1. Introduction
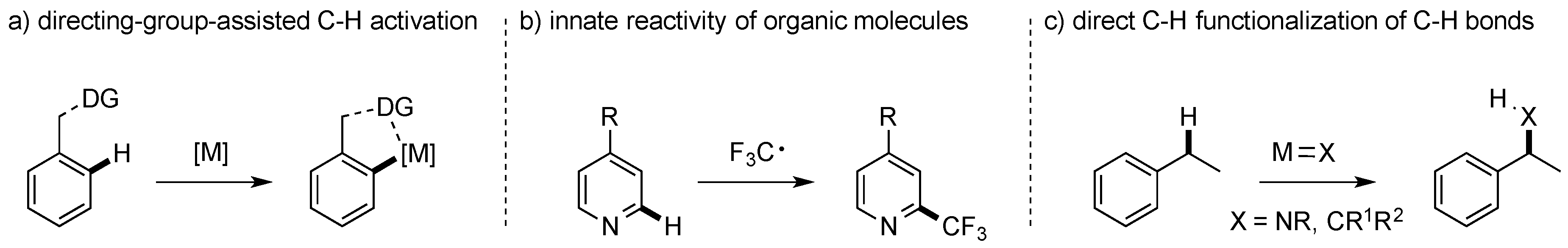
C-H bonds belong to the most common motifs in organic molecules and their direct functionalization is one of the main challenges in synthesis methodology, which impacts on the step economy and sustainability of chemical processes. In recent decades, this research area has flourished and different approaches have been realized to enable C-H functionalization reactions via different strategies: (a) by the directing group-assisted C-H activation, (b) via the innate reactivity of organic molecules, or (c) via direct C-H functionalization (
Scheme 1) [
1,
2,
3,
4]. While the introduction and subsequent removal of directing groups is a prerequisite for directed C-H activation, direct C-H functionalization allows the introduction of new functional groups into organic molecules without the need for directing groups. It thus represents the most efficient and step-economic strategy by which to conduct C-H functionalization reaction. The challenge of this strategy lies within the selective activation of only one type of C-H bond; elegant methods have been developed in recent years, mainly relying on the use of expensive and mostly toxic precious metal catalysts based on rhodium, iridium, palladium, and others to direct the C-H functionalization reaction as either the catalyst or the substrate [
5,
6,
7].
High costs of catalysts, limited resources, and toxicity of precious metals are the main drivers in the exploration of new synthesis methods based on third-row transition metals. Among these, iron plays a pivotal role. It is the second most abundant metal in the Earth’s crust following aluminum, and catalysts based on iron are currently emerging as new and important catalysts able to improve the environmental impact of chemical processes [
8,
9,
10,
11,
12,
13]. In nature, iron-containing enzymes play an important role, e.g., in the hemoglobin or myoglobin enzymes that are important for oxygen transport in mammals. They can also be found in cytochrome P450 enzymes or iron-sulfur clusters, amongst others, and are pivotal for the detoxification, e.g., by C-H oxidation reactions, of xenobiotics [
14]. Against this background, the development of C-H functionalization reactions with iron catalysts has received significant attention over the years with the goal of reducing the economic and environmental footprint of organic synthesis methodologies, but also that of accessing new reactivity that is unique to iron. In this review, we discuss the advances made in this research area with a focus on C-H functionalization reactions with carbenes.
2. Iron in Carbene-Transfer Reactions
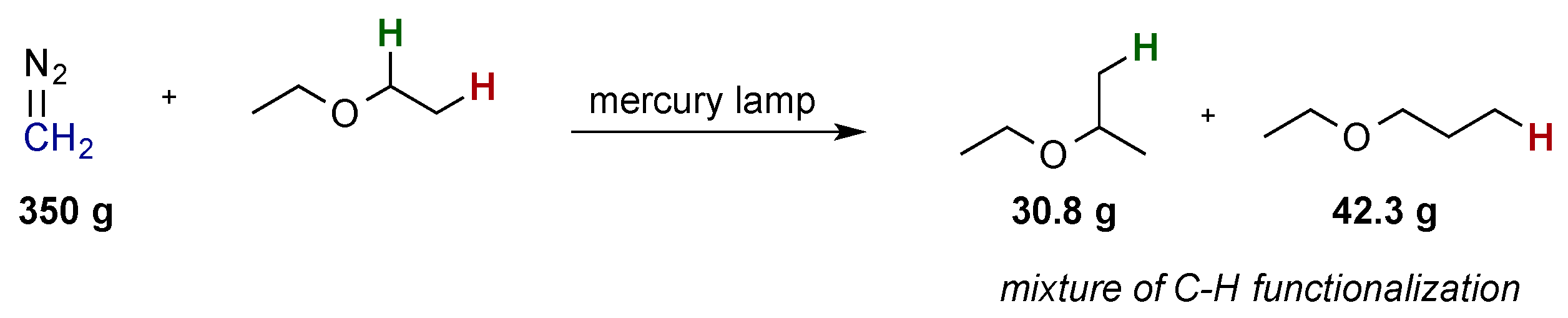
Carbene-transfer reactions are one of the key strategies used today to conduct highly efficient and selective C-H functionalization reactions. The earliest examples date back to reports by Meerwein and Doering from 1942 and 1959, in which they described metal-free photochemical C-H functionalization reactions with diazoalkanes using high-energy UV light via free carbene intermediates (
Scheme 2) [
15,
16]. However, the high reactivity of the free carbene intermediate led to unselective reactions and, as a consequence, a lack of applications in organic synthesis. In the subsequent decades, these shortcomings led to the development of metal-catalyzed carbine-transfer reactions using noble metals such as Rh(II), Ru(II), Ir(III), Au(I), Pd(II), or Cu(I) [
17,
18,
19,
20,
21].
