1. Introduction
KYNA (kynurenic acid) is an endogenous product of the tryptophan (TRP) metabolism, a pathway known to be responsible for the production of nicotinamide adenine dinucleotide (NAD
+) and NAD phosphate (NADP
+) [
1,
2]. In this pathway, TRP is converted into various compounds, including L-kynurenine (L-KYN), which can be metabolised in two separate ways. One furnishes KYNA, whereas the other gives 3-hydroxykynurenine (3-OH-KYN) and quinolinic acid (QUIN), the precursors of NAD [
3,
4].
Among the important features of KYNA, one is that it is one of the few known endogenous excitatory amino acid receptor blockers with a broad spectrum of antagonistic properties in supraphysiological concentrations. One of its confirmed sites of action is the α-7-nicotinic acetylcholine (α-7-nACh) receptor and, interestingly, the other, identified recently, is a higher-affinity positive modulatory binding site at the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor [
5].
Since KYNA is a neuroprotective agent able to prevent neuronal loss following excitotoxic, ischemia-induced, and infectious neuronal injuries [
6,
7], there has recently been increasing interest in the synthesis and pharmacological studies of KYNA derivatives. The substitution of KYNA at positions 5–8 were achieved by starting from the corresponding aniline via the modified Conrad–Limpach method [
8,
9,
10]. The hydroxy group at position 4 was transformed to ether [
10,
11,
12] or amine functions [
13,
14,
15], while the carboxylic function at position 2 was mostly modified into the corresponding esters [
10,
11,
12] or amides [
16,
17,
18,
19,
20,
21].
Formally, KYNA can be considered to be a nitrogen-containing 1-naphthol derivative. In our previous studies, 1-naphthol and its
N-containing analogues were successfully applied in the modified Mannich reaction (
mMr) [
22] leading to the corresponding aminonaphthols [
23], aminoquinolinols or aminoisoquinolinols [
24,
25]. A similar transformation starting from xanthurenic acid has been described by Schmitt et al. [
26]. They managed to perform regioselective aminoalkylation at position 3 on this substrate, by using benzyl-protection of 8-hydroxyl group. Our present aim was to examine the possibility of aminoalkylation of KYNA derivatives at position 3 via a
mMr approach. Since it is postulated that the reactivity of KYNA derivatives is influenced by tautomerism, we also envisaged identifying the tautomeric form dominant in solution. Finally, the reactivity of representative KYNA amides, carrying
N-dialkylaminoalkyl-type side-chain possibly protonated under the standard reaction conditions, was also planned to be tested as these substrates with slightly decreased electron density at C-3 might display a decreased reactivity compared to that produced by a KYNA ester.
2. Results
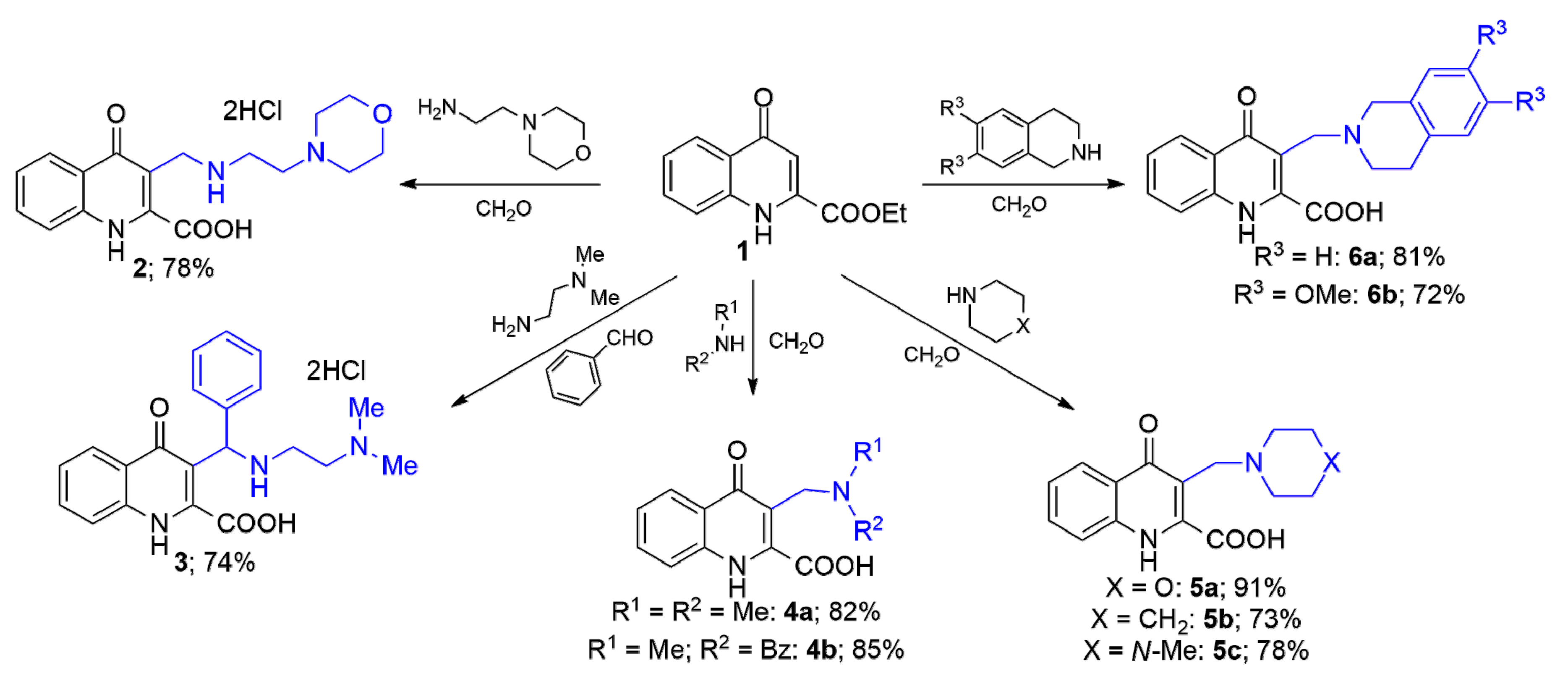
In our first experiments, ethyl 4-oxo-1,4-dihydroquinoline-2-carboxylate (1) was reacted with 2-morpholinoethylamine in the presence of aqueous formaldehyde (22% solution). The reaction was conducted in different solvents (acetonitrile, N,N-dimethylformamide, EtOH, and 1,4-dioxane) at different temperatures (60 °C, 80 °C, and 110 °C). The optimised reaction conditions were found to be reflux temperature, using 1,4-dioxane as solvent, and 5 h reaction time. All reaction conditions led to the formation of 3-(((2-morpholinoethyl)amino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (2) as the single product. It is interesting to note that the media was basic enough to lead to the hydrolysis of the ester function.
To explore the scope and limitation of the transformation,
1 was first reacted with
N,
N-dimethylethane-1,2-diamine in the presence of benzaldehyde. The desired amino acid derivative
3 was isolated in a yield of 74%. Next, starting from dimethylamine or
N-benzylmethylamine in the
mMr, the insertion of cationic centers in one-carbon distance at position 3 can be achieved. The structures of the formed amino acids
4a and
4b are depicted in
Scheme 1.
