Identification of Secondary Metabolites from Aspergillus pachycristatus by Untargeted UPLC-ESI-HRMS/MS and Genome Mining


Abstract
1. Introduction
2. Results and Discussion
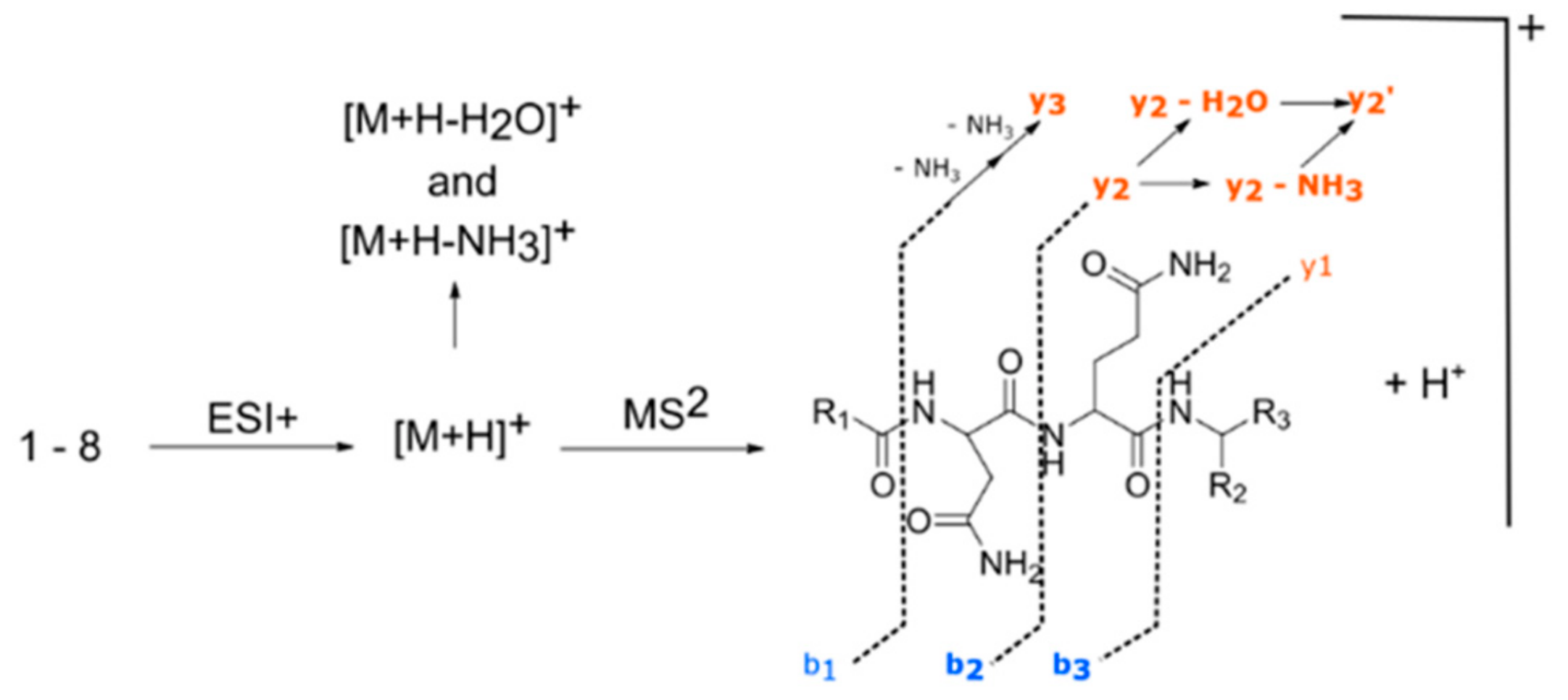
2.1. Metabolic Diversity in Aspergillus pachycristatus
2.2. Confirmation of Homologous Gene Clusters in A. pachycristatus for the Identified Metabolites
3. Materials and Methods
3.1. Construction of Gene Deletion and Marker-Recycling Strains
3.2. Growth and Sample Preparation
3.3. HPLC-MS Analysis
3.4. UPLC-HRMS Analysis
3.5. Metabolic Profiling, Molecular Networking and Compound Dereplication
3.6. Comparison of Secondary Metabolic Biosynthestic Gene Clusters of A. pachycristatus NRRL 11440 and A. nidulans FGSC A4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tormo, J.R.; Asensio, F.J.; Bills, G.F. Manipulating filamentous fungus chemical phenotypes by growth on nutritional arrays. Methods Mol. Biol. 2012, 944, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Bouslimani, A.; Sanchez, L.M.; Garg, N.; Dorrestein, P.C. Mass spectrometry of natural products: current, emerging and future technologies. Nat. Prod. Rep. 2014, 31, 718–729. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.J.; Frisvad, J.C.; Sun, B.D.; Varga, J.; Kocsube, S.; Dijksterhuis, J.; Kim, D.H.; Hong, S.B.; Houbraken, J.; Samson, R.A. Aspergillus section Nidulantes (formerly Emericella): Polyphasic taxonomy, chemistry and biology. Stud. Mycol. 2016, 84, 1–118. [Google Scholar] [CrossRef] [PubMed]
- Tóth, V.; Nagy, C.T.; Miskei, M.; Pócsi, I.; Emri, T. Polyphasic characterization of “Aspergillus nidulans var. roseus” ATCC 58397. Folia. Microbiol. (Praha) 2011, 56, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Boeck, L.D. New antibiotics: Antifungals from Aspergillus. In Frontiers in Industrial Mycology; Leatham, G., Ed.; Chapman & Hall: New York, NY, USA, 1992; pp. 54–65. [Google Scholar]
- Boeck, L.D.; Kastner, R.E. Method of producing the A-30912 antibiotics. U.S. Patent 4,288,549, 21 January 1981. [Google Scholar]
- Emri, T.; Majoros, L.; Toth, V.; Pocsi, I. Echinocandins: Production and applications. Appl. Microbiol. Biotechnol. 2013, 97, 3267–3284. [Google Scholar] [CrossRef]
- Andersen, M.R.; Nielsen, J.B.; Klitgaard, A.; Petersen, L.M.; Zachariasen, M.; Hansen, T.J.; Blicher, L.H.; Gotfredsen, C.H.; Larsen, T.O.; Nielsen, K.F.; et al. Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proc. Natl. Acad. Sci. USA 2013, 110, E99–E107. [Google Scholar] [CrossRef]
- De Vries, R.P.; Riley, R.; Wiebenga, A.; Aguilar-Osorio, G.; Amillis, S.; Uchima, C.A.; Anderluh, G.; Asadollahi, M.; Askin, M.; Barry, K.; et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017, 18, 28. [Google Scholar] [CrossRef]
- Lan, N.; Yue, Q.; An, Z.; Bills, G.F. Apc.LaeA and Apc.VeA of the velvet complex govern secondary metabolism and morphological development in the echinocandin-producing fungus Aspergillus pachycristatus. J. Ind. Microbiol. Biotechnol. 2019. [Google Scholar] [CrossRef]
