohol with orientated porous structure. Carbohydr. Polym. 2017, 159, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Eke, G.; Mangir, N.; Hasirci, N.; MacNeil, S.; Hasirci, V. Development of a UV crosslinked biodegradable hydrogel containing adipose derived stem cells to promote vascularization for skin wounds and tissue engineering. Biomaterials 2017, 129, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Rezaeeyazdi, M.; Colombani, T.; Memic, A.; Bencherif, S.A. Injectable Hyaluronic Acid-co-Gelatin Cryogels for Tissue-Engineering Applications. Materials (Basel) 2018, 11, 1374. [Google Scholar] [CrossRef]
- Brown, G.C.J.; Lim, K.S.; Farrugia, B.L.; Hooper, G.J.; Woodfield, T.B.F. Covalent Incorporation of Heparin Improves Chondrogenesis in Photocurable Gelatin-Methacryloyl Hydrogels. Macromol. Biosci. 2017, 17. [Google Scholar] [CrossRef]
- Pezeshki-Modaress, M.; Mirzadeh, H.; Zandi, M.; Rajabi-Zeleti, S.; Sodeifi, N.; Aghdami, N.; Mofrad, M.R.K. Gelatin/chondroitin sulfate nanofibrous scaffolds for stimulation of wound healing: In-vitro and in-vivo study. J. Biomed. Mater. Res. A 2017, 105, 2020–2034. [Google Scholar] [CrossRef]
- Sawatjui, N.; Damrongrungruang, T.; Leeanansaksiri, W.; Jearanaikoon, P.; Hongeng, S.; Limpaiboon, T. Silk fibroin/gelatin-chondroitin sulfate-hyaluronic acid effectively enhances in vitro chondrogenesis of bone marrow mesenchymal stem cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2015, 52, 90–96. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Mead, I.; Bishop, C.; Shupe, T.; Lee, S.J.; Jackson, J.; Yoo, J.; Soker, S.; et al. A hydrogel bioink toolkit for mimicking native tissue biochemical and mechanical properties in bioprinted tissue constructs. Acta Biomater. 2015, 25, 24–34. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Crowell, K.; Mack, D.; Atala, A.; Soker, S. A tunable hydrogel system for long-term release of cell-secreted cytokines and bioprinted in situ wound cell delivery. J. Biomed. Mater. Res. B Appl Biomater 2017, 105, 1986–2000. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Zhang, J.; McCoard, L.; Xu, X.; Oottamasathien, S.; Prestwich, G.D. Photocrosslinkable hyaluronan-gelatin hydrogels for two-step bioprinting. Tissue Eng. Part A 2010, 16, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qu, T.; Ding, C.; Ma, C.; Sun, H.; Li, S.; Liu, X. Injectable gelatin derivative hydrogels with sustained vascular endothelial growth factor release for induced angiogenesis. Acta Biomater. 2015, 13, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Sun, J.S.; Wu, H.C.; Tsuang, Y.H.; Wang, W.H.; Lin, F.H. The effect of gelatin-chondroitin sulfate-hyaluronic acid skin substitute on wound healing in SCID mice. Biomaterials 2006, 27, 5689–5697. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.W.; Liu, C.; Wu, J.H.; Lin, L.X.; Fan, H.M.; Zhao, D.H.; Zhuang, Y.Q.; Sun, Y.L. In situ injectable hyaluronic acid/gelatin hydrogel for hemorrhage control. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 98, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lee, S.S.; Bae, S.; Lee, H.; Hwang, N.S. Heparin Functionalized Injectable Cryogel with Rapid Shape-Recovery Property for Neovascularization. Biomacromolecules 2018, 19, 2257–2269. [Google Scholar] [CrossRef]
- Djabourov, M.; Leblond, J.; Papon, P. Gelation of aqueous gelatin solutions. I. Structural investigation. J. De Phys. 1988, 49, 319–332. [Google Scholar] [CrossRef]
- Djabourov, M.; Papon, P. Influence of thermal treatments on the structure and stability of gelatin gels. Polymer 1983, 24, 537–542. [Google Scholar] [CrossRef]
- Le Thi, P.; Lee, Y.; Nguyen, D.H.; Park, K.D. In situ forming gelatin hydrogels by dual-enzymatic cross-linking for enhanced tissue adhesiveness. J. Mater. Chem. B 2017, 5, 757–764. [Google Scholar] [CrossRef]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef]
- Yue, K.; Trujillo-de Santiago, G.; Alvarez, M.M.; Tamayol, A.; Annabi, N.; Khademhosseini, A. Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials 2015, 73, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, C.G.; Berner, A.; Koch, M.; Krutsch, W.; Kujat, R.; Angele, P.; Nerlich, M.; Zellner, J. Higher Ratios of Hyaluronic Acid Enhance Chondrogenic Differentiation of Human MSCs in a Hyaluronic Acid-Gelatin Composite Scaffold. Materials (Basel) 2016, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, H.; Wei, K.; Yang, Y.; Zheng, R.Y.; Kim, I.S.; Zhang, K.Q. A Review of Structure Construction of Silk Fibroin Biomaterials from Single Structures to Multi-Level Structures. Int. J. Mol. Sci. 2017, 18, 237. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Han, G.; Wang, Q.; Zhang, S.; You, R.; Luo, Z.; Xu, A.; Li, X.; Li, M.; Zhang, Q.; et al. Directed assembly of robust and biocompatible silk fibroin/hyaluronic acid composite hydrogels. Compos. Part B: Eng. 2019, 176. [Google Scholar] [CrossRef]