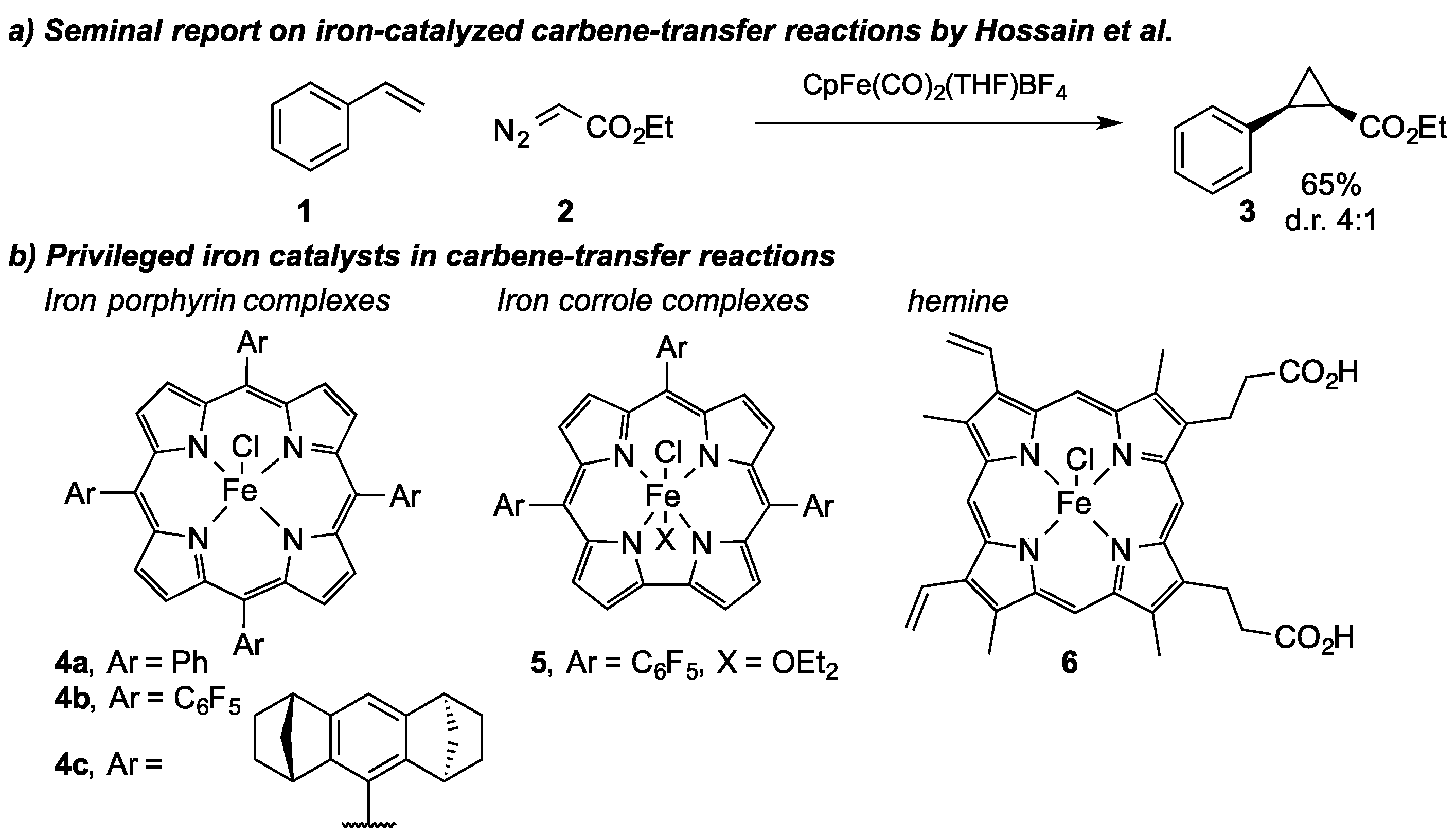
More recently, carbene-transfer reactions with iron complexes have gained significant attention in organic synthesis methodology for their potential to overcome the limitations of precious metal complexes. In 1992, Hossain et al. described the first iron-catalyzed carbene-transfer reaction using the cationic CpFe(CO)
2(THF)BF
4 complex in cyclopropanation reactions of styrene
1 and ethyl diazoacetate
2 (
Scheme 3a) [
22]. Following this strategy, different groups have since reported on their efforts to conduct iron-catalyzed carbene-transfer reactions [
11,
12,
13].
In the following years, several groups reported their efforts in the field of iron-catalyzed carbine-transfer reactions. Woo et al. reported an important milestone in this research area when they uncovered the cyclopropanation of styrenes with ethyl diazoacetate
2 using an iron porphyrin complex (
4a) in 1995 [
23]. In this report and in further reports by the Che (
4c) and Aviv (
5) groups, the fine-tuning of iron porphyrin complexes by the axial ligand and/or electronics of the porphyrin ring system was demonstrated (
Scheme 3b) [
24,
25,
26]. Today, iron porphyrin complexes and derivatives thereof are important cheap and readily available catalysts to enable carbine-transfer reactions under mild conditions.
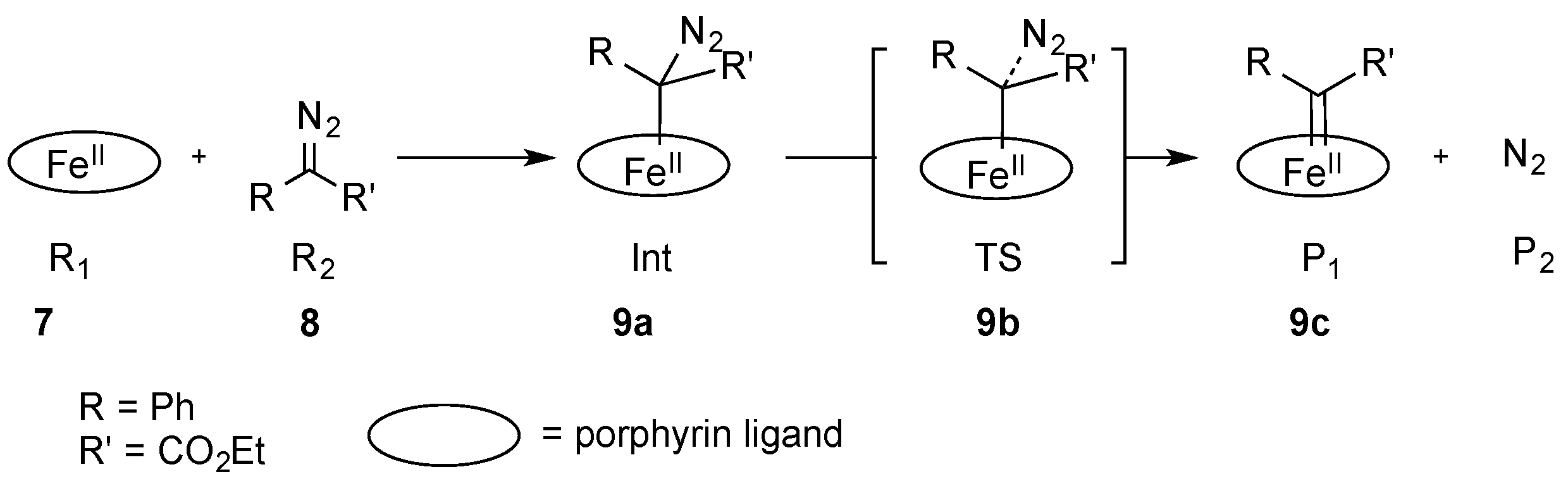
Khade and Zhang studied the formation of iron carbene complexes via quantum chemical studies using ethyl phenyldiazoacetate (
8) as a model compound. They were able to identify a reaction intermediate (
9a) in which ethyl phenyldiazoacetate (
8) coordinated to the iron porphyrin complex
7 via the carbon atom. In a next step, nitrogen was expelled via transition state
9b and the iron carbene complex
9c was formed. In these studies, the authors were also able to show that
N-methyl imidazole reduced both the reaction energy ΔG and the activation energy ΔG
TS (
Scheme 4) [
27].