The extension possibility of the reaction was further tested by starting from cyclic secondary amines such as morpholine, piperidine or N-methylpiperazine leading to 5a, 5b or 5c, respectively. As last representative amines 1,2,3,4-tetrahydroisoquinoline and its dimethoxy analogue were chosen as aromatic fused cyclic secondary amines. In these cases, relatively long reaction times (8 h and 10 h) led to the formation of 3-((3,4-dihydroisoquinolin-2(1H)-yl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (6a) and 3-((6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (6b).
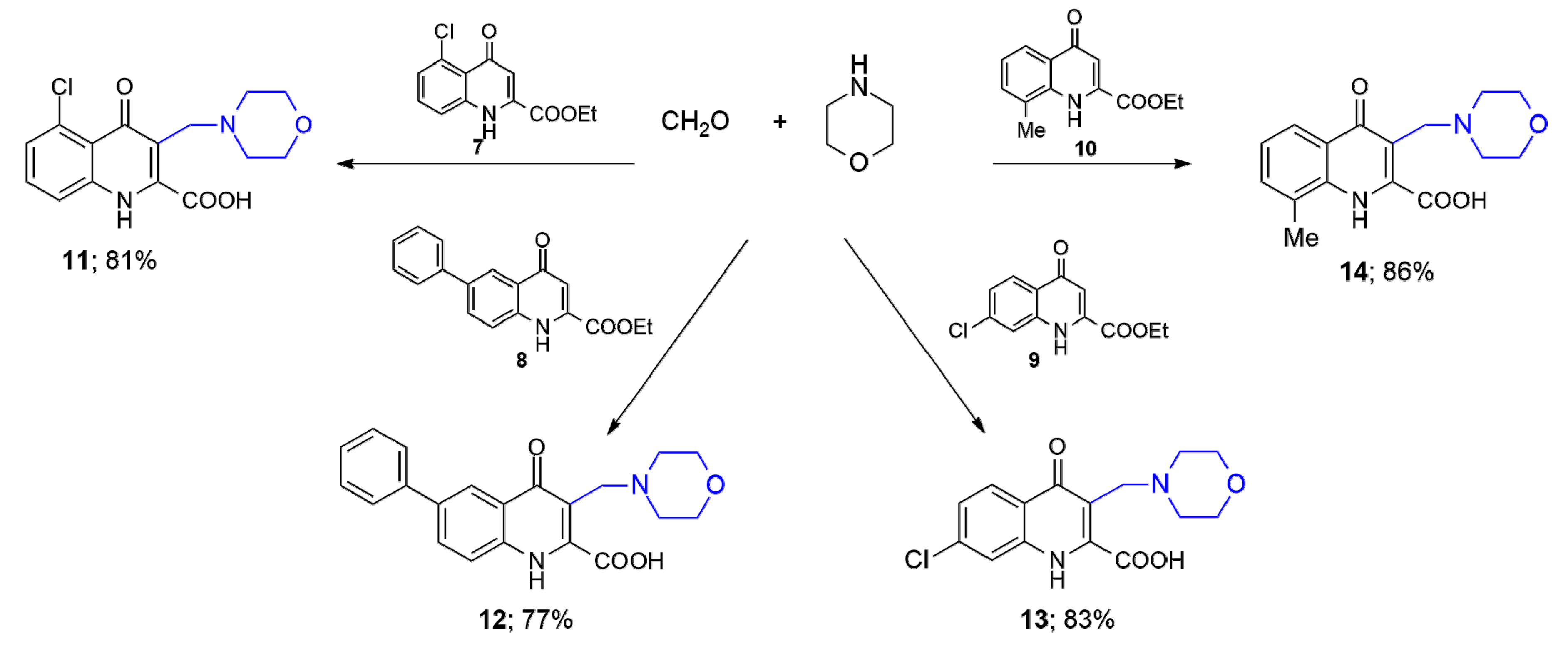
To test the effect of substituent at 5-, 6-, 7- or 8-positions morpholine as a representative secondary cyclic amine was selected. First, the initial KYNA analogues (
7–
10) were synthesised with the Conrad–Limpach method applying the following optimization: a) the intermediate was purified by column chromatography using n-hexane:EtOAc as eluent and b) ring closure was carried out utilizing 1,2-dichlorobenzene instead of diphenyl ether used in the literature [
8]. Reactions leading to the formation of
11–
14 were performed starting from the corresponding ethyl ester and morpholine in the presence of formaldehyde (
Scheme 2). It can be concluded that aryl/alkyl substituents at position 6 or 8, and the halogen at position 5 or 7 have no significant influence on the substitution at position 3.
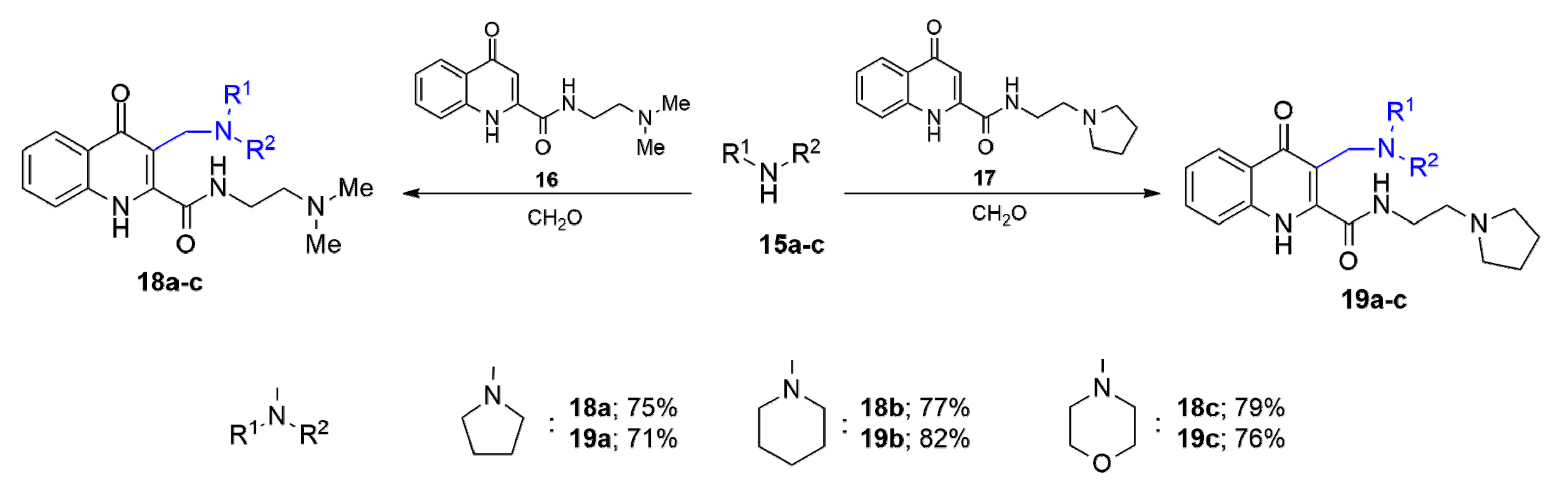
Since KYNA amides
16 and
17—containing cationic side-chain—proved to be the most effective analogues [
27,
28], our attention focused on the substitution of these compounds at position 3 via the
mMr. In this reaction, different secondary amines such as pyrrolidine, piperidine, and morpholine were tested. Substrates
N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (
16) and 4-oxo-
N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (
17), synthesised according to a literature method [
21], were aminoalkylated with
15a–
c in the presence of formaldehyde, resulting in aminoalkylated KYNA derivatives
18a–
c and
19a–
c, respectively (
Scheme 3).