- Yan, S.; Wang, Q.; Tariq, Z.; You, R.; Li, X.; Li, M.; Zhang, Q. Facile preparation of bioactive silk fibroin/hyaluronic acid hydrogels. Int. J. Biol. Macromol. 2018, 118, 775–782. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, S.; You, R.; Tariq, Z.; Huang, J.; Li, M.; Yan, S. Silk fibroin/hyaluronic acid porous scaffold for dermal wound healing. Fibers Polym. 2017, 18, 1056–1063. [Google Scholar] [CrossRef]
- Tavsanli, B.; Okay, O. Mechanically robust and stretchable silk/hyaluronic acid hydrogels. Carbohydr. Polym. 2019, 208, 413–420. [Google Scholar] [CrossRef]
- Naeimi, M.; Fathi, M.; Rafienia, M.; Bonakdar, S. Silk fibroin-chondroitin sulfate-alginate porous scaffolds: Structural properties andin vitrostudies. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Yang, W.; Xu, H.; Lan, Y.; Zhu, Q.; Liu, Y.; Huang, S.; Shi, S.; Hancharou, A.; Tang, B.; Guo, R. Preparation and characterisation of a novel silk fibroin/hyaluronic acid/sodium alginate scaffold for skin repair. Int. J. Biol. Macromol. 2019, 130, 58–67. [Google Scholar] [CrossRef]
- Gokila, S.; Gomathi, T.; Vijayalakshmi, K.; Sukumaran, A.; Sudha, N.S. Development of 3D scaffolds using nanochitosan/silk-fibroin/hyaluronic acid biomaterials for tissue engineering applications. Int. J. Biol. Macromol. 2018, 120, 876–885. [Google Scholar]
- Zhou, J.; Zhang, B.; Liu, X.; Shi, L.; Zhu, J.; Wei, D.; Zhong, J.; Sun, G.; He, D. Facile method to prepare silk fibroin/hyaluronic acid films for vascular endothelial growth factor release. Carbohydr. Polym. 2016, 143, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Jaipaew, J.; Wangkulangkul, P.; Meesane, J.; Raungrut, P.; Puttawibul, P. Mimicked cartilage scaffolds of silk fibroin/hyaluronic acid with stem cells for osteoarthritis surgery: Morphological, mechanical, and physical clues. Mater. Sci. Eng.: C 2016, 64, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fuentes, M.; Giger, E.; Meinel, L.; Merkle, H.P. The effect of hyaluronic acid on silk fibroin conformation. Biomaterials 2008, 29, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Malay, Ö.; Yalçın, D.; Batıgün, A.; Bayraktar, O. Characterization of silk fibroin/hyaluronic acid polyelectrolyte complex (PEC) films. J. Therm. Anal. Calorim. 2008, 94, 749–755. [Google Scholar] [CrossRef]
- Raia, N.R.; Partlow, B.P.; McGill, M.; Kimmerling, E.P.; Ghezzi, C.E.; Kaplan, D.L. Enzymatically crosslinked silk-hyaluronic acid hydrogels. Biomaterials 2017, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
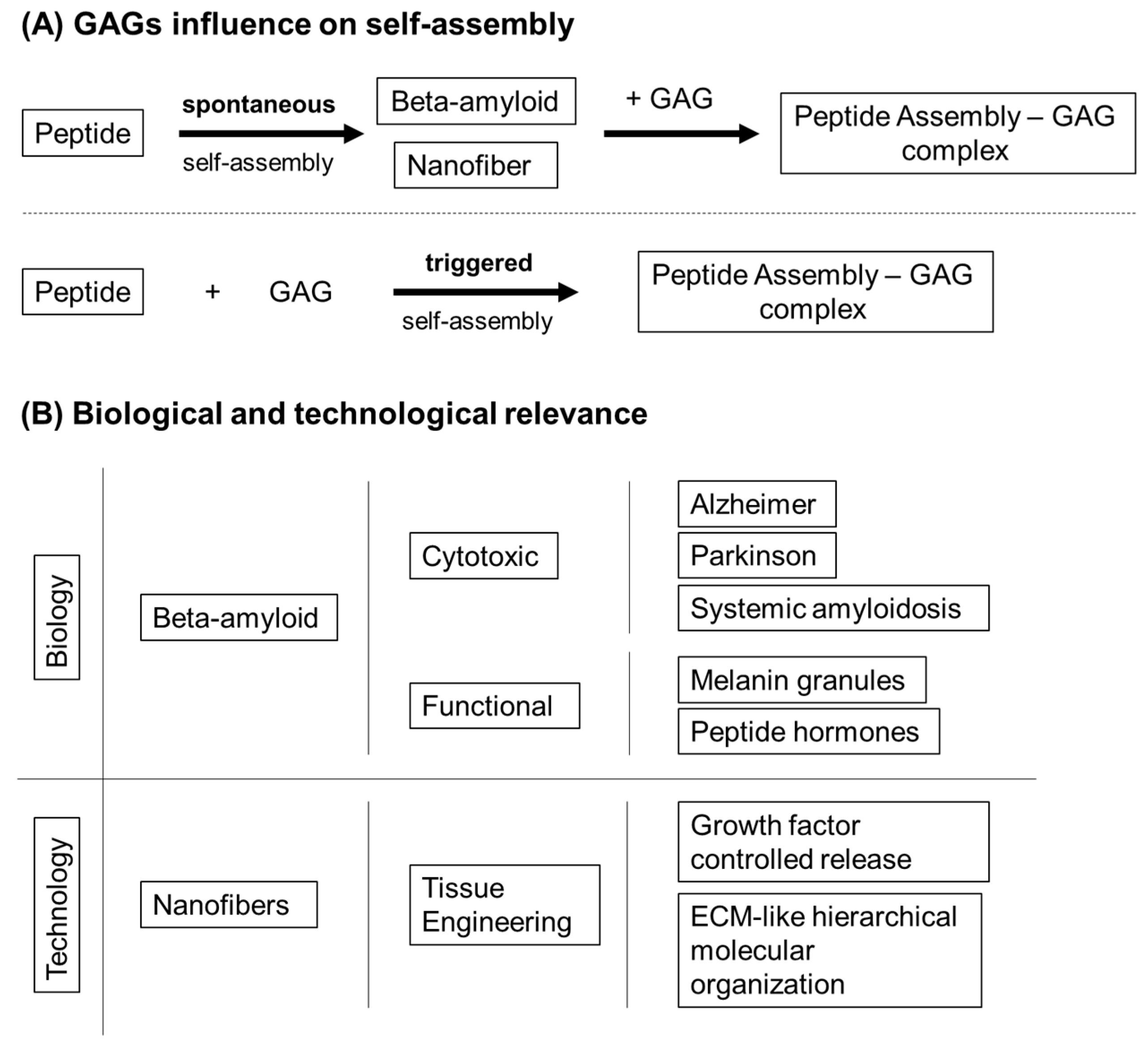
- Quittot, N.; Sebastiao, M.; Bourgault, S. Modulation of amyloid assembly by glycosaminoglycans: From mechanism to biological significance. Biochem. Cell Biol. 2017, 95, 329–337. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Sebastiao, M.; Quittot, N.; Marcotte, I.; Bourgault, S. Glycosaminoglycans Induce Amyloid Self-Assembly of a Peptide Hormone by Concerted Secondary and Quaternary Conformational Transitions. Biochemistry 2019, 58, 1214–1225. [Google Scholar] [CrossRef]
- Dharmadana, D.; Reynolds, N.P.; Dekiwadia, C.; Conn, C.E.; Valery, C. Heparin assisted assembly of somatostatin amyloid nanofibrils results in disordered precipitates by hindrance of protofilaments interactions. Nanoscale 2018, 10, 18195–18204. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Davies, H.A.; Hughes, E.; Middleton, D.A. Heparin promotes the rapid fibrillization of a peptide with low intrinsic amyloidogenicity. Biochemistry 2013, 52, 8984–8992. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Berns, E.J.; Stupp, S.I. Supramolecular Nanofibers of Peptide Amphiphiles for Medicine. Isr J. Chem. 2013, 53, 530–554. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, N.; Ortony, J.H.; Stupp, S.I. Self-assembly for the synthesis of functional biomaterials. Acta Mater. 2013, 61, 912–930. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.B.; Stupp, S.I. Self-assembling peptide scaffolds for regenerative medicine. Chem. Commun. (Camb. Engl.) 2012, 48, 26–33. [Google Scholar] [CrossRef]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef]