- Bayram, O.; Braus, G.H. Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol. Rev. 2012, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Keller, N.P. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot Cell 2004, 3, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Soukup, A.A.; Chadwick, E.; Chiang, Y.M.; Wang, C.C.; Keller, N.P. VeA and MvlA repression of the cryptic orsellinic acid gene cluster in Aspergillus nidulans involves histone 3 acetylation. Mol. Microbiol. 2013, 89, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.P.; Milde, L.; Trapp, M.K.; Frisvad, J.C.; Keller, N.P.; Bok, J.W. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal. Genet. Biol. 2008, 45, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Lind, A.L.; Smith, T.D.; Saterlee, T.; Calvo, A.M.; Rokas, A. Regulation of secondary metabolism by the velvet complex is temperature-responsive in Aspergillus. G3 Genes Genomes Genetics 2016, 6, 4023–4033. [Google Scholar] [CrossRef] [PubMed]
- Oakley, C.E.; Ahuja, M.; Sun, W.W.; Entwistle, R.; Akashi, T.; Yaegashi, J.; Guo, C.J.; Cerqueira, G.C.; Russo Wortman, J.; Wang, C.C.; et al. Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 2017, 103, 347–365. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, M.; Martin, J.; Gonzalez-Menendez, V.; Perez-Victoria, I.; Moreno, C.; Tormo, J.R.; El Aouad, N.; Guarro, J.; Vicente, F.; Reyes, F.; et al. Chemical and physical modulation of antibiotic activity in emericella species. Chem. Biodivers 2012, 9, 1095–1113. [Google Scholar] [CrossRef]
- Schrettl, M.; Bignell, E.; Kragl, C.; Sabiha, Y.; Loss, O.; Eisendle, M.; Wallner, A.; Arst, H.N., Jr.; Haynes, K.; Haas, H. Distinct roles for intra- and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog. 2007, 3, 1195–1207. [Google Scholar] [CrossRef]
- De Lorenzo, V.; Bindereif, A.; Paw, B.H.; Neilands, J.B. Aerobactin biosynthesis and transport genes of plasmid ColV-K30 in Escherichia coli K-12. J. Bacteriol. 1986, 165, 570–578. [Google Scholar] [CrossRef]
- Martin, J.D.; Ito, Y.; Homann, V.V.; Haygood, M.G.; Butler, A. Structure and membrane affinity of new amphiphilic siderophores produced by Ochrobactrum sp. SP18. J. Biol. Inorg. Chem. 2006, 11, 633–641. [Google Scholar] [CrossRef]
- McMahon, M.D.; Rush, J.S.; Thomas, M.G. Analyses of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 2809–2818. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, Y.; Lu, Y.; Wang, B.; Sun, J.; Zhang, H.; Wang, H. Chemistry and biology of siderophores from narine microbes. Mar. Drugs 2019, 17, 562. [Google Scholar] [CrossRef] [PubMed]
- Haas, H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Hilder, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Products Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef]
- Renshaw, J.C.; Robson, G.D.; Trinci, A.P.J.; Wiebe, M.G.; Livens, F.R.; Collison, D.; Taylor, R.J. Fungal siderophores: Structures, functions and applications. Mycol. Res. 2002, 106, 1123–1142. [Google Scholar] [CrossRef]
- Lee, Y.M.; Dang, H.T.; Hong, J.; Lee, C.O.; Bae, K.S.; Kim, D.K.; Jung, J.H. A cytotoxic lipopeptide from the sponge-derived fungus Aspergillus versicolor. Bull. Korean Chem. Soc. 2010, 31, 205–208. [Google Scholar] [CrossRef]