4. C-H Functionalization Reactions of Aromatic C-H Bonds
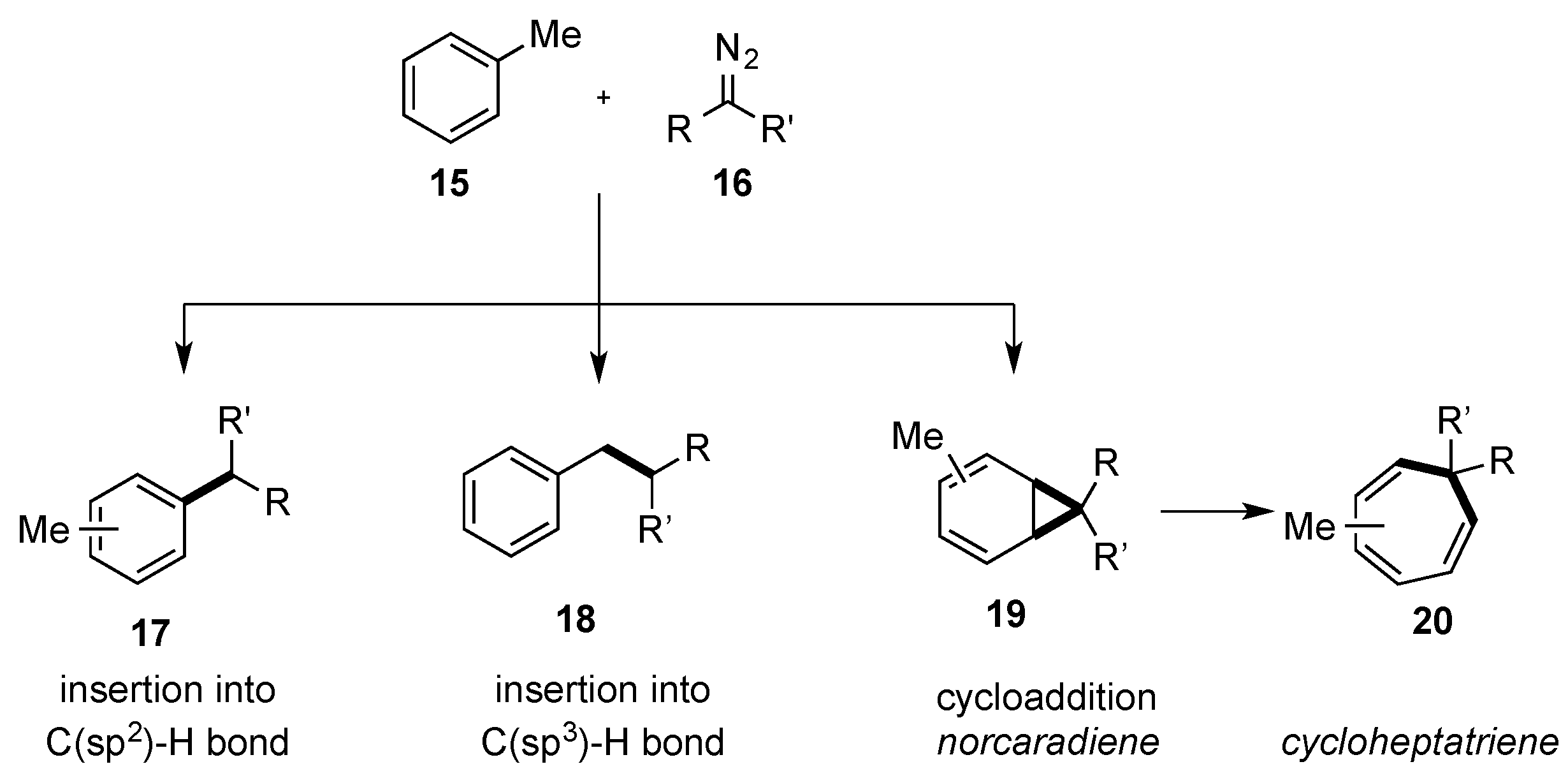
The direct C-H functionalization of benzene or alkyl benzenes (
15) via carbene or metal-carbene intermediates can give access to three different products, which arise from (a) insertion into a C(sp
2)-H bond (
17), (b) insertion into a side-chain C(sp
3)-H bond (
18), or (c) a cycloaddition reaction with the aromatic system to give a norcaradiene
19 that can undergo a Buchner reaction to give a cycloheptatriene
20 (
Scheme 6). The challenge lies in the chemoselective differentiation of these three reaction pathways. Moreover, a C-H insertion into a C(sp
2)-H bond may give rise to different regioisomers.
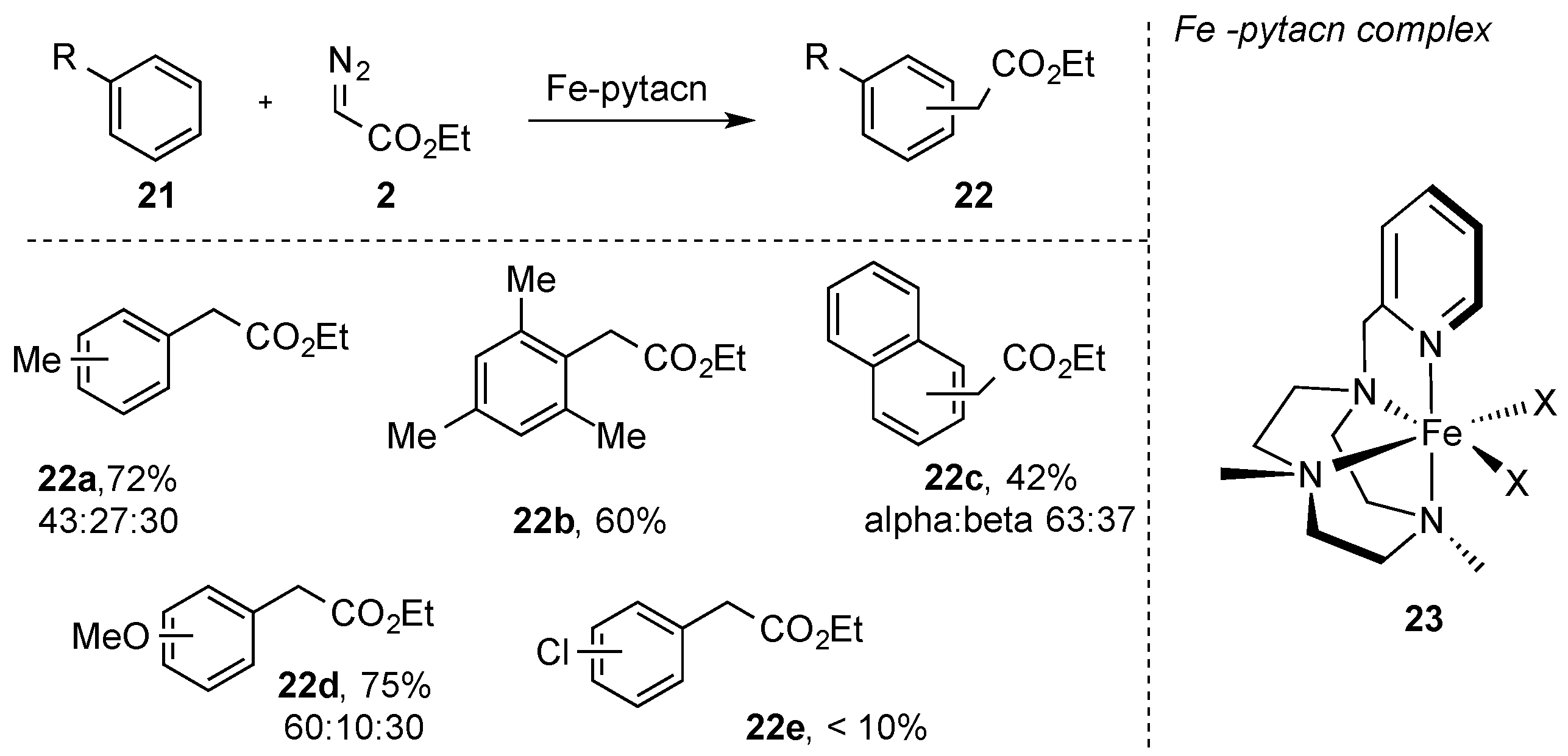
In this context, Luis, Perez, and coworkers reported on the use of iron(II)-complex (
23) bearing a pytacn ligand in the reaction of ethyl diazoacetate
2 with aromatic hydrocarbons (
21). Under these reaction conditions, ethyl diazoacetate (
2) underwent a chemoselective C-H functionalization of the aromatic C(sp
2)-H bond of benzene and alkyl benzene derivatives, and only trace amounts of the norcaradiene/cycloheptatriene reaction pathway were observed [
31]. The substrate scope of benzene derivatives was further studied by the same authors and, in all cases, exclusive C(sp
2)-H bond insertion occurred in moderate to good yields as a mixture of regioisomers. Electron-withdrawing groups (e.g., Cl) had a detrimental effect on the C-H functionalization reaction, and the corresponding products were obtained with reduced yields (
Scheme 7) [
32].