- Miles, D.E.; Mitchell, E.A.; Kapur, N.; Beales, P.A.; Wilcox, R.K. Peptide: Glycosaminoglycan hybrid hydrogels as an injectable intervention for spinal disc degeneration. J. Mater. Chem. B 2016, 4, 3225–3231. [Google Scholar] [CrossRef]
- Aggeli, A.; Bell, M.; Boden, N.; Keen, J.N.; Knowles, P.F.; McLeish, T.C.; Pitkeathly, M.; Radford, S.E. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric beta-sheet tapes. Nature 1997, 386, 259–262. [Google Scholar] [CrossRef]
- Yan, D.; Lin, X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol. 2009, 1, a002493. [Google Scholar] [CrossRef]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Martinez-Burgo, B.; Sepuru, K.M.; Rajarathnam, K.; Kirby, J.A.; Sheerin, N.S.; Ali, S. Regulation of Chemokine Function: The Roles of GAG-Binding and Post-Translational Nitration. Int. J. Mol. Sci. 2017, 18, 1692. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, R.; Hoffmann, E.; Sebald, W. Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem. 1996, 237, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.J.; Mulloy, B.; Gallagher, J.T.; Stringer, S.E. VEGF165-binding sites within heparan sulfate encompass two highly sulfated domains and can be liberated by K5 lyase. J. Biol. Chem. 2006, 281, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Makarenkova, H.P.; Hoffman, M.P.; Beenken, A.; Eliseenkova, A.V.; Meech, R.; Tsau, C.; Patel, V.N.; Lang, R.A.; Mohammadi, M. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci. Signal. 2009, 2, ra55. [Google Scholar] [CrossRef] [PubMed]
- Rajangam, K.; Behanna, H.A.; Hui, M.J.; Han, X.; Hulvat, J.F.; Lomasney, J.W.; Stupp, S.I. Heparin binding nanostructures to promote growth of blood vessels. Nano Lett. 2006, 6, 2086–2090. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arterioscler. (DallasTex.) 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Rajangam, K.; Arnold, M.S.; Rocco, M.A.; Stupp, S.I. Peptide amphiphile nanostructure-heparin interactions and their relationship to bioactivity. Biomaterials 2008, 29, 3298–3305. [Google Scholar] [CrossRef]
- Stendahl, J.C.; Wang, L.J.; Chow, L.W.; Kaufman, D.B.; Stupp, S.I. Growth factor delivery from self-assembling nanofibers to facilitate islet transplantation. Transplantation 2008, 86, 478–481. [Google Scholar] [CrossRef]
- Chow, L.W.; Wang, L.J.; Kaufman, D.B.; Stupp, S.I. Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets. Biomaterials 2010, 31, 6154–6161. [Google Scholar] [CrossRef]
- Webber, M.J.; Han, X.; Murthy, S.N.; Rajangam, K.; Stupp, S.I.; Lomasney, J.W. Capturing the stem cell paracrine effect using heparin-presenting nanofibres to treat cardiovascular diseases. J. Tissue Eng. Regen. Med. 2010, 4, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Ghanaati, S.; Webber, M.J.; Unger, R.E.; Orth, C.; Hulvat, J.F.; Kiehna, S.E.; Barbeck, M.; Rasic, A.; Stupp, S.I.; Kirkpatrick, C.J. Dynamic in vivo biocompatibility of angiogenic peptide amphiphile nanofibers. Biomaterials 2009, 30, 6202–6212. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Huang, B.J.; Kaltz, S.R.; Sur, S.; Newcomb, C.J.; Stock, S.R.; Shah, R.N.; Stupp, S.I. Bone regeneration with low dose BMP-2 amplified by biomimetic supramolecular nanofibers within collagen scaffolds. Biomaterials 2013, 34, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Capito, R.M.; Azevedo, H.S.; Velichko, Y.S.; Mata, A.; Stupp, S.I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar] [CrossRef] [PubMed]
- Helen Zha, R.; Velichko, Y.S.; Bitton, R.; Stupp, S.I. Molecular design for growth of supramolecular membranes with hierarchical structure. Soft Matter 2016, 12, 1401–1410. [Google Scholar] [CrossRef]
- Chow, L.W.; Bitton, R.; Webber, M.J.; Carvajal, D.; Shull, K.R.; Sharma, A.K.; Stupp, S.I. A bioactive self-assembled membrane to promote angiogenesis. Biomaterials 2011, 32, 1574–1582. [Google Scholar] [CrossRef]
- Bitton, R.; Chow, L.W.; Zha, R.H.; Velichko, Y.S.; Pashuck, E.T.; Stupp, S.I. Electrostatic Control of Structure in Self-Assembled Membranes. Small (Weinh. Der Bergstr. Ger.) 2014, 10, 500–505. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lee, H.P.; Chan, H.Y.; Sung, L.Y.; Chen, H.C.; Hu, Y.C. Composite chondroitin-6-sulfate/dermatan sulfate/chitosan scaffolds for cartilage tissue engineering. Biomaterials 2007, 28, 2294–2305. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, H.C.; Chan, H.Y.; Chuang, C.K.; Chang, Y.H.; Hu, Y.C. Co-conjugating chondroitin-6-sulfate/dermatan sulfate to chitosan scaffold alters chondrocyte gene expression and signaling profiles. Biotechnol. Bioeng. 2008, 101, 821–830. [Google Scholar] [CrossRef]