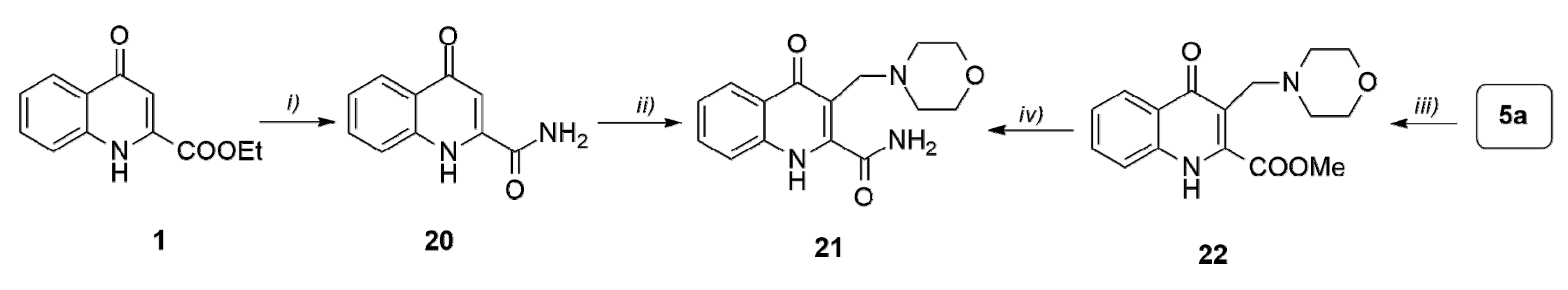
Summarizing these results and those of a previous study [
29], the synthesis of this type of KYNA derivatives containing an amide moiety at position 2 and an aminoalkyl group at position 3 can be achieved by applying two different synthetic pathways: amidation followed by aminoalkylation (route A) and a reverse reaction sequence (route B). Our results show that route A, that is amidation followed by aminoalkylation is more favorable, since it affords higher yields. For further investigation, the synthesis of
21 as representative aminoalkylated amide has been selected.
A comparison of the overall yields to obtain
21 by using the two approaches shows that amidation followed by aminoalkylation (route A) resulted in the formation of the desired compounds in slightly higher yield (
Scheme 4).
In the frame of our previous work, KYNA derivatives containing cationic centers (5a, 16, 18c, 17, 19c) have been surveyed in blood-brain-barrier models. It was concluded that 5a, 18c, and 19c have higher permeability towards the brain compared with amides 16 and 17. In the case of the permeability measured from the brain to the outer compartment, compounds 5a and 18c showed the most promising results [P1]. Further investigation of the transport mechanism is still in progress.
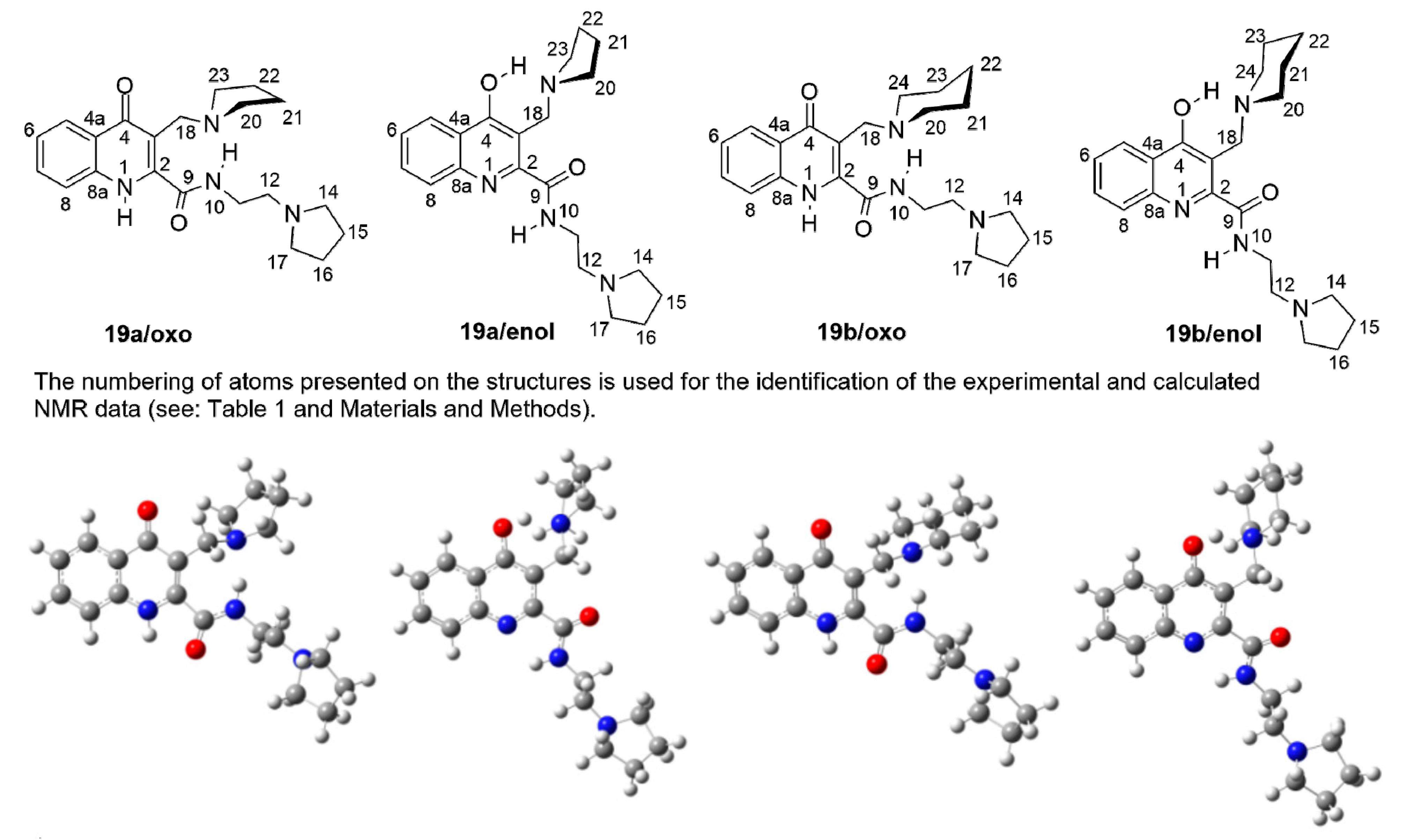
Finally, on two selected aminoalkylated KYNA amides
19a and
19b with closely related structures we studied the possible oxo-enol tautomerisation associated with the intramolecular hydrogen-bond framework that might exert significant influence on the intermolecular binding properties thus, on the drug-like character of these highly fuctionalised heterocycles. On the basis of the cross-peak correlations discernible in the 2D-COSY-, 2D-HSQC- and 2D-HMBC spectra, complete assignment of
1H- and
13C-NMR data was performed for the two investigated compounds assumed first to have oxo structures (
Figure 1). The highly similar
1H- and
13C-NMR spectra of these compounds registered in DMSO-
d6 solution refer to their practically identical structural features also including oxo-enol tautomerism. One of the two downfield broadened signals could be identified as 10-NH amide resonance coupled with the quartet originated from the adjacent CH
2 group as disclosed by a COSY measurement carried out for
19b that gives somewhat sharper signals than does
19a. Regarding tautomerisation, it is of pronounced importance that in the NOESY spectrum of
19b weak, but clearly discernible cross peaks connecting H-8 doublet with the most downfielded broadened signal (separated from the amide 10-NH signal by ca. 0.4 ppm) strongly suggest the presence of the oxo tautomer. Thus, this signal must be originated from the skeletal 1-NH proton being in the proximity of H-8. In order to get an additional support for the exclusive presence or dominance of the oxo tautomers over the enol forms, theoretical
1H- and
13C-NMR chemical shifts were calculated for both
19a,
b and their enol forms (
Figure 1,
Table 1) by GIAO method [
30] using B3LYP fuctional [
31,
32,
33] and the extended 6-311++G(2d,p) basis set [
34]. The calculations were carried out on the structures optimised by the same functional with 6-31+G(d,p) basis set [
35] and IEFPCM solvent model [
36] employing dielectric constant of DMSO (ε = 46.7) that represents the conditions of NMR measurements. The comparision of the diagnostic chemical shifts of the skeletal and directly attached atoms calculated for the tautomer pairs with the corresponding experimental
1H- and
13C-NMR chemical shifts (
Table 1) supports at least the dominance of the oxo tautomers over the enol forms. Finally, the relative thermodynamic stability obtained for the tautomeric pairs by B3LYP/6-31 + G(d,p) method also confirm the dominance of the oxo tautomers over the enol counterparts [∆G(
19a/
enol −
19a/
oxo) = +0.57 kcal/mol, ∆G(
19b/