- Lee, Y.M.; Dang, H.T.; Li, J.; Zhang, P.; Hong, J.; Lee, C.O.; Jung, J.H. A cytotoxic fellutamide analogue from the sponge-derived fungus Aspergillus versicolor. Bull. Korean Chem. Soc. 2011, 32, 3817–3820. [Google Scholar] [CrossRef]
- Shigemori, H.; Wakuri, S.; Yazawa, K.; Nakamura, T.; Sasaki, T.; Kobayashi, J.I. Fellutamides A and B, cytotoxic peptides from a marine fish-possessing fungus Penicillium fellutanum. Tetrahedron 1991, 47, 8529–8534. [Google Scholar] [CrossRef]
- Xu, D.; Ondeyka, J.; Harris, G.H.; Zink, D.; Kahn, J.N.; Wang, H.; Bills, G.; Platas, G.; Wang, W.; Szewczak, A.A.; et al. Isolation, structure, and biological activities of fellutamides C and D from an undescribed Metulocladosporiella (Chaetothyriales) using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2011, 74, 1721–1730. [Google Scholar] [CrossRef]
- Wu, C.J.; Li, C.W.; Cui, C.B. Seven new and two known lipopeptides as well as five known polyketides: The activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1815–1838. [Google Scholar] [CrossRef]
- Bok, J.W.; Hoffmeister, D.; Maggio-Hall, L.A.; Murillo, R.; Glasner, J.D.; Keller, N.P. Genomic mining for Aspergillus natural products. Chem. Biol. 2006, 13, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Mead, M.E.; Knowles, S.L.; Raja, H.A.; Beattie, S.R.; Kowalski, C.H.; Steenwyk, J.L.; Silva, L.P.; Chiaratto, J.; Ries, L.N.A.; Goldman, G.H.; et al. Characterizing the pathogenic, genomic, and chemical traits of Aspergillus fischeri, a close relative of the major human fungal pathogen Aspergillus fumigatus. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Albright, J.C.; Henke, M.T.; Soukup, A.A.; McClure, R.A.; Thomson, R.J.; Keller, N.P.; Kelleher, N.L. Large-scale metabolomics reveals a complex response of Aspergillus nidulans to epigenetic perturbation. ACS Chem. Biol. 2015, 10, 1535–1541. [Google Scholar] [CrossRef]
- Chiang, Y.M.; Szewczyk, E.; Nayak, T.; Davidson, A.D.; Sanchez, J.F.; Lo, H.C.; Ho, W.Y.; Simityan, H.; Kuo, E.; Praseuth, A.; et al. Molecular genetic mining of the Aspergillus secondary metabolome: discovery of the emericellamide biosynthetic pathway. Chem. Biol. 2008, 15, 527–532. [Google Scholar] [CrossRef]
- Henke, M.T.; Soukup, A.A.; Goering, A.W.; McClure, R.A.; Thomson, R.J.; Keller, N.P.; Kelleher, N.L. New aspercryptins, lipopeptide natural products, revealed by HDAC inhibition in Aspergillus nidulans. ACS Chem. Biol. 2016, 11, 2117–2123. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Larsen, T.O. The importance of mass spectrometric dereplication in fungal secondary metabolite analysis. Front. Microbiol. 2015, 6, 71. [Google Scholar] [CrossRef]
- Oh, D.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Induced production of emericellamides A and B from the marine-derived fungus Emericella sp. in competing co-culture. J. Nat. Prod. 2007, 70, 515–520. [Google Scholar] [CrossRef]