- Soriano-Ruiz, J.L.; Galvez-Martin, P.; Lopez-Ruiz, E.; Suner-Carbo, J.; Calpena-Campmany, A.C.; Marchal, J.A.; Clares-Naveros, B. Design and evaluation of mesenchymal stem cells seeded chitosan/glycosaminoglycans quaternary hydrogel scaffolds for wound healing applications. Int. J. Pharm. 2019, 570, 118632. [Google Scholar] [CrossRef]
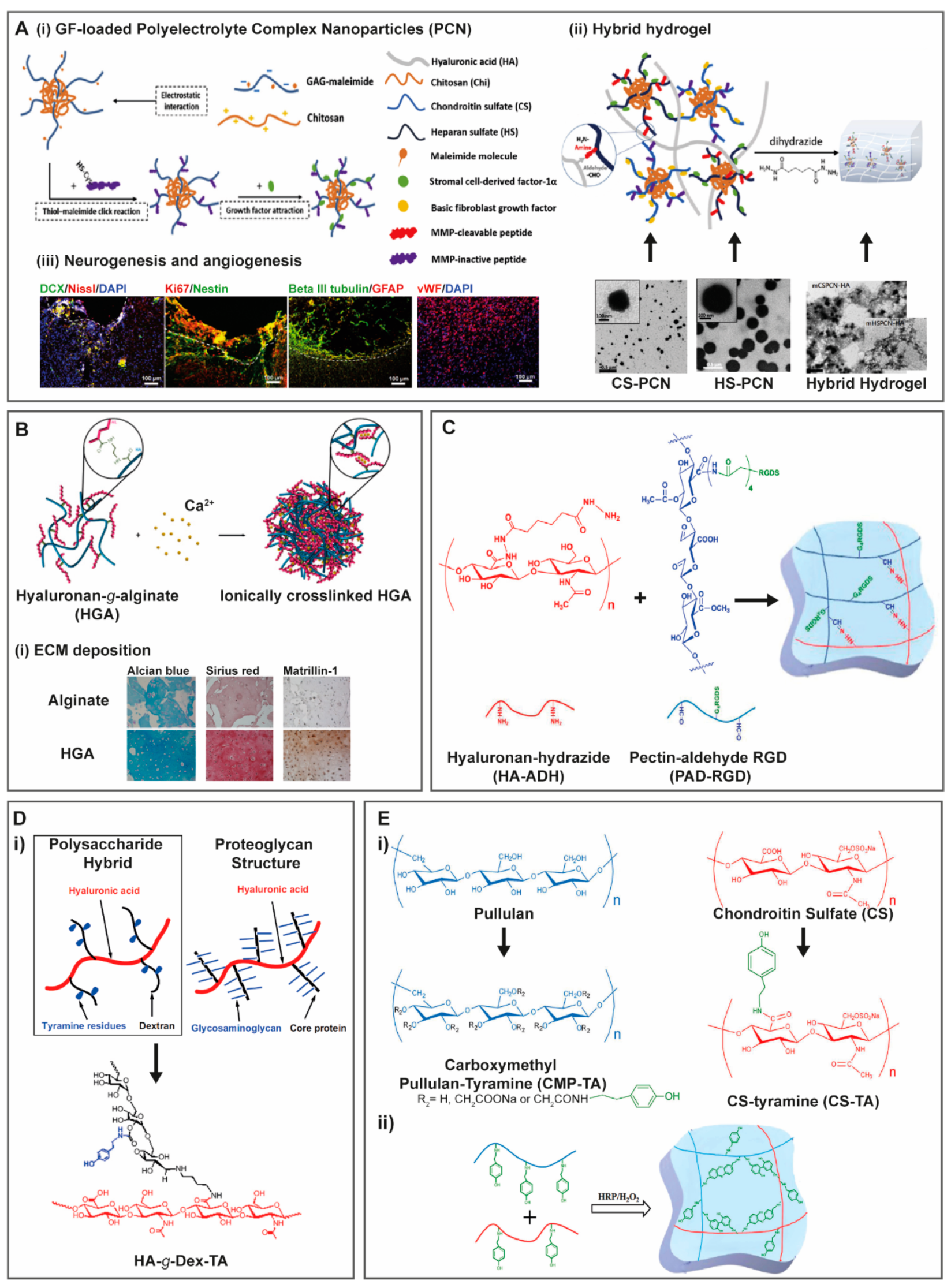
- Jian, W.H.; Wang, H.C.; Kuan, C.H.; Chen, M.H.; Wu, H.C.; Sun, J.S.; Wang, T.W. Glycosaminoglycan-based hybrid hydrogel encapsulated with polyelectrolyte complex nanoparticles for endogenous stem cell regulation in central nervous system regeneration. Biomaterials 2018, 174, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, B.; Sionkowska, A.; Osyczka, A.M. Scaffolds based on chitosan and collagen with glycosaminoglycans cross-linked by tannic acid. Polym. Test. 2018, 65, 163–168. [Google Scholar] [CrossRef]
- Kaczmarek, B.; Sionkowska, A.; Łukowicz, K.; Maria Osyczka, A. The cells viability study on the composites of chitosan and collagen with glycosaminoglycans isolated from fish skin. Mater. Lett. 2017, 206, 166–168. [Google Scholar] [CrossRef]
- Yar, M.; Shahzad, S.; Shahzadi, L.; Shahzad, S.A.; Mahmood, N.; Chaudhry, A.A.; Rehman, I.U.; MacNeil, S. Heparin binding chitosan derivatives for production of pro-angiogenic hydrogels for promoting tissue healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 74, 347–356. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, H.C.; Lee, H.P.; Chan, H.Y.; Hu, Y.C. Rational development of GAG-augmented chitosan membranes by fractional factorial design methodology. Biomaterials 2006, 27, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Uygun, B.E.; Stojsih, S.E.; Matthew, H.W. Effects of immobilized glycosaminoglycans on the proliferation and differentiation of mesenchymal stem cells. Tissue Eng. Part A 2009, 15, 3499–3512. [Google Scholar] [CrossRef]
- Nunes, C.S.; Rufato, K.B.; Souza, P.R.; de Almeida, E.; da Silva, M.J.V.; Scariot, D.B.; Nakamura, C.V.; Rosa, F.A.; Martins, A.F.; Muniz, E.C. Chitosan/chondroitin sulfate hydrogels prepared in [Hmim][HSO4] ionic liquid. Carbohydr. Polym. 2017, 170, 99–106. [Google Scholar] [CrossRef]
- Daley, E.L.; Coleman, R.M.; Stegemann, J.P. Biomimetic microbeads containing a chondroitin sulfate/chitosan polyelectrolyte complex for cell-based cartilage therapy. J. Mater. Chem. B 2015, 3, 7920–7929. [Google Scholar] [CrossRef]
- Sharma, S.; Swetha, K.L.; Roy, A. Chitosan-Chondroitin sulfate based polyelectrolyte complex for effective management of chronic wounds. Int. J. Biol. Macromol. 2019, 132, 97–108. [Google Scholar] [CrossRef]
- Piai, J.F.; Rubira, A.F.; Muniz, E.C. Self-assembly of a swollen chitosan/chondroitin sulfate hydrogel by outward diffusion of the chondroitin sulfate chains. Acta Biomater. 2009, 5, 2601–2609. [Google Scholar] [CrossRef]
- Rodrigues, M.N.; Oliveira, M.B.; Costa, R.R.; Mano, J.F. Chitosan/Chondroitin Sulfate Membranes Produced by Polyelectrolyte Complexation for Cartilage Engineering. Biomacromolecules 2016, 17, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Ma, Y.; Tan, H.; Jia, Y.; Zou, S.; Guo, S.; Zhao, M.; Huang, H.; Ling, Z.; Chen, Y.; et al. Covalent and injectable chitosan-chondroitin sulfate hydrogels embedded with chitosan microspheres for drug delivery and tissue engineering. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 71, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Dawlee, S.; Sugandhi, A.; Balakrishnan, B.; Labarre, D.; Jayakrishnan, A. Oxidized Chondroitin Sulfate-Cross-Linked Gelatin Matrixes: A New Class of Hydrogels. Biomacromolecules 2005, 6, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-D.; Zhai, P.; Schreyer, D.J.; Zheng, R.-S.; Sun, X.-D.; Cui, F.-Z.; Chen, X.-B. Novel crosslinked alginate/hyaluronic acid hydrogels for nerve tissue engineering. Front. Mater. Sci. 2013, 7, 269–284. [Google Scholar] [CrossRef]