enol −
19b/
oxo) = +0.59 kcal/mol].
4. Materials and Methods
The
1H and
13C-NMR spectra were recorded in DMSO-
d6, CDCl
3 and D
2O solutions in 5 mm tubes at room temperature (RT), on a Bruker DRX-500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H, 13C). The 2D-COSY, NOESY, HSQC, and HMBC spectra of
19a and
19b were obtained by using the standard Bruker pulse programs (cosygpppqf (2D COSY with gradient pulses for selection and purge pulses before relaxation delay d1) for COSY, noesygpphpp (2D phase sensitive NOESY with gradient pulses in mixing time and purge pulses before relaxation delay d1 for NOESY), hsqcetgp (2D phase sensitive HSQC using Echo/Antiecho-TPPI gradient selection with decoupling during acquisition and using trim pulses in inept transfer) for HSQC and hmbcgpndqf (2D H-1/X HMBC optimised on long range couplings, no decoupling during acquisition using gradient pulses for selection) for HMBC, Bruker Biospin, Karlsruhe, Baden Württemberg, Germany). All calculations were carried out by using Gaussian 09 software package [
37]. The optimised structures are available from the authors.
Melting points were determined on a Hinotek X-4 melting point apparatus. Elemental analyses were performed with a Perkin-Elmer 2400 CHNS elemental analyser. Merck Kieselgel 60F254 plates were used for TLC.
The synthesis of compounds
1 [
38] and
5a [
29] was based on the method known from the literature.
1H- and
13C-NMR chemical shifts were calculated for both
19a,
b and their enol forms (
Figure 1,
Table 1) by GIAO method [
30] using B3LYP functional [
31,
32,
33] and the extended 6-311++G(2d,p) basis set [
34]. The calculations were carried out on the structures optimised by the same functional with 6-31+G(d,p) basis set [
35] and IEFPCM solvent model [
36] employing dielectric constant of DMSO (ε = 46.7) that represents the conditions of NMR measurements.
4.1. General Procedure for the Synthesis of C-3 Substituted Kynurenic Acid Derivatives (2, 3, 4a,b, 5b, 5c, 6a,b)
4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (1, 326 mg, 1.5 mmol), primary or secondary amine (2.0 mmol), and aldehyde (3.0 mmol) were placed in a 50 mL round-bottom flask. The mixture was treated at reflux temperature in 1,4-dioxane (30 mL) for 0.5–4 h. After the evaporation of the solvent the residue was dissolved in H2O (40 mL) and extracted with DCM (3 × 30 mL). The collected organic phases were dried (Na2SO4), the solvent was evaporated, and the residue was crystallised from a n-hexane:EtOAc (95:5) mixture (10 mL).
4.1.1. 3-(((2-Morpholinoethyl)amino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (2)
Preparation according to general procedure, using 2-morpholinoethylamine (260 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 1 h. Yield: 473 mg (78%); M.p. 300–303 °C.
1H NMR (DMSO-
d6); 3.07–3.2 (2H, m); 3.47–3,56 (2H, m); 3.79 (2H, t,
J = 12.2 Hz); 3.92–4.04 (4H, m); 4.47 (2H, s); 7.4 (1H, t,
J = 7.5 Hz); 7.73 (1H, t,
J = 8.1 Hz); 7.82 (1H, d,
J = 8.3 Hz); 8.20 (1H, d,
J = 8.0 Hz); 10.86 (1H, br ~s);
13C NMR (DMSO-
d6); 37.5; 47.8; 51.9; 54.2; 63.8; 120.3; 121.6; 124.5; 125.8; 127.1; 133.2; 141.2; 141.3; 165.4; 174.0; (
Figures S1 and S2 in Supplementary Materials).
4.1.2. 3-(((2-(Dimethylamino)ethyl)amino)(phenyl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (3)
Preparation according to general procedure, using
N,
N-dimethylethylene diamine (176 mg, 2.0 mmol) and benzaldehyde (318 mg, 3.0 mmol); reflux time: 0.5 h. Yield: 487 mg (74%); M.p. 193–195 °C.
1H NMR (DMSO-
d6); 2.81 (6H, s); 2.85–2.94 (1H, m); 3.12-3.22 (1H, m); 3.44–3.54 (1H, m); 4.12–4.23 (1H, m); 5.84 (1H, s); 7.30–7.41 (6H, m); 7.73 (1H, t,
J = 7.8 Hz); 7.86 (1H, d,
J = 8.5 Hz); 8.07 (1H, d,
J = 8.1 Hz); 10.01 (1H, br ~s); 12.85 (1H, br ~s);
13C NMR (DMSO-
d6); 35.7; 42.1; 44.2; 54.7; 61.2; 120.3; 124.6; 125.0; 125.8; 127.8; 128.9; 129.4; 129.7; 133.2; 136.0; 141.1; 141.2; 165.2; 173.3; (
Figures S3 and S4).