- Scherlach, K.; Schuemann, J.; Dahse, H.M.; Hertweck, C. Aspernidine A and B, prenylated isoindolinone alkaloids from the model fungus Aspergillus nidulans. J. Antibiot. (Tokyo) 2010, 63, 375–377. [Google Scholar] [CrossRef]
- Yaegashi, J.; Praseuth, M.B.; Tyan, S.W.; Sanchez, J.F.; Entwistle, R.; Chiang, Y.M.; Oakley, B.R.; Wang, C.C. Molecular genetic characterization of the biosynthesis cluster of a prenylated isoindolinone alkaloid aspernidine A in Aspergillus nidulans. Org. Lett. 2013, 15, 2862–2865. [Google Scholar] [CrossRef]
- Yeh, H.H.; Ahuja, M.; Chiang, Y.M.; Oakley, C.E.; Moore, S.; Yoon, O.; Hajovsky, H.; Bok, J.W.; Keller, N.P.; Wang, C.C.; et al. Resistance gene-guided genome mining: Serial promoter exchanges in Aspergillus nidulans reveal the biosynthetic athway for fellutamide B, a proteasome inhibitor. ACS Chem. Biol. 2016, 11, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de Los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic. Acids. Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic. Acids. Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Yu, J.H.; Kelkar, H.S.; Fernandes, M.; Nesbitt, T.C.; Keller, N.P.; Adams, T.H.; Leonard, T.J. Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 1996, 93, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- David, H.; Özçelik, I.Ş.; Hofmann, G.; Nielsen, J. Analysis of Aspergillus nidulans metabolism at the genome-scale. BMC Genomics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.F.; Chiang, Y.M.; Szewczyk, E.; Davidson, A.D.; Ahuja, M.; Elizabeth Oakley, C.; Woo Bok, J.; Keller, N.; Oakley, B.R.; Wang, C.C. Molecular genetic analysis of the orsellinic acid/F9775 gene cluster of Aspergillus nidulans. Mol. Biosyst. 2010, 6, 587–593. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
Sample Availability: Samples of the crude extracts are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Molecule | R. Time | Exact Mass [M + H]+ | m/z Found | Error (ppm) | Class |
|---|---|---|---|---|---|---|
| 1 | Alternariol b | 4.08 | 259.0607 | 259.0603 | −1.5 | Polyketide |
| 2 | F9775-A b | 3.40 | 397.0923 | 397.0921 | −0.5 | Polyketide |
| 3 | F9775-B b | 3.91 | 397.0923 | 397.0920 | −0.8 | Polyketide |
| 4 | Cyclo (Leu-Phe) a | 3.89 | 261.1603 | 261.1600 | −1.2 | Diketopiperazine |
| 5 | Aspernidine A b | 7.25 | 400.2488 | 400.2487 | −0.3 | Prenylated alkaloid |
| 6 | Aspernidine B b | 6.70 | 386.2331 | 386.2324 | −1.8 | Prenylated alkaloid |
| 7 | Aspernidine C b | 7.29 | 414.2644 | 414.2638 | −1.5 | Prenylated alkaloid |
| 8 | Aspercryptin A1 b | 5.54 | 758.5392 | 758.5383 | −1.2 | Lipopeptide |
| 9 | Aspercryptin A2 b | 5.60 | 742.5443 | 742.5432 | −1.5 | Lipopeptide |
| 10 | Antibiotic 1656G b | 6.40 | 584.4018 | 584.4023 | 0.9 | Lipopeptide |
| 11 | Antibiotic 3127 b | 5.58 | 542.3548 | 542.3557 | 1.6 | Lipopeptide |
| 12 | Fellutamide B b | 5.68 | 556.3705 | 556.3706 | 0.2 | Lipopeptide |
| 13 | Fellutamide C b | 5.47 | 558.3861 | 558.3860 | −0.2 | Lipopeptide |
| 14 | Emericellamide A a | 6.64 | 610.4174 | 610.4177 | 0.5 | Cyclic lipopeptide |
| 15 | Emericellamide C/D b,c | 6.09 | 596.4018 | 596.4022 | 0.7 | Cyclic lipopeptide |