- Ansari, S.; Diniz, I.M.; Chen, C.; Sarrion, P.; Tamayol, A.; Wu, B.M.; Moshaverinia, A. Human Periodontal Ligament- and Gingiva-derived Mesenchymal Stem Cells Promote Nerve Regeneration When Encapsulated in Alginate/Hyaluronic Acid 3D Scaffold. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Peng, C.; Huang, T.; Zhou, H.; Ou, B.; Chen, J.; Liu, Q.; He, S.; Cao, D.; et al. Fabrication and Physical Properties of Gelatin/Sodium Alginate/Hyaluronic Acid Composite Wound Dressing Hydrogel. J. Macromol. Sci. Part. A 2014, 51, 318–325. [Google Scholar] [CrossRef]
- Catanzano, O.; D’Esposito, V.; Pulcrano, G.; Maiolino, S.; Ambrosio, M.R.; Esposito, M.; Miro, A.; Ungaro, F.; Formisano, P.; Catania, M.R.; et al. Ultrasmall silver nanoparticles loaded in alginate–hyaluronic acid hybrid hydrogels for treating infected wounds. Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 626–634. [Google Scholar] [CrossRef]
- Ansari, S.; Diniz, I.M.; Chen, C.; Aghaloo, T.; Wu, B.M.; Shi, S.; Moshaverinia, A. Alginate/hyaluronic acid hydrogel delivery system characteristics regulate the differentiation of periodontal ligament stem cells toward chondrogenic lineage. J. Mater. Sci. Mater. Med. 2017, 28, 162. [Google Scholar] [CrossRef]
- Mahapatra, C.; Jin, G.Z.; Kim, H.W. Alginate-hyaluronic acid-collagen composite hydrogel favorable for the culture of chondrocytes and their phenotype maintenance. Tissue Eng Regen Med. 2016, 13, 538–546. [Google Scholar] [CrossRef]
- Ma, F.; Pang, X.; Tang, B. Alginate/chondroitin sulfate based hybrid hydrogel with different molecular weight and its capacity to regulate chondrocytes activity. Carbohydr. Polym. 2019, 206, 229–237. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, S.H.; Kim, I.G.; Lee, J.H.; Kwon, S.K. Injectable basic fibroblast growth factor-loaded alginate/hyaluronic acid hydrogel for rejuvenation of geriatric larynx. Acta Biomater. 2019, 89, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Forsythe, S.; He, Y.; Liu, Q.; Xiong, G.; Wei, S.; Li, G.; Atala, A.; Skardal, A.; Zhang, Y. Tissue-specific extracellular matrix promotes myogenic differentiation of human muscle progenitor cells on gelatin and heparin conjugated alginate hydrogels. Acta Biomater. 2017, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Yoshikawa, M.; Maeda, H. Hard Tissue-Forming Ability and Ultra-Micro Structure of Newly Developed Sponges as Scaffolds Made with Sodium Alginate Gel and Chondroitin Sulfate. J. Biomed. Sci. Eng. 2018, 11, 289–306. [Google Scholar] [CrossRef][Green Version]
- Park, H.; Woo, E.K.; Lee, K.Y. Ionically cross-linkable hyaluronate-based hydrogels for injectable cell delivery. J. Control. Release 2014, 196, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.; Lee, J.W.; Lee, K.Y. Magnetic field-responsive release of transforming growth factor beta 1 from heparin-modified alginate ferrogels. Carbohydr. Polym. 2016, 151, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Zhong, N.; Huang, Y.; He, K.; Ye, X. Injectable in situ dual-crosslinking hyaluronic acid and sodium alginate based hydrogels for drug release. J. Biomater. Sci. Polym. Ed. 2019, 30, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Noreen, A.; Nazli, Z.I.; Akram, J.; Rasul, I.; Mansha, A.; Yaqoob, N.; Iqbal, R.; Tabasum, S.; Zuber, M.; Zia, K.M. Pectins functionalized biomaterials; a new viable approach for biomedical applications: A review. Int. J. Biol. Macromol. 2017, 101, 254–272. [Google Scholar] [CrossRef]
- Ji, F.; Li, J.; Qin, Z.; Yang, B.; Zhang, E.; Dong, D.; Wang, J.; Wen, Y.; Tian, L.; Yao, F. Engineering pectin-based hollow nanocapsules for delivery of anticancer drug. Carbohydr. Polym. 2017, 177, 86–96. [Google Scholar] [CrossRef]
- Bayón, B.; Bucalá, V.; Castro, G.R. Development of antimicrobial hybrid mesoporous silver phosphate-pectin microspheres for control release of levofloxacin. Microporous Mesoporous Mater. 2016, 226, 71–78. [Google Scholar] [CrossRef]
- Giusto, G.; Vercelli, C.; Comino, F.; Caramello, V.; Tursi, M.; Gandini, M. A new, easy-to-make pectin-honey hydrogel enhances wound healing in rats. BMC Complementary Altern. Med. 2017, 17, 1–7. [Google Scholar] [CrossRef]
- Neves, S.C.; Gomes, D.B.; Sousa, A.; Bidarra, S.J.; Petrini, P.; Moroni, L.; Barrias, C.C.; Granja, P.L. Biofunctionalized pectin hydrogels as 3D cellular microenvironments. J. Mater. Chem. B 2015, 3, 2096–2108. [Google Scholar] [CrossRef]