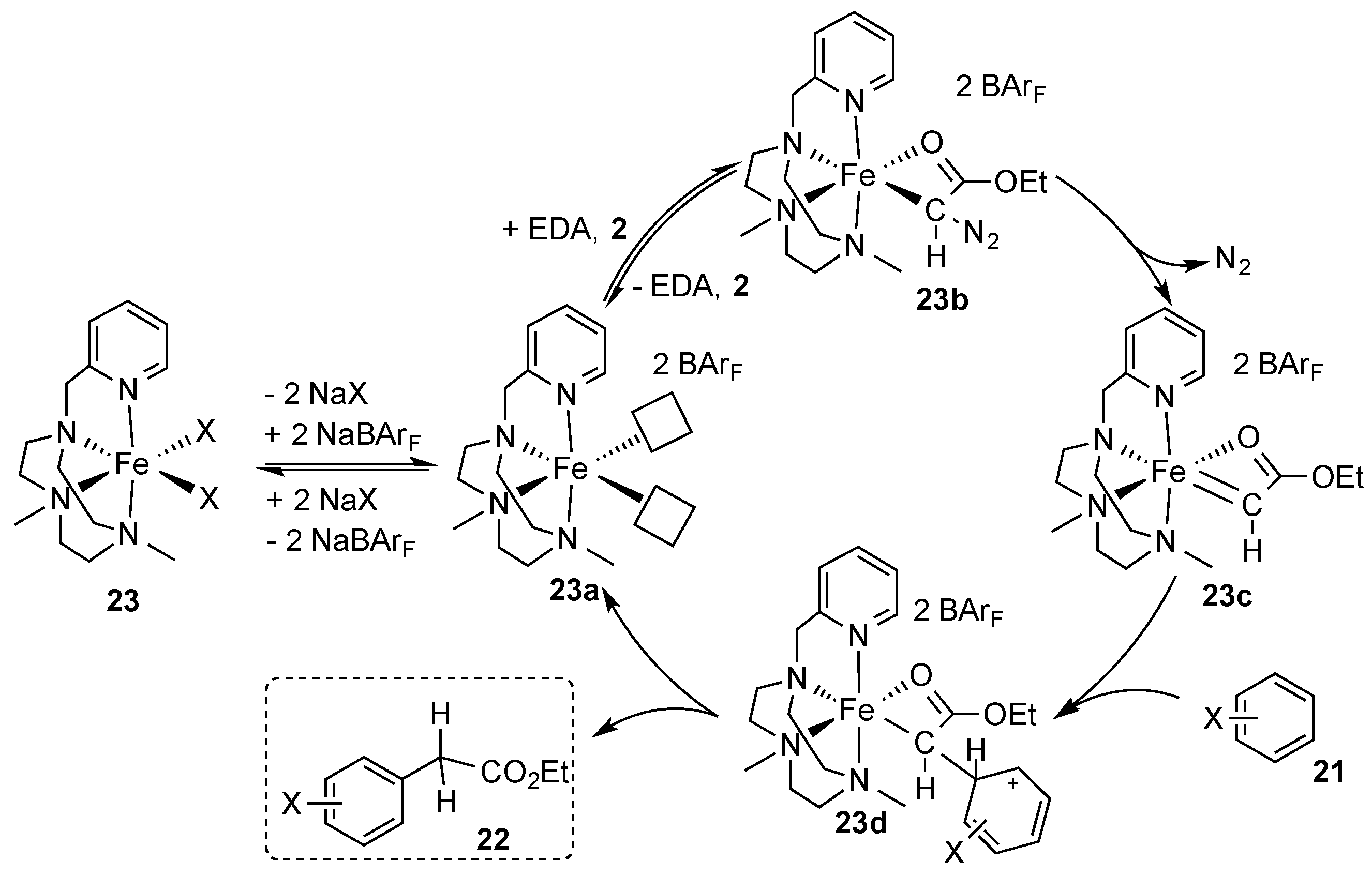
The reaction mechanism of this reaction was studied in detail by the authors. In a first step, the iron complex
23 undergoes a counterion exchange with NaBAr
F to give the dicationic complex
23a. The latter undergoes formation of the initial coordination of ethyl diazoacetate (
23b), which leads to the formation of an iron carbene complex
23c upon extrusion of nitrogen. Next, the arene
21 undergoes nucleophilic addition to
23c under formation of a Wheland-type intermediate
23d, which undergoes a 1,2-H shift to release product
22 and the active catalyst
23a (
Scheme 8) [
32]. The proposed mechanism was in line with a previous report by Doyle, Padwa, and coworkers [
33].
The direct C-H functionalization reaction of aromatic compounds (used as solvents) was also studied by Woo and coworkers using dimethyl diazomalonate
25 and an iron(III) porphyrin complex (
4a) as a catalyst at elevated reaction temperatures. However, only moderate chemoselectivity of C(sp
2)-H bond vs. C(sp
3)-H bond insertion could be achieved in this reaction (
26:
27, 2:1 to 10:1). Different substituted aromatic systems such as toluene, mesitylene, 4-chloro-toluene, or anisole were shown to be compatible with the present reaction conditions. In contrast to the previously described reaction, the product from the C(sp
3)-H bond insertion reaction was obtained as the major product (
Scheme 9) [
34].