| 16 | Emericellamide E b | 6.82 | 624.4331 | 624.4327 | −0.6 | Cyclic lipopeptide |
| 17 | Emericellamide F b | 6.89 | 624.4331 | 624.4332 | 0.2 | Cyclic lipopeptide |
| 18 | Emericellamide G b | 5.90 | 582.3861 | 582.3862 | 0.2 | Cyclic lipopeptide |
| 19 | Emericellamide H b | 7.08 | 638.4487 | 638.4488 | 0.2 | Cyclic lipopeptide |
| 20 | Sterigmatocystin b | 5.76 | 325.0712 | 325.0706 | −1.9 | Polyketide |
| 21 | N,N’,N’’-triacetylfusarinine a | 3.82 | 853.4189 | 853.4197 | 0.9 | Hydroxamate siderophore |
| N° | Name | Molecular Formula | [M + H]+ Ion | R1 | R2 | R3 |
|---|---|---|---|---|---|---|
| 10 | Antibiotic 1656G | C29H53N5O7 | 584.4023 | C11H23CH(OH)CH2 | CHO | i-Bu |
| 11 | Antibiotic 3127 | C26H49N5O7 | 542.3557 | C9H19CH(OH)CH2 | CHO | i-Pr |
| 12 | Fellutamide B | C27H49N5O7 | 556.3706 | C9H19CH(OH)CH2 | CHO | i-Bu |
| 13 | Fellutamide C | C27H51N5O7 | 558.3860 | C9H19CH(OH)CH2 | CH2OH | i-Bu |
| 25 | Fellutamide derivative 1 | C29H55N5O7 | 586.4175 | C11H23CH(OH)CH2 | CH2OH | i-Bu |
| 26 | Fellutamide derivative 2 | C27H53N5O6 | 542.3914 | C11H23 | CH2OH | i-Bu |
| 27 | Fellutamide derivative 3 | C28H51N5O7 | 570.3862 | C10H21CH(OH)CH2 | CHO | i-Bu |
| 28 | Fellutamide derivative 4 | C28H53N5O7 | 572.4018 | C10H21CH(OH)CH2 | CH2OH | i-Bu |
| Gene in A. pachycristatus NRRL 11440 | Gene Name A. Nidulans | Corresponding Gene FGSC A4 | Product | Type | Similarity % |
|---|---|---|---|---|---|
| g7715 | atnA | AN7884 | Aspercryptin | NRPS | 92.2 |
| g5866 | easA | AN2545 | Emericellamide | NRPS | 91.1 |
| g5867 | easB | AN2547 | Emericellamide | PKS | 93.7 |
| g7876 | inpA | AN3495 | Fellutamide | NRPS | 91.3 |
| g7875 | inpB | AN3496 | Fellutamide | NRPS | 92.2 |
| g4770 | sidD | AN6236 | N’,N’’,N’’’-triacetylfusarinine C | NRPS | 95.6 |
| g8711 | orsA | AN7909 | Orsellinic acid/F9775 | PKS | 93.2 |
| g4218 | pkfA | AN3230 | Aspernidine | PKS | 94.9 |
| g5606 | pkgA | AN7071 | Alternariol | PKS | 79.2 |
| g7351 | stcA | AN7825 | Sterigmatocystin | PKS | 93.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perlatti, B.; Lan, N.; Jiang, Y.; An, Z.; Bills, G. Identification of Secondary Metabolites from Aspergillus pachycristatus by Untargeted UPLC-ESI-HRMS/MS and Genome Mining. Molecules 2020, 25, 913. https://doi.org/10.3390/molecules25040913
Perlatti B, Lan N, Jiang Y, An Z, Bills G. Identification of Secondary Metabolites from Aspergillus pachycristatus by Untargeted UPLC-ESI-HRMS/MS and Genome Mining. Molecules. 2020; 25(4):913. https://doi.org/10.3390/molecules25040913
Chicago/Turabian StylePerlatti, Bruno, Nan Lan, Yongying Jiang, Zhiqiang An, and Gerald Bills. 2020. "Identification of Secondary Metabolites from Aspergillus pachycristatus by Untargeted UPLC-ESI-HRMS/MS and Genome Mining" Molecules 25, no. 4: 913. https://doi.org/10.3390/molecules25040913
APA StylePerlatti, B., Lan, N., Jiang, Y., An, Z., & Bills, G. (2020). Identification of Secondary Metabolites from Aspergillus pachycristatus by Untargeted UPLC-ESI-HRMS/MS and Genome Mining. Molecules, 25(4), 913. https://doi.org/10.3390/molecules25040913