- Pereira, R.F.; Barrias, C.C.; Bartolo, P.J.; Granja, P.L. Cell-instructive pectin hydrogels crosslinked via thiol-norbornene photo-click chemistry for skin tissue engineering. Acta Biomater. 2018, 66, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Liu, Y.; Yang, F.; Xiu, Z.; Ma, X.; Sun, G. Preparation and optimization of calcium pectate beads for cell encapsulation. J. Appl. Polym. Sci. 2018, 135. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Li, X.; Xiao, S.; Zheng, D.; Zhu, P.; Li, C.; Liu, J.; He, J.; Lei, J.; et al. A novel self-assembled nanoparticle platform based on pectin-eight-arm polyethylene glycol-drug conjugates for co-delivery of anticancer drugs. Mater. Sci. Eng. C 2018, 86, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Sabra, R.; Billa, N.; Roberts, C.J. An augmented delivery of the anticancer agent, curcumin, to the colon. React. Funct. Polym. 2018, 123, 54–60. [Google Scholar] [CrossRef]
- Munarin, F.; Tanzi, M.C.; Petrini, P. Advances in biomedical applications of pectin gels. Int. J. Biol. Macromol. 2012, 51, 681–689. [Google Scholar] [CrossRef]
- Braccini, I.; Perez, S. Molecular basis of Ca2+-induced gelation in alginates and pectins: The egg-box model revisited. Biomacromolecules 2001, 2, 1089–1096. [Google Scholar] [CrossRef]
- Siew, C.K.; Williams, P.A.; Young, N.W.G. New Insights into the Mechanism of Gelation of Alginate and Pectin: Charge Annihilation and Reversal Mechanism. Biomacromolecules 2005, 6, 963–969. [Google Scholar] [CrossRef]
- Kohn, R. Ion binding on polyuronates - alginate and pectin. Pure Appl. Chem. 1975, 42, 371. [Google Scholar] [CrossRef]
- Chen, F.; Ni, Y.; Liu, B.; Zhou, T.; Yu, C.; Su, Y.; Zhu, X.; Yu, X.; Zhou, Y. Self-crosslinking and injectable hyaluronic acid/RGD-functionalized pectin hydrogel for cartilage tissue engineering. Carbohydr. Polym. 2017. [Google Scholar] [CrossRef]
- Luo, Y.; Kobler, J.B.; Heaton, J.T.; Jia, X.; Zeitels, S.M.; Langer, R. Injectable hyaluronic acid-dextran hydrogels and effects of implantation in ferret vocal fold. J. Biomed. Mater. Res. B Appl. Biomater. 2010, 93, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Teixeira, L.S.; Dijkstra, P.J.; van Blitterswijk, C.A.; Karperien, M.; Feijen, J. Enzymatically-crosslinked injectable hydrogels based on biomimetic dextran-hyaluronic acid conjugates for cartilage tissue engineering. Biomaterials 2010, 31, 3103–3113. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.X.; O’Rear, E.A. Modified dextran, heparin-based triggered release microspheres for cardiovascular delivery of therapeutic drugs using protamine as a stimulus. J. Microencapsul. 2017, 34, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Cao, L.J.; Shi, Y.; Wang, P.; Chen, J.H. In situ supramolecular hydrogel based on hyaluronic acid and dextran derivatives as cell scaffold. J. Biomed. Mater. Res. A 2016, 104, 2263–2270. [Google Scholar] [CrossRef]
- Pescosolido, L.; Schuurman, W.; Malda, J.; Matricardi, P.; Alhaique, F.; Coviello, T.; van Weeren, P.R.; Dhert, W.J.; Hennink, W.E.; Vermonden, T. Hyaluronic acid and dextran-based semi-IPN hydrogels as biomaterials for bioprinting. Biomacromolecules 2011, 12, 1831–1838. [Google Scholar] [CrossRef]
- Budtova, T.; Navard, P. Cellulose in NaOH–water based solvents: A review. Cellulose 2015, 23, 5–55. [Google Scholar] [CrossRef]
- Medronho, B.; Lindman, B. Brief overview on cellulose dissolution/regeneration interactions and mechanisms. Adv Colloid Interface Sci. 2015, 222, 502–508. [Google Scholar] [CrossRef]
- Minnick, D.L.; Flores, R.A.; DeStefano, M.R.; Scurto, A.M. Cellulose Solubility in Ionic Liquid Mixtures: Temperature, Cosolvent, and Antisolvent Effects. J. Phys. Chem. B 2016, 120, 7906–7919. [Google Scholar] [CrossRef]
- Domingues, R.M.; Silva, M.; Gershovich, P.; Betta, S.; Babo, P.; Caridade, S.G.; Mano, J.F.; Motta, A.; Reis, R.L.; Gomes, M.E. Development of Injectable Hyaluronic Acid/Cellulose Nanocrystals Bionanocomposite Hydrogels for Tissue Engineering Applications. Bioconj. Chem. 2015, 26, 1571–1581. [Google Scholar] [CrossRef]
- Jia, Y.; Zhu, W.; Zheng, M.; Huo, M.; Zhong, C. Bacterial cellulose/hyaluronic acid composite hydrogels with improved viscoelastic properties and good thermodynamic stability. Plast. Rubber Compos. 2018, 47, 165–175. [Google Scholar] [CrossRef]
- Karimi Dehkordi, N.; Minaiyan, M.; Talebi, A.; Akbari, V.; Taheri, A. Nanocrystalline cellulose-hyaluronic acid composite enriched with GM-CSF loaded chitosan nanoparticles for enhanced wound healing. Biomed. Mater. 2019, 14, 035003. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Liu, L.; Xu, W.; Fan, L.; Nie, M. Preparation and characterization of aminated hyaluronic acid/oxidized hydroxyethyl cellulose hydrogel. Carbohydr. Polym. 2018, 199, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Law, N.; Doney, B.; Glover, H.; Qin, Y.; Aman, Z.M.; Sercombe, T.B.; Liew, L.J.; Dilley, R.J.; Doyle, B.J. Characterisation of hyaluronic acid methylcellulose hydrogels for 3D bioprinting. J. Mech. Behav. Biomed. Mater. 2018, 77, 389–399. [Google Scholar] [CrossRef]