4.1.3. 3-((Dimethylamino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (4a)
Preparation according to general procedure, using dimethylamine (90 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 1 h; Yield: 303 mg (82%); M.p. 246–249 °C.
1H NMR (DMSO-
d6); 2.75 (6H, s); 4.36 (2H, s); 7.36 (1H, t,
J = 7.7 Hz); 7.68 (1H, t,
J = 7.8 Hz); 8.01 (1H, d,
J = 7.2 Hz); 8.12 (1H, d,
J = 7.6 Hz); 11.05 (1H, br ~s); 11.75 (1H, br ~s);
13C NMR (DMSO-
d6); 42.0; 52.6; 109.3; 120.3; 124.6; 125.0; 126.0; 133.0; 139.7; 149.1; 164.7; 178.0; (
Figures S5 and S6).
4.1.4. 3-((Benzyl(methyl)amino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (4b)
Preparation according to general procedure, using N-benzylmethylamine (242 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 2 h. Yield: 411 mg (85%); M.p. 235–237 °C.
1H NMR (DMSO-
d6); 2.43–2.49 (4H, m); 4.16–4.58 (5H, m); 7.35 (1H, t,
J = 7.3 Hz); 7.41–7.55 (5H, m); 7.67 (1H, t,
J = 7.6 Hz); 7.99 (1H, d,
J = 8.4 Hz); 8.10 (1H, d,
J = 8.0 Hz); 11.82 (1H, br ~s); 12.75 (1H, br ~s);
13C NMR (DMSO-
d6); 37.0; 50.5; 57.7; 108.7; 119.9; 124.3; 124.5; 125.6; 129.4; 129.7; 131.0; 131.7; 132.6; 139.4; 148.6; 164.9; 177.5; (
Figures S7 and S8).
4.1.5. 4-Oxo-3-(piperidin-1-ylmethyl)-1,4-dihydroquinoline-2-carboxylic acid (5b)
Preparation according to general procedure, using piperidine (170 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 1 h. Yield: 353 mg (73%); M.p. 215–217 °C.
1H NMR (DMSO-
d6); 1.38–1.52 (1H, m); 1.59–1.77 (3H, m); 1.78–1.86 (2H, m); 3.07 (2H, t,
J = 12.63 Hz); 3.34 (2H, d,
J = 11.93 Hz); 4.50 (2H, s); 7.44 (1H, t,
J = 7.6 Hz); 7.76 (1H, t,
J = 7.8 Hz); 8.05 (1H, d,
J = 8.5 Hz); 8.14 (1H, d,
J = 8.0 Hz); 9.35 (1H, br ~s); 12.20 (1H, br ~s);
13C NMR (DMSO-
d6); 22.0; 23.1; 51.6; 53.3; 111.4; 120.5; 125.2; 125.6; 125.9; 133.8; 139.9; 142.6; 164.6; 178.2; (
Figures S9 and S10).
4.1.6. 3-((4-Methylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (5c)
Preparation according to general procedure, using 1-methylpiperazine (200 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 0.5 h. Yield: 352 mg (78%); M.p. 229–231 °C.
1H NMR (DMSO-
d6); 2.22 (3H, s); 2.74–3.11 (4H, m); 3.35–3.41 (4H, m); 4.35 (2H, s); 7.34 (1H, t,
J = 7.4 Hz); 7.66 (1H, t,
J = 7.6 Hz); 7.97 (1H, d,
J = 8.4 Hz); 8.09 (1H, d,
J = 8.0 Hz); 11.75 (1H, br ~s);
13C NMR (DMSO-
d6); 45.8; 50.1; 50.3; 52.7; 108.7; 120.3; 124.6; 124.9; 126.0; 133.0; 139.8; 148.9; 165.2; 177.9; (
Figures S11 and S12).
4.1.7. 3-((3,4-Dihydroisoquinolin-2(1H)-yl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (6a)
Preparation according to general procedure, using 1,2,3,4-tetrahydroisoquinoline (266 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 3 h. Yield: 406 mg (81%); M.p. 275–278 °C.
1H NMR (DMSO-
d6); 3.01–3.16 (2H, m); 3.36–3.57 (2H, m); 4.32–4.46 (2H, m); 4.51 (2H, s); 7.17–7.32 (4H, m); 7.36 (1H, t,
J = 7.6 Hz); 7.68 (1H, t,
J = 7.4 Hz); 8.00 (1H, d,
J = 8.4 Hz); 8.12 (1H, d,
J = 8.0 Hz); 11.77 (1H, br ~s); 13.06 (1H, br ~s);
13C NMR (DMSO-
d6); 25.9; 47.5; 49.8; 51.7; 108.9; 120.4; 124.7; 124.9; 126.0; 127.5; 127.8; 128.5; 129.5; 129.7; 132.4; 133.0; 139.8; 148.8; 165.2; 178.0; (
Figures S13 and S14).
4.1.8. 3-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (6b)
Preparation according to general procedure, using 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (382 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol); reflux time: 4 h. The crystals were purified in EtOH (10 mL). Yield: 355 mg (60%); M.p. 248–250 °C.
1H NMR (DMSO-
d6); 2.90−3.08 (2H, m); 3.50−3.60 (2H, m); 3.69 (3H, s); 3.74 (3H, s); 4.16–4.37 (2H, m); 4.49 (2H, s); 6.78 (1H, s); 6.81 (1H, s); 7.37 (1H, t,
J = 7.7 Hz); 7.67 (1H, t,
J = 7.4 Hz); 8.00 (1H, d,
J = 8.8 Hz); 8.13 (1H, d,
J = 7.8 Hz); 11.75 (1H, br ~s); 13.03 (1H, br ~s);
13C NMR (DMSO-
d6); 25.3; 47.4; 49.4; 50.8; 56.0; 63.1; 108.5; 110.4; 112.0; 119.9; 120.8; 123.7; 124.3; 124.5; 125.6; 132.7; 139.4; 148.1; 148.4; 148.8; 164.8; 177.6; (
Figures S15 and S16).
4.2. General Procedure for the Synthesis of 5-, 6-, 7-, and 8-Substituted Kynurenic Acid Derivatives (7–10)
Diethyl acetylenedicarboxylate (510 mg, 3.0 mmol) and 2.0 mmol of the corresponding aniline derivatives (4-aminobiphenyl, 338 mg; 3-chloroaniline, 255 mg; 2-methylaniline 214 mg) were dissolved in EtOH (60 mL). The mixture was treated at reflux temperature for 1–8 h. After the evaporation of the solvent, the intermediate enamine was purified using column-chromatography (eluent: n-hexane:EtOAc 1:1). After the purification the intermediate was mixed with 1,2-dichlorobenzene (40 mL) and then was treated at reflux temperature for 10–24 h. After the solvent was removed by evaporation, the residue was crystallised from Et2O:EtOAc 95:5 mixture (10 mL).