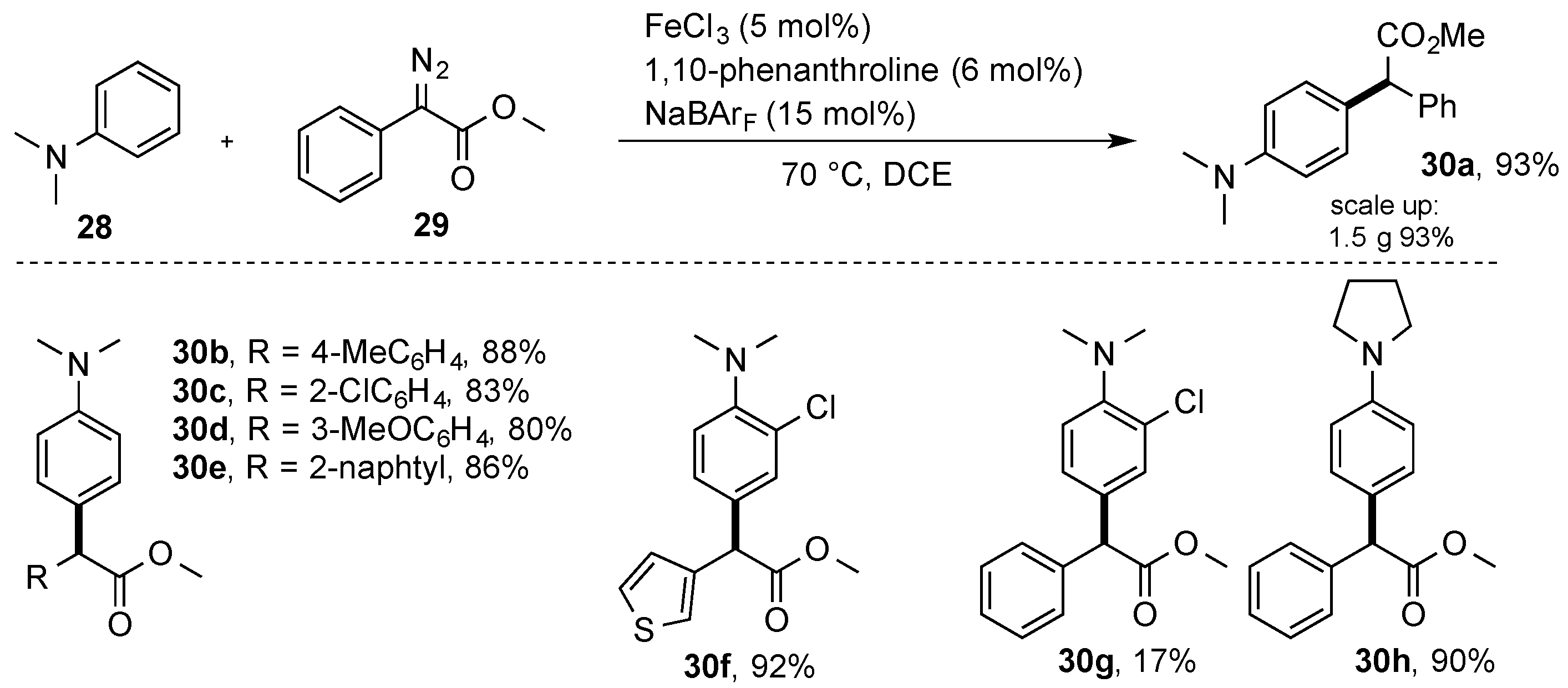
In 2015, Zhou and coworkers studied the reaction of donor–acceptor diazo compounds
29 with electron-rich aromatics
28. Using a simple, non-porphyrin iron complex, the authors were able to demonstrate the C(sp
2)-H functionalization reaction of
N,
N-dialkyl aniline derivatives (
28) with high yield and selectivity. In all cases, selective C-H functionalization in the 4 position occurred, which could be rationalized by the high nucleophilicity of
N,
N-dialkyl aniline derivatives in the
para position. From the viewpoint of the catalyst, a very simple iron catalyst was used that could be obtained in situ from FeCl
3, 1,10-phenanthroline, and NaBAr
F (
Scheme 10) [
35]. More recently, Deng and coworkers reported on a similar transformation using a
bis(imino)pyridine iron complex, which could be used in the direct C-H functionalization of
N,
N-dimethylaniline
28 to give
30a with a 71% yield [
36].
5. C-H Functionalization Reactions of Heteroaromatic C-H Bonds
N-heterocycles like indoles or pyrroles are privileged motifs in drugs and natural products, and a broad variety of strategies for their construction or their functionalization has been described in recent decades. A particularly intriguing transformation is the direct C-H bond functionalization of heterocycles, which would streamline current synthesis strategies of drugs or natural products. Furthermore, it would also allow the introduction of molecular diversity into late-stage functionalization reactions. This approach, in combination with the benefits of iron catalysts, would enable important applications of C-H functionalization strategies in medicinal chemistry, agrochemistry, or total synthesis.
In 2006, Woo et al. reported on the iron-catalyzed functionalization of
N-heterocycles with ethyl diazoacetate
2 as a carbene precursor. While no reaction was observed with indole
31, the closely related pyrrole
32 underwent C-H functionalization without concomitant N-H functionalization to give
33 in a 37% yield (
Scheme 11) [
37].
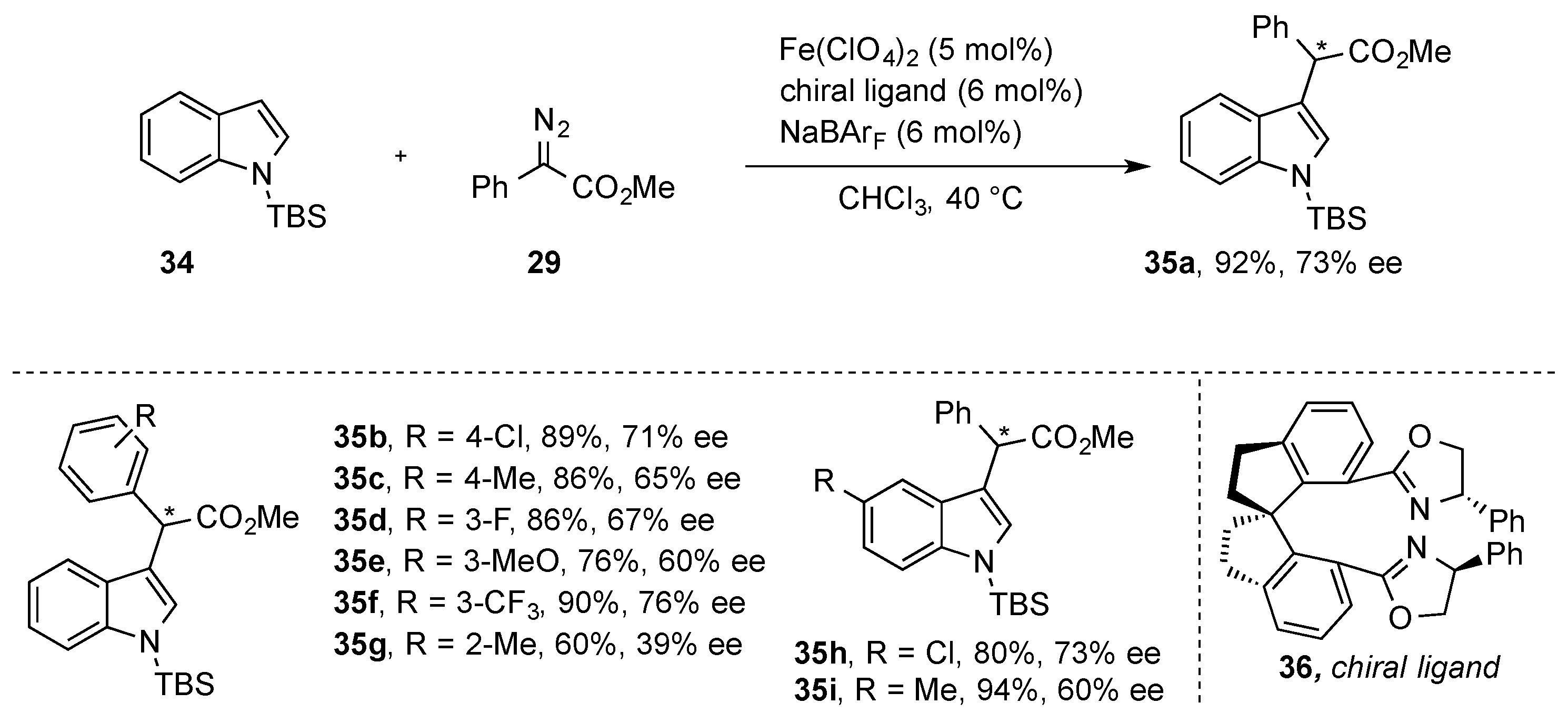
Thereafter, Zhou and-coworkers reported on an enantioselective C-H functionalization of indole heterocycles. For this purpose, they studied the reaction of silyl-protected indole heterocycles (
34) with α-aryl-α-diazoesters (
29) in the presence of Fe(ClO
4)
2 and a chiral spirobisoxazoline ligand
36. This strategy allowed the C-H functionalization of indole heterocycles under mild reaction conditions with moderate enantioselectivity. Importantly, different substituents at the indole heterocycles were tolerated, such as chlorine and methyl (
Scheme 12) [
38].