- Deng, S.; Li, X.; Yang, W.; He, K.; Ye, X. Injectable in situ cross-linking hyaluronic acid/carboxymethyl cellulose based hydrogels for drug release. J. Biomater. Sci. Polym. Ed. 2018, 29, 1643–1655. [Google Scholar] [CrossRef]
- Singh, R.S.; Kaur, N.; Rana, V.; Kennedy, J.F. Pullulan: A novel molecule for biomedical applications. Carbohydr. Polym. 2017, 171, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xue, Y.; Jia, B.; Bai, Y.; Zuo, Y.; Wang, S.; Zhao, Y.; Yang, W.; Tang, H. The preparation of hyaluronic acid grafted pullulan polymers and their use in the formation of novel biocompatible wound healing film. Carbohydr. Polym. 2018, 188, 92–100. [Google Scholar] [CrossRef]
- Chen, F.; Yu, S.; Liu, B.; Ni, Y.; Yu, C.; Su, Y.; Zhu, X.; Yu, X.; Zhou, Y.; Yan, D. An Injectable Enzymatically Crosslinked Carboxymethylated Pullulan/Chondroitin Sulfate Hydrogel for Cartilage Tissue Engineering. Sci Rep. 2016, 6, 20014. [Google Scholar] [CrossRef]
- Li, T.; Song, X.; Weng, C.; Wang, X.; Sun, L.; Gong, X.; Yang, L.; Chen, C. Self-crosslinking and injectable chondroitin sulfate/pullulan hydrogel for cartilage tissue engineering. Appl. Mater. Today 2018, 10, 173–183. [Google Scholar] [CrossRef]
- Kim, Y.S.; Majid, M.; Melchiorri, A.J.; Mikos, A.G. Applications of decellularized extracellular matrix in bone and cartilage tissue engineering. Bioeng. Transl. Med. 2019, 4, 83–95. [Google Scholar] [CrossRef]
- Ye, X.; Wang, H.; Gong, W.; Li, S.; Li, H.; Wang, Z.; Zhao, Q. Impact of decellularization on porcine myocardium as scaffold for tissue engineered heart tissue. J. Mater. Sci.: Mater. Med. 2016, 27, 70. [Google Scholar] [CrossRef]
- Roosens, A.; Somers, P.; De Somer, F.; Carriel, V.; Van Nooten, G.; Cornelissen, R. Impact of Detergent-Based Decellularization Methods on Porcine Tissues for Heart Valve Engineering. Ann. Biomed. Eng. 2016, 44, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Beachley, V.; Ma, G.; Papadimitriou, C.; Gibson, M.; Corvelli, M.; Elisseeff, J. Extracellular matrix particle-glycosaminoglycan composite hydrogels for regenerative medicine applications. J. Biomed. Mater. Res. A 2018, 106, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lyu, Z.; Chen, G.; Wang, H.; Yuan, Y.; Ding, K.; Yu, Q.; Yuan, L.; Chen, H. A new avenue to the synthesis of GAG-mimicking polymers highly promoting neural differentiation of embryonic stem cells. Chem. Commun. 2015, 51, 15434–15437. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel, J.; Novoa-Carballal, R.; Pérez-Martín, R.I.; Reis, R.L.; Vázquez, J.A. Glycosaminoglycans from marine sources as therapeutic agents. Biotechnol. Adv. 2017, 35, 711–725. [Google Scholar] [CrossRef]
- Regan, J.R.; Bruno, J.G.; Chang, M.N.; Sabatino, R.; D’Alisa, R.; Ben-Sasson, S.A.; Eilat, D. Mimicry of Biological Macromolecules by Polyaromatic Anionic Compounds. J. Bioact. Compat. Polym. 1993, 8, 317–337. [Google Scholar] [CrossRef]
- Benezra, M.; Ishai-Michaeli, R.; Ben-Sasson, S.A.; Vlodavsky, I. Structure–activity relationships of heparin-mimicking compounds in induction of bFGF release from extracellular matrix and inhibition of smooth muscle cell proliferation and heparanase activity. J. Cell. Physiol. 2002, 192, 276–285. [Google Scholar] [CrossRef]
- Liekens, S.; Leali, D.; Neyts, J.; Esnouf, R.; Rusnati, M.; Dell’Era, P.; Maudgal, P.C.; De Clercq, E.; Presta, M. Modulation of Fibroblast Growth Factor-2 Receptor Binding, Signaling, and Mitogenic Activity by Heparin-Mimicking Polysulfonated Compounds. Mol. Pharmacol. 1999, 56, 204–213. [Google Scholar] [CrossRef]
- Christman, K.L.; Vázquez-Dorbatt, V.; Schopf, E.; Kolodziej, C.M.; Li, R.C.; Broyer, R.M.; Chen, Y.; Maynard, H.D. Nanoscale Growth Factor Patterns by Immobilization on a Heparin-Mimicking Polymer. J. Am. Chem. Soc. 2008, 130, 16585–16591. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Kim, S.-H.; Decker, C.G.; Wong, D.Y.; Loo, J.A.; Maynard, H.D. A heparin-mimicking polymer conjugate stabilizes basic fibroblast growth factor. Nat. Chem. 2013, 5, 221. [Google Scholar] [CrossRef]