4.2.1. Ethyl 5-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (7)
Reflux time: 8 h (for enamine formation), 24 h (for ring closure). The crystals were recrystallised from EtOAc (5 mL). Yield: 131 mg (26%); M.p. 200–205, (lit [
39]: 197–198 °C).
1H NMR (DMSO-
d6); 1.37 (3H, t,
J = 7. Hz); 4.42 (2H, q,
J = 7.2 Hz); 6.58 (1H, s); 7.32 (1H, d,
J = 7.3 Hz); 7.59 (1H, t,
J = 8.1 Hz); 7.91 (1H, d,
J = 8.4 Hz); 11.93 (1H, br ~s);
13C NMR (DMSO-
d6); 14.4; 63.1; 112.8; 119.4; 122.4; 126.9; 132.3; 132.7; 137.4; 143.2; 162.4; 194.2; (
Figures S17 and S18).
4.2.2. Ethyl 4-oxo-6-phenyl-1,4-dihydroquinoline-2-carboxylate (8)
Reflux time: 1 h (for enamine formation) and 10 h (for ring closure). The crystals were recrystallised from EtOAc (8 mL) Yield: 422 mg (72%); M.p. 263–265 °C, (lit [
40]: 261–261.5 °C).
1H NMR (CDCl
3); 1.39 (3H, t,
J = 7.2 Hz); 4.45 (2H, q,
J = 7.0 Hz); 6.68 (1H, s); 7.40 (1H, t,
J = 7.1 Hz); 7.50 (2H, t,
J = 7.5 Hz); 7.74 (2H, d,
J = 7.5 Hz); 8.02–8.10 (2H, m); 12.10 (1H, br ~s);
13C NMR (CDCl
3); 14.1; 63.4; 111.3; 118.8; 123.9; 126.4; 127.1; 127.8; 129.0; 132.4; 136.6; 137.8; 138.3; 139.5; 162.8; 179.3; (
Figures S19 and S20).
4.2.3. Ethyl 7-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (9)
Reflux time: 8 h (for enamine formation), 20 h (for ring closure). Yield: 211 mg (42%); M.p. 258–259 °C, (lit [
39]: 250–251 °C).
1H NMR (DMSO-
d6); 1.37 (3H, t,
J = 7.0 Hz); 4.43 (2H, q,
J = 7.2 Hz); 6.65 (1H, s); 7.39 (1H, d,
J = 8.0 Hz); 8.01 (1H, s); 8.07 (1H, d,
J = 8.8 Hz); 12.09 (1H, br ~s);
13C NMR (DMSO-
d6); 14.4; 63.3; 111.2; 119.1; 124.9; 124.9; 127.6; 137.7; 138.8; 141.2; 162.4; 177.5; (
Figures S21 and S22).
4.2.4. Ethyl 4-oxo-8-methyl-1,4-dihydroquinoline-2-carboxylate (10)
Reflux time: 3 h (for enamine formation), 14 h (for ring closure). Yield: 337 mg (73%); M.p. 138–143 °C, (lit [
41]: 139–139.5 °C).
1H NMR (CDCl
3); 1.46 (3H, t,
J = 7,1 Hz) 2.58 (3H, s); 4.51 (2H, q,
J = 7.0 Hz); 7.11 (1H, s); 7.31 (1H, t,
J = 7.6 Hz); 7.53 (1H, d,
J = 7.0 Hz); 8.23 (1H, d,
J = 8.2 Hz); 9.03 (1H, br ~s);
13C NMR (CDCl
3); 14.0; 16.5; 63.4; 111.4; 124.3; 124.4; 125.2; 126.3; 133.8; 136.1; 137.8; 163.1; 179.8; (
Figures S23 and S24).
4.3. General Procedure for the Synthesis of B Ring Substituted C-3 Morpholinomethyl Kynurenic Acid Derivatives (11–14)
The correspondingly substituted kynurenic acid ethyl esters (7–10 1.5 mmol), morpholine (175 mg, 2.0 mmol), and aqueous formaldehyde (22%, 410 mg, 3.0 mmol) were mixed in 1,4-dioxane (30 mL) and then hetead at reflux temperature for 6–12 h. After the evaporation of the solvent the residue was dissolved in H2O (40 mL), and extracted with DCM (3 × 30 mL). The collected organic phases were dried (Na2SO4), the solvent was removed by evaporation, and the residue was crystallised from n- hexane:EtOAc 95:5 mixture (10 mL).
4.3.1. 5-Chloro-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (11)
Preparation according to general procedure, using ethyl 5-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (
7, 377 mg, 1.5 mmol; reflux time: 6 h. The crystals were purified in EtOH (10 mL). Yield: 368 mg (70%); M.p. 265–270 °C (decomposition).
1H NMR (DMSO-
d6); 2.97–3.32 (4H, m); 3.48–3.77 (2H, m); 3.78–4.10 (2H, m); 4.35 (2H, s); 7.30 (1H, d,
J = 7.9 Hz); 7.55 (1H, t,
J = 8.12 Hz); 7.96 (1H, d,
J = 8.5 Hz); 11,69 (1H, br ~s); 12.60 (1H, br ~s);
13C NMR (DMSO-
d6); 50.1; 50.6; 64.3; 109.9; 119.4; 120.6; 126.8; 132.4; 132.4; 137.7; 142.0; 147.5; 164.4; 177.0; (
Figures S25 and S26).
4.3.2. 3-(Morpholinomethyl)-4-oxo-6-phenyl-1,4-dihydroquinoline-2-carboxylic acid (12)
Preparation according to general procedure, using ethyl 4-oxo-6-phenyl-1,4-dihydroquinoline-2-carboxylate (
9, 440 mg, 1.5 mmol); reflux time: 12 h. Yield: 420 mg (77%); M.p. 225–227 °C.
1H NMR (DMSO-
d6); 3.02–3.20 (2H, m); 3.20–3.37 (2H, m); 3.52–3.71 (2H, m); 3.89–4.08 (2H, m); 4.44 (2H, s); 7.40 (1H, t,
J = 7.2 Hz); 7.51 (2H, t,
J = 7.9 Hz); 7.74 (2H, d,
J = 7.7 Hz); 8.03 (1H, d,
J = 8.6 Hz); 8.10 (1H, d,
J = 8.8 Hz); 8.34 (1H, s); 11.94 (1H, br ~s); 12.72 (1H, br ~s);
13C NMR (DMSO-
d6); 50.0; 50.6; 64.3; 108.3; 120.8; 122.8; 124.7; 127.1; 128.1; 129.6; 131.4; 136.1; 138.8; 139.8; 148.4; 164.8; 177.6; (
Figures S27 and S28).
4.3.3. 7-Chloro-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (13)
Preparation according to general procedure, using ethyl 7-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (
11, 377 mg, 1.5 mmol); reflux time: 6 h. Yield: 402 mg (83%); M.p. 278–281 °C.