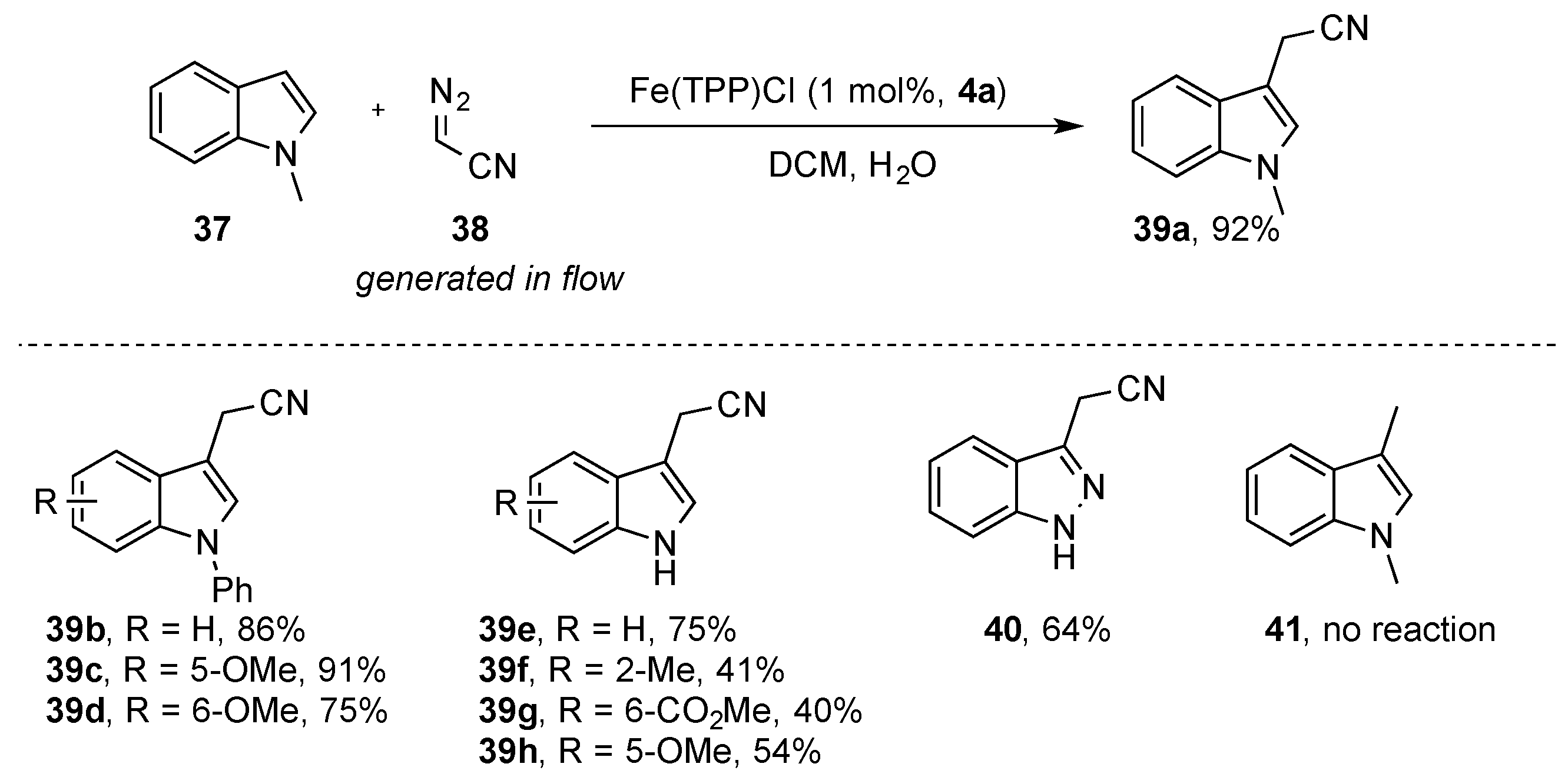
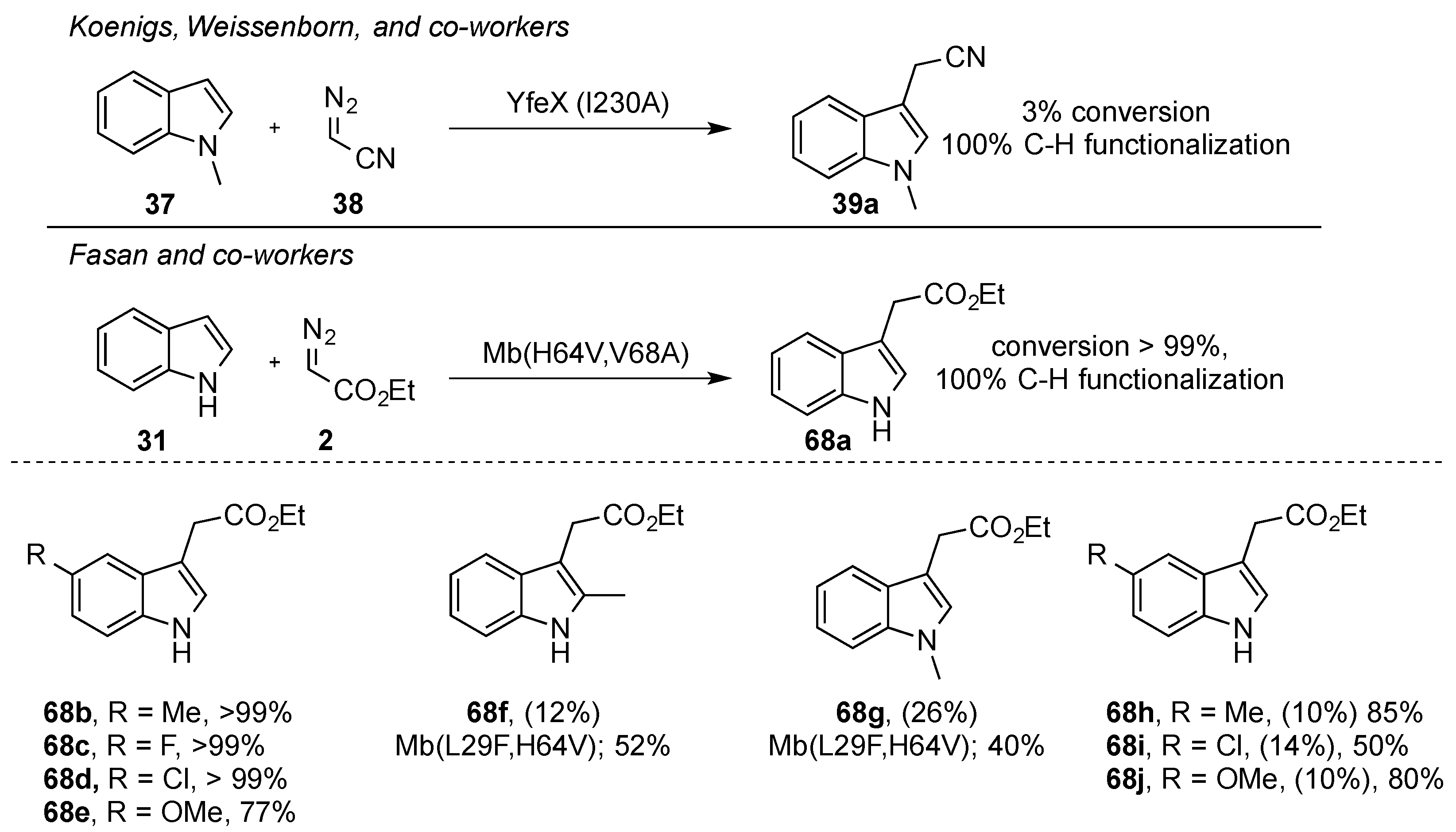
As described by Woo and coworkers, the direct functionalization of indole heterocycles in the C3 position does not readily occur when using ethyl diazoacetate [
37]. The direct functionalization of indole with acceptor-only diazoalkanes would open up avenues towards the efficient synthesis of tryptamines and related alkaloids. In recent years, different groups have focused their efforts on identifying iron catalysts and/or reaction conditions that would allow this important transformation. In 2019, Koenigs and Weissenborn et al. reported the iron-catalyzed C-H functionalization reaction of indoles
37 in the C3 position using the same catalyst as the Woo group, but using diazoacetonitrile
38. Using this methodology, protected and unprotected indole and indazole heterocycles underwent selective functionalization in the C3 position. Importantly, no reaction was observed when blocking the C3 position (
Scheme 13). This approach has now enabled a two-step approach towards the synthesis of important tryptamine derivatives [
39].