- Grande, D.; Baskaran, S.; Baskaran, C.; Gnanou, Y.; Chaikof, E.L. Glycosaminoglycan-Mimetic Biomaterials. 1. Nonsulfated and Sulfated Glycopolymers by Cyanoxyl-Mediated Free-Radical Polymerization. Macromolecules 2000, 33, 1123–1125. [Google Scholar] [CrossRef]
- Sun, X.-L.; Grande, D.; Baskaran, S.; Hanson, S.R.; Chaikof, E.L. Glycosaminoglycan Mimetic Biomaterials. 4. Synthesis of Sulfated Lactose-Based Glycopolymers That Exhibit Anticoagulant Activity. Biomacromolecules 2002, 3, 1065–1070. [Google Scholar] [CrossRef]
- Lee, S.-G.; Brown, J.M.; Rogers, C.J.; Matson, J.B.; Krishnamurthy, C.; Rawat, M.; Hsieh-Wilson, L.C. End-functionalized glycopolymers as mimetics of chondroitin sulfate proteoglycans. Chem. Sci. 2010, 1, 322–325. [Google Scholar] [CrossRef]
- Li, G.; Chen, S.; Wang, Y.; Xue, Y.; Chang, Y.; Li, Z.; Wang, J.; Xue, C. A novel glycosaminoglycan-like polysaccharide from abalone Haliotis discus hannai Ino: Purification, structure identification and anticoagulant activity. Int. J. Biol. Macromol. 2011, 49, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Cláudia, N.; Manuel, A.C. The Potential of Fucose-Containing Sulfated Polysaccharides As Scaffolds for Biomedical Applications. Curr. Med. Chem. 2019, 26, 1–13. [Google Scholar]
- Senni, K.; Gueniche, F.; Foucault-Bertaud, A.; Igondjo-Tchen, S.; Fioretti, F.; Colliec-Jouault, S.; Durand, P.; Guezennec, J.; Godeau, G.; Letourneur, D. Fucoidan a sulfated polysaccharide from brown algae is a potent modulator of connective tissue proteolysis. Arch. Biochem. Biophys. 2006, 445, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Merceron, C.; Portron, S.; Vignes-Colombeix, C.; Rederstorff, E.; Masson, M.; Lesoeur, J.; Sourice, S.; Sinquin, C.; Colliec-Jouault, S.; Weiss, P.; et al. Pharmacological Modulation of Human Mesenchymal Stem Cell Chondrogenesis by a Chemically Oversulfated Polysaccharide of Marine Origin: Potential Application to Cartilage Regenerative Medicine. STEM CELLS 2012, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Mauzac, M.; Aubert, N.; Jozefonvicz, J. Antithrombic activity of some polysaccharide resins. Biomaterials 1982, 3, 221–224. [Google Scholar] [CrossRef]
- Paluck, S.J.; Nguyen, T.H.; Maynard, H.D. Heparin-Mimicking Polymers: Synthesis and Biological Applications. Biomacromolecules 2016, 17, 3417–3440. [Google Scholar] [CrossRef]
- Peschel, D.; Zhang, K.; Fischer, S.; Groth, T. Modulation of osteogenic activity of BMP-2 by cellulose and chitosan derivatives. Acta Biomater. 2012, 8, 183–193. [Google Scholar] [CrossRef]
- Ding, K.; Wang, Y.; Wang, H.; Yuan, L.; Tan, M.; Shi, X.; Lyu, Z.; Liu, Y.; Chen, H. 6-O-Sulfated Chitosan Promoting the Neural Differentiation of Mouse Embryonic Stem Cells. ACS Appl. Mater. Interfaces 2014, 6, 20043–20050. [Google Scholar] [CrossRef]
- Fan, L.; Jiang, L.; Xu, Y.; Zhou, Y.; Shen, Y.; Xie, W.; Long, Z.; Zhou, J. Synthesis and anticoagulant activity of sodium alginate sulfates. Carbohydr. Polym. 2011, 83, 1797–1803. [Google Scholar] [CrossRef]
- Ronghua, H.; Yumin, D.; Jianhong, Y. Preparation and in vitro anticoagulant activities of alginate sulfate and its quaterized derivatives. Carbohydr. Polym. 2003, 52, 19–24. [Google Scholar] [CrossRef]
- Mhanna, R.; Becher, J.; Schnabelrauch, M.; Reis, R.L.; Pashkuleva, I. Sulfated Alginate as a Mimic of Sulfated Glycosaminoglycans: Binding of Growth Factors and Effect on Stem Cell Behavior. Adv. Biosyst. 2017, 1, 1700043. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, M.I.; Araújo, M.; Moroni, L.; da Silva, R.M.P.; Barrias, C.C. Glycosaminoglycan-Inspired Biomaterials for the Development of Bioactive Hydrogel Networks. Molecules 2020, 25, 978. https://doi.org/10.3390/molecules25040978
Neves MI, Araújo M, Moroni L, da Silva RMP, Barrias CC. Glycosaminoglycan-Inspired Biomaterials for the Development of Bioactive Hydrogel Networks. Molecules. 2020; 25(4):978. https://doi.org/10.3390/molecules25040978
Chicago/Turabian StyleNeves, Mariana I., Marco Araújo, Lorenzo Moroni, Ricardo M.P. da Silva, and Cristina C. Barrias. 2020. "Glycosaminoglycan-Inspired Biomaterials for the Development of Bioactive Hydrogel Networks" Molecules 25, no. 4: 978. https://doi.org/10.3390/molecules25040978
APA StyleNeves, M. I., Araújo, M., Moroni, L., da Silva, R. M. P., & Barrias, C. C. (2020). Glycosaminoglycan-Inspired Biomaterials for the Development of Bioactive Hydrogel Networks. Molecules, 25(4), 978. https://doi.org/10.3390/molecules25040978