1H NMR (DMSO-
d6); 2.97–3.35 (4H, m); 3.51–3.71 (2H, m); 3.81–4.03 (2H, m); 4.39 (2H, s); 7.36 (1H, d,
J = 8.7 Hz); 8.06-8.11 (2H, m); 11.85 (1H, br ~s); 12.57 (1H, br ~s);
13C NMR (DMSO-
d6); 29.5; 50.2; 50.5; 64.4; 66.0; 119.0; 123.1; 124.6; 127.9; 137.2; 140.1; 148.9; 164.4; 177.2; (
Figures S29 and S30).
4.3.4. 8-Methyl-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (14)
Preparation according to general procedure, using ethyl 4-oxo-8-methyl-1,4-dihydroquinoline-2-carboxylate (
13, 347 mg, 1.5 mmol); reflux time: 8 h. Yield: 390 mg (86%); M.p. 255–258 °C.
1H NMR (DMSO-
d6); 2.54 (3H, s); 3.12–3.28 (4H, m); 3.63–3.95 (4H, m); 4.51 (2H, s); 7.29 (1H, t,
J = 7.8 Hz); 7.58 (1H, d,
J = 7.2 Hz); 7.99 (1H, d,
J = 8.1 Hz); 10.22 (1H, br ~s);
13C NMR (DMSO-
d6); 16.9; 50,8; 50,9; 64.5; 109.2; 124.3; 124.6; 124.8; 127.1; 134.0; 137.4; 146.4; 164.4; 178.6; (
Figures S31 and S32).
4.4. General Procedure for the Synthesis of C-3 Morpholinomethyl Kynurenic Acid Amides (18a–c, 19a–c)
The corresponding kynurenic acid amides (16, 17, 1.5 mmol), and secondary amines (15a–c, 2.0 mmol), were mixed with formaldehyde (22%, 410 mg, 3.0 mmol) in a 50 mL round-bottom flask. The mixture was heated at reflux temperature in 1,4-dioxane (30 mL) for 1–8 h. After the evaporation of the solvent the residue was crystallised from Et2O (10 mL).
4.4.1. N-(2-(Dimethylamino)ethyl)-4-oxo-3-(pyrrolidin-1-ylmethyl)-1,4-dihydroquinoline-2-carboxamide (18a)
Preparation according to general procedure, using
N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (
16, 389 mg, 1,5 mmol), pyrrolidine (
15a 142 mg, 2.0 mmol); reflux time: 1 h. Yield: 385 mg (75%); M.p. > 350 °C.
1H NMR (D
2O); 2.12 (4H, m); 2.38 (6H, s); 2.76 (2H, t,
J = 5.5 Hz); 3.41 (4H, m); 3.63 (2H, t,
J = 6.3 Hz); 4.48 (2H, s) 7.52 (1H, t,
J = 7.2 Hz); 7.75 (1H, t,
J = 6.8 Hz); 7.82 (1H, d,
J = 8.1 Hz); 8.23 (1H, d,
J = 8.0 Hz);
13C NMR (D
2O); 22.8; 36.9; 43.9; 51.6; 53.3; 56.8; 107.5; 123.9; 125.1; 126.0; 127.2; 131.0; 147.9; 152.6; 169.9; 174.5; (
Figures S33 and S34).
4.4.2. N-(2-(Dimethylamino)ethyl)-4-oxo-3-(piperidin-1-ylmethyl)-1,4-dihydroquinoline-2-carboxamide (18b)
Preparation according to general procedure, using
N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (
16, 389 mg, 1,5 mmol) and piperidine (
15b 170 mg, 2.0 mmol); reflux time: 1 h. Yield: 412 mg (77%); M.p. > 350 °C.
1H NMR (D
2O); 1.62–1.76 (2H, m); 1.76–1.91 (4H, m); 2.38 (6H, s); 2.75 (2H, t,
J = 6.0 Hz); 3.24 (4H, m); 3.62 (2H, t,
J = 6.0 Hz); 4.40 (2H, s); 7.53 (1H, t,
J = 7.2 Hz); 7.75 (1H, t,
J = 7.2 Hz); 7.82 (1H, d,
J = 8.5 Hz); 8.22 (1H, d,
J = 8.2 Hz);
13C NMR (D
2O); 21.5; 23.2; 36.9; 43.9; 52.3; 53.4; 56.8; 106.2; 123.8; 125.2; 125.8; 127.2; 131.1; 147.8, 152.9; 170.1; 174.6; (
Figures S35 and S36).
4.4.3. N-(2-(Dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (18c)
Preparation according to general procedure, using
N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (
16, 389 mg, 1,5 mmol) and morpholine (
15c 174 mg, 2.0 mmol); reflux time: 8 h. Yield: 425 mg (79%); M.p. > 350 °C.
1H NMR (DMSO-
d6); 2.19 (6H, s); 2.41–2.48 (6H, m); 3.45–3.52 (2H, m); 3.53–3.59 (4H, m); 3.61 (2H, s) 7.32 (1H, t,
J = 7.3 Hz); 7.64 (1H, t,
J = 7.7 Hz); 7.86 (1H, d,
J = 8.3 Hz); 8.08 (1H, d,
J = 8.1 Hz); 10.63 (1H, br ~s);
13C NMR (DMSO-
d6); 38.4; 46.0; 51.1; 52.6; 59.1; 67.0; 113.7; 120.4; 124.4; 124.8; 126.0; 131.0; 132.8; 140.5; 163.9; 177.8; (
Figures S37 and S38).
4.4.4. 4-Oxo-N-(2-(pyrrolidin-1-yl)ethyl)-3-(pyrrolidin-1-ylmethyl)-1,4-dihydroquinoline-2-carboxamide (19a)
Preparation according to general procedure, using 4-oxo-
N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (
17, 428 mg, 1,5 mmol) and pyrrolidine (
15a, 142 mg, 2.0 mmol); reflux time: 1 h. Yield: 392 mg (71%); M.p. > 350 °C.
1H NMR (DMSO-
d6); 1.64 (4H, br ~s, H-15,16); 1.67 (4H, br ~s, H-21,22); 2.42 (4H, br ~s, H-14,17); 2.54 (4H, br ~s, H-20,23); 2.55 (2H, t,
J = 5.8 Hz, H-12); 3.44 (2H, qa,
J = 5.8 Hz, H-11); 3.68 (2H, br ~s, H-18); 7.26 (1H, d,
J = 7.8 Hz, H-6); 7.58 (1H, br ~t,
J = 8 Hz, H-7); 7.81 (1H, br ~d,
J = 8 Hz, H-8); 8.05 (1H, br d,
J = 7.8 Hz, H-5); 11.30 (1H, br ~s, 10-NH); 11.64 (1H, br ~s, 1-NH);
13C NMR (DMSO-
d6); 23.6 (two coalesced lines, C-15,16,21,22); 39.4 (merged in the septet signal of the solvent, C-11); 52.2 (C-20,23); 47.5 (C-18); 52.2 (C-20,23); 54.1 (C-14,17); 55.2 (C-12); 115.1 (C-3); 119.6 (two coalesced lines, C-4a,8); 123.9 (C-6); 125.6 (C-5); 132.5 (C-7); 139.6 (C-8a); 143.5 (C-2); 163.3 (C-9); 177.3 (C-4); (
Figures S39–S41).