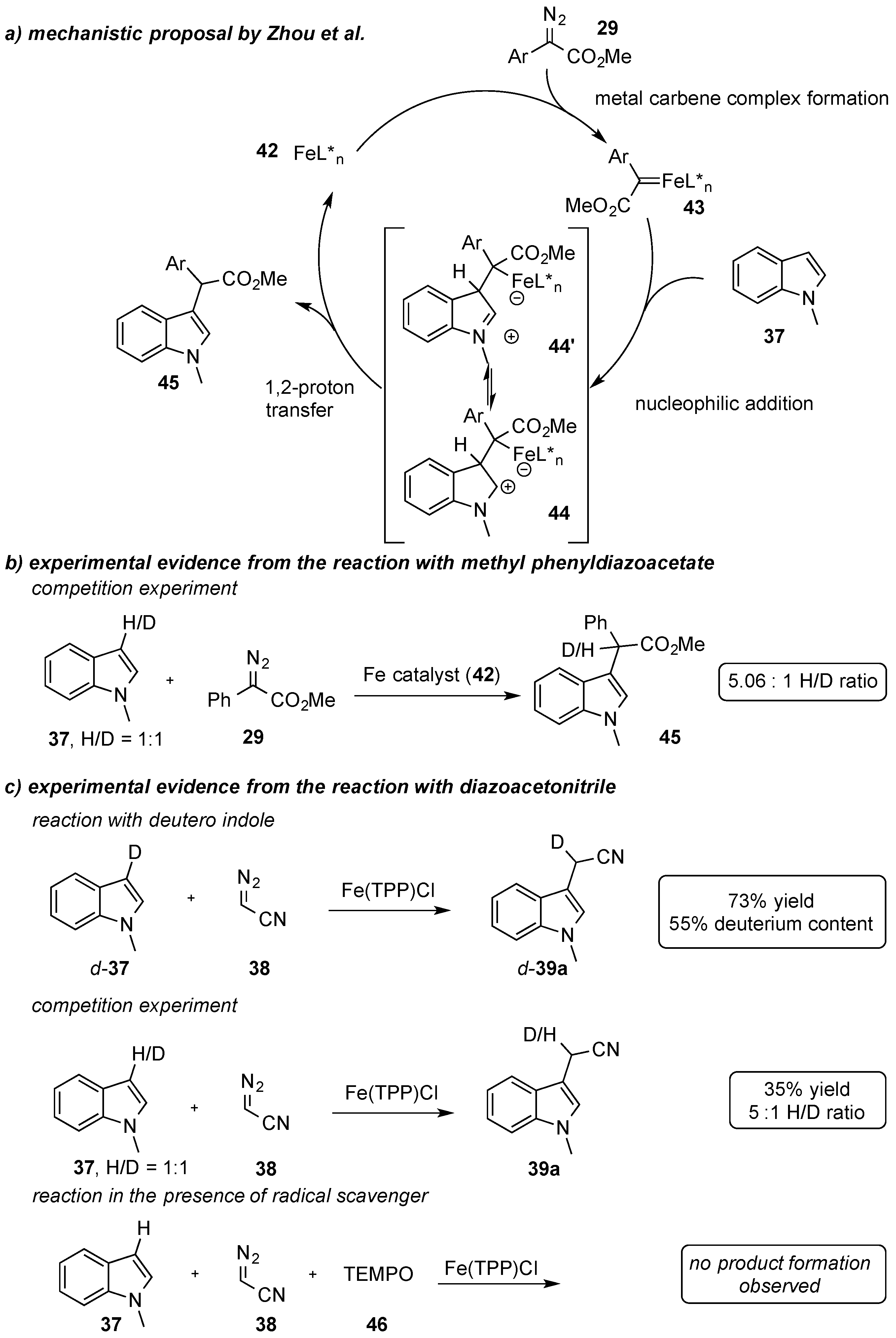
To date, various proposals for the C-H functionalization of indole heterocycles have been reported in the literature. One important mechanistic proposal suggests the initial formation of an iron carbene complex
43 that undergoes nucleophilic addition of an indole followed by a [
1,
2]-proton transfer reaction (
Scheme 14a). Zhou and coworkers reported a KIE (= kinetic isotope effect) of 5.06 in the reaction of
N-methyl indole and deuterated
N-methyl indole, which led them to conclude a proton-transfer reaction to be the rate-determining step (
Scheme 14b) [
38]. Koenigs, Weissenborn, and coworkers observed a similar trend in the reaction of
N-methyl indole and deuterated
N-methyl indole with diazoacetonitrile using both a synthetic and an enzymatic iron catalyst. However, investigations using TEMPO
46 as a radical scavenger revealed a complete inhibition of the C-H functionalization reaction. Based on these experimental data, the authors assumed a radical reaction, yet it was not yet clear in which particular reaction step radical intermediates were involved (
Scheme 14c) [
39].