4.4.5. 4-Oxo-3-(piperidin-1-ylmethyl)-N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (19b)
Preparation according to general procedure, using 4-oxo-
N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (
17, 428 mg, 1,5 mmol) and piperidine (
15b, 170 mg, 2.0 mmol); reflux time: 1 h. Yield: 470 mg (82%); M.p. > 350 °C.
1H NMR (DMSO-
d6); 1,38 (2H, br ~s, H-22); 1.44 (4H, br ~s, H-21,23); 1.63 (4H, br ~s, H-15,16); 2.40 (4H, br ~s, H-20,24); 2.43 (4H, br ~s, H-14,17); 2.57 (2H, t,
J = 5.8 Hz, H-12); 3.44 (2H, qa,
J = 5.8 Hz, H-11); 3.52 (2H, br ~s, H-18); 7.28 (1H, t,
J = 7.8 Hz, H-6); 7.60 (1H, br ~t,
J = 8 Hz, H-7); 7.84 (1H, br ~d,
J = 8 Hz, H-8); 8.03 (1H, br d,
J = 7.8 Hz, H-5); 11.30 (1H, br ~s, 10-NH); 11.69 (1H, br ~s, 1-NH);
13C NMR (DMSO-
d6); 23.6 (C-15,16); 24.3 (C-22); 25.9 (C-21, 23); 39.4 (C-11); 50.8 (C-18); 52.5 (C-20,24); 54.2 (C-14,17); 55.4 (C-12); 114.2 (C-3); 119.6 (C-4a); 119.6 (C-8); 124.1 (C-6); 125.6 (C-5); 132.6 (C-7); 139.7 (C-8a); 143.9 (C-2); 163.5 (C-9); 177.8 (C-4); (
Figures S42–S45).
4.4.6. 4-Oxo-3-(morpholinomethyl)-N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (19c)
Preparation according to general procedure, using 4-oxo-
N-(2-(pyrrolidin-1-yl)ethyl)-1,4-dihydroquinoline-2-carboxamide (
17, 428 mg, 1,5 mmol) and morpholine (
15c, 174 mg, 2.0 mmol); reflux time: 8 h. Yield: 438 mg (76%); M.p. > 350 °C.
1H NMR (DMSO-
d6); 1.63–1.73 (4H, m); 2.46–2.52 (6H, m); 2.63 (2H, t,
J = 6.2 Hz); 3.48–3.63 (10H, m); 7.34 (1H, t,
J = 7.1 Hz); 7.66 (1H, t,
J = 7.2 Hz); 7.88 (1H, d,
J = 8.0 Hz); 8.09 (1H, d,
J = 8.0 Hz); 10.82 (1H, br ~s); 11.78 (1H, br ~s);
13C NMR (DMSO-
d6); 23.6; 50.6; 52.1; 54.1; 55.4; 66.6; 113.4; 119.6; 124.2; 124.3; 125.7; 132.6; 139.7; 144.3; 163.2; 177.7; (
Figures S46 and S47).
4.5. 4-Oxo-1,4-dihydroquinoline-2-carboxamide (20)
Ethyl 4-oxo-1,4-dihydroquinoline-2-carboxylate (1, 652 mg, 3.0 mmol) and NH3/MeOH (20%, 5 mL) were placed in a pressure-resistant vessel of 10 mL. The mixture was kept at 100 °C for 30 min with a CEM Discover SP microwave reactor. Following the removal of the solvent the residue was crystallised from Et2O (10 mL) and recrystallised from EtOH (8 mL).
Yield: 355 mg (63%); M.p. 300–303 °C (lit [
42]: 295–297 °C).
1H NMR (DMSO-
d6); 6.75 (1H, s); 7.33 (1H, t,
J = 7.6 Hz); 7.67 (1H, t,
J = 7.7 Hz); 7.96 (1H, d,
J = 8.4 Hz); 8.03 (1H, s); 8.07 (1H, d,
J = 8.2 Hz); 8.44 (1H, s) 11.69 (1H, br ~s);
13C NMR (DMSO-
d6); 107.8; 120.0; 124.1; 125.1; 125.9; 132.7; 140.3; 141.6; 164.1; 178.4; (
Figures S48 and S49).
4.6. Methyl 3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylate (22)
In a 50 mL round-bottom flask 576 mg (2.0 mmol) 3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (5a) was dissolved in 20 mL MeOH. The mixture was reacted with 1 mL Et2O solution of CH2N2 at room temperature for 5 h. Following the removal of the solvent the residue was crystallised from n-hexane:EtOAc (95:5; 10 mL).
Yield: 405 mg (67%); M.p. 122–123 °C.
1H NMR (DMSO-
d6); 1.23 (3H, s); 2.99–3.19 (2H, m); 2.19–3.35 (2H, m); 3.50–3.69 (2H, m); 3.87–4.04 (2H, m); 4,40 (2H, s); 7.35 (1H, t,
J = 7.5 Hz); 7.67 (1H, t,
J = 7.8 Hz); 7.99 (1H, d,
J = 8.3 Hz); 8.09 (1H, d,
J = 8.1 Hz); 11.81 (1H, br ~s); 12.71 (1H, br ~s);
13C NMR (DMSO-
d6); 29.5; 49.9; 50.6; 64.3; 108.1; 119.9; 124.3; 124.5; 125.5; 132.6; 139.4; 148.5; 164.8; 177.6; (
Figures S52 and S53).
4.7. 3-(Morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (21)
Scheme 4: (ii): Carboxamide
20 (188 mg, 1.0 mmol), morpholine (174 mg, 2.0 mmol) and aqueous formaldehyde (22%, 410 mg, 3.0 mmol) were heated at reflux temperature in 1,4-dioxane for 3 h. After evaporation of the solvent the residue was crystallised with Et
2O (10 mL). Yield A: 202 mg (87%); M.p. 144–145 °C.
Scheme 4: (iv): Methyl 3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylate (
22, 151 mg, 0.5 mmol) and NH
3/MeOH (20%, 5 mL) were placed and stirred in a pressurised reaction vial. After a reaction of 4 h, the solvent was evaporated, and the residue was crystallised with Et
2O (10 mL). Yield B: 126 mg (88%) M.p. 143–146 °C.
1H NMR (DMSO-
d6); 2.38–2.49 (4H, m); 3.48–3.58 (4H, m); 3.61 (2H, s); 7.34 (1H, t,
J = 6.8 Hz); 7.66 (1H, t,
J = 7.3 Hz); 7.87 (1H, d,
J = 8.2 Hz); 8.09 (1H, d,
J = 7.2 Hz); 8.15 (1H, br ~s); 10.02 (1H, br ~s); 11.73 (1H, br ~s);
13C NMR (DMSO-
d6); 51.3; 52.7; 67.0; 114.0; 119.9; 124.5; 124.8; 126.1; 133.0; 140.0; 144.9; 165.4; 178.1; (
Figures S50 and S51).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}