The reaction mechanism of the C-H functionalization reaction of indole heterocycles with iron carbene intermediates still remains unclear. It would be valuable to investigate this reaction via DFT calculations to gain knowledge of the exact spin state of the participating iron catalyst and to identify the exact reaction mechanism.
6. C-H Functionalization Reactions of Aliphatic C-H Bonds
The direct C-H functionalization reaction of aliphatic C-H bonds is an important strategy for the introduction of new functional groups onto a hydrocarbon skeleton; the difficulties lie in the differentiation of chemically very similar C-H bonds. Over the years, metal-catalyzed insertion reactions of carbene fragments have emerged as a promising strategy for this purpose, and a variety of precious-metal-catalyzed C(sp
3)-H bond functionalization reactions have been described in the literature, ranging from site-selective C-H functionalization to late-stage functionalization of complex molecules [
5,
40,
41].
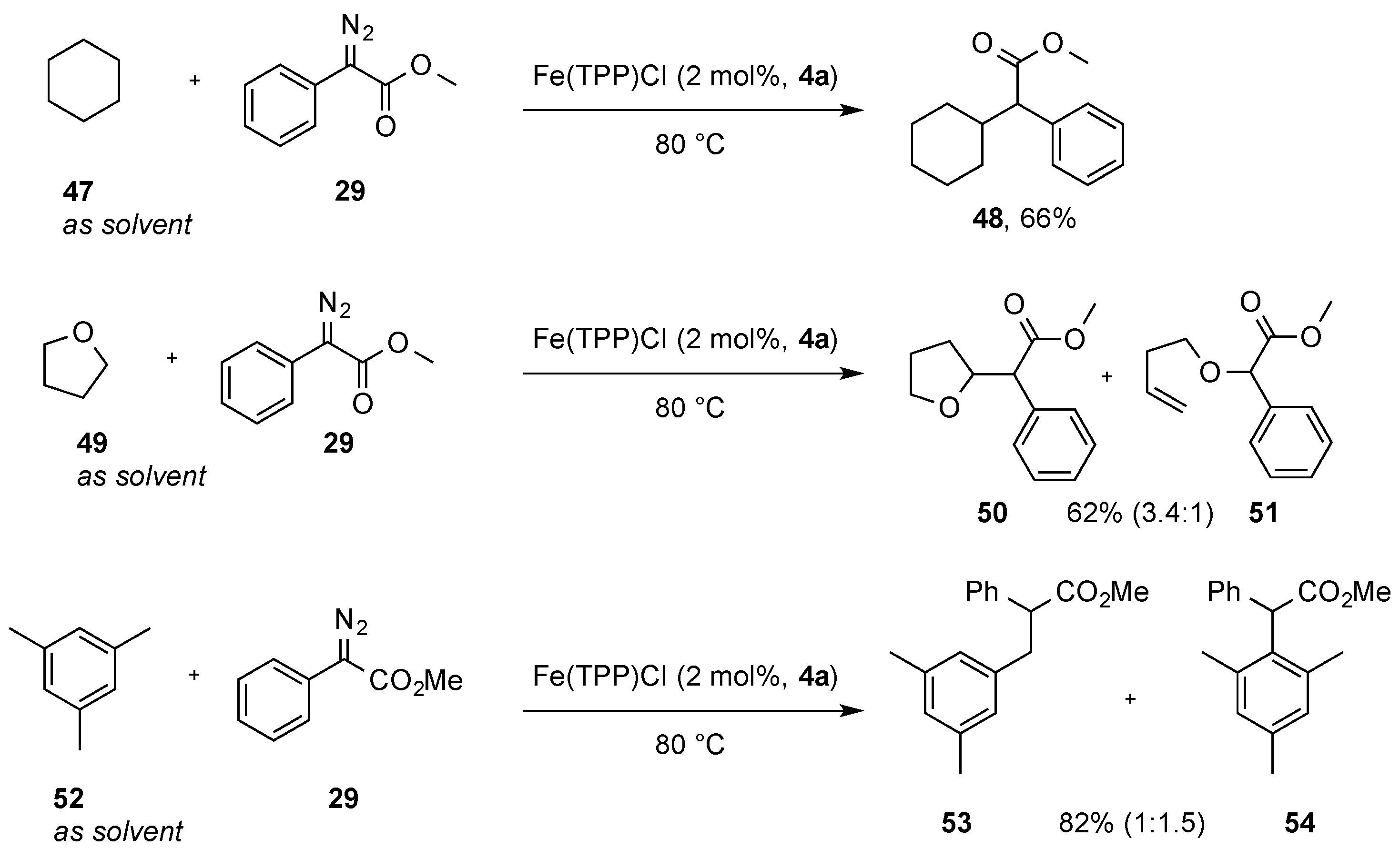
The application of non-toxic and highly active iron catalysts has blossomed in the last few years, and today, iron-catalyzed carbine-transfer reactions have emerged as an important, environmentally benign strategy by which to conduct C(sp
3)-H bond functionalization reactions. In this context, Woo and coworkers reported their studies in C-H functionalization reactions of cyclohexane
47 and tetrahydrofuran
49 as substrates. Using a donor–acceptor diazoalkanes (
29) as a carbene precursor and a Fe(TPP)Cl (
4a) catalyst, the authors demonstrated proof-of-concept studies in this research area. Cyclohexane
47 underwent smooth C-H functionalization to give product
48 with a 66% yield. No C-H functionalization was observed when ethyl diazoacetate
2 or dimethyl diazomalonate
25 were used as carbene precursors. Similarly, tetrahydrofuran
49 underwent selective C-H functionalization reaction in the β position (
50). A byproduct arising from ring-opening of the THF ring was observed as a minor reaction product
51 with ~20% yield. In a competition experiment with mesitylene (
52) as a substrate, the authors obtained the product from C(sp
3)-H bond and C(sp
2)-H bond in a 1.5:1 ratio (
Scheme 15) [
34].
The C-H functionalization of cyclohexane represents one of the simplest examples, as all C-H bonds within cyclohexane are identical. Contrarily, linear or branched hydrocarbons are more challenging, as different C-H bonds are present that need to be differentiated by the catalyst. Zhu et al. studied the reaction of these substrates with donor–acceptor diazoalkanes using an iron catalyst. When
n-hexane
56 was used as a substrate, selective C-H functionalization of secondary C-H groups occurred, but without any meaningful selectivity of the C2 vs. C3 positon. When studying 2-metylpentane
59, the C-H functionalization occurred preferentially at the tertiary C-H group. No C-H functionalization of primary C-H bonds was observed, and the reactivity order for the C-H functionalization decreased from tertiary > secondary > allylic/benzylic >> primary C-H bonds (
Scheme 16). It is of note that this catalyst can be applied in the C-H functionalization of cyclohexane with turnover numbers of up to 690 on gram scale. In this reaction, the authors observed a KIE of 2.0 for k
H/k
D [
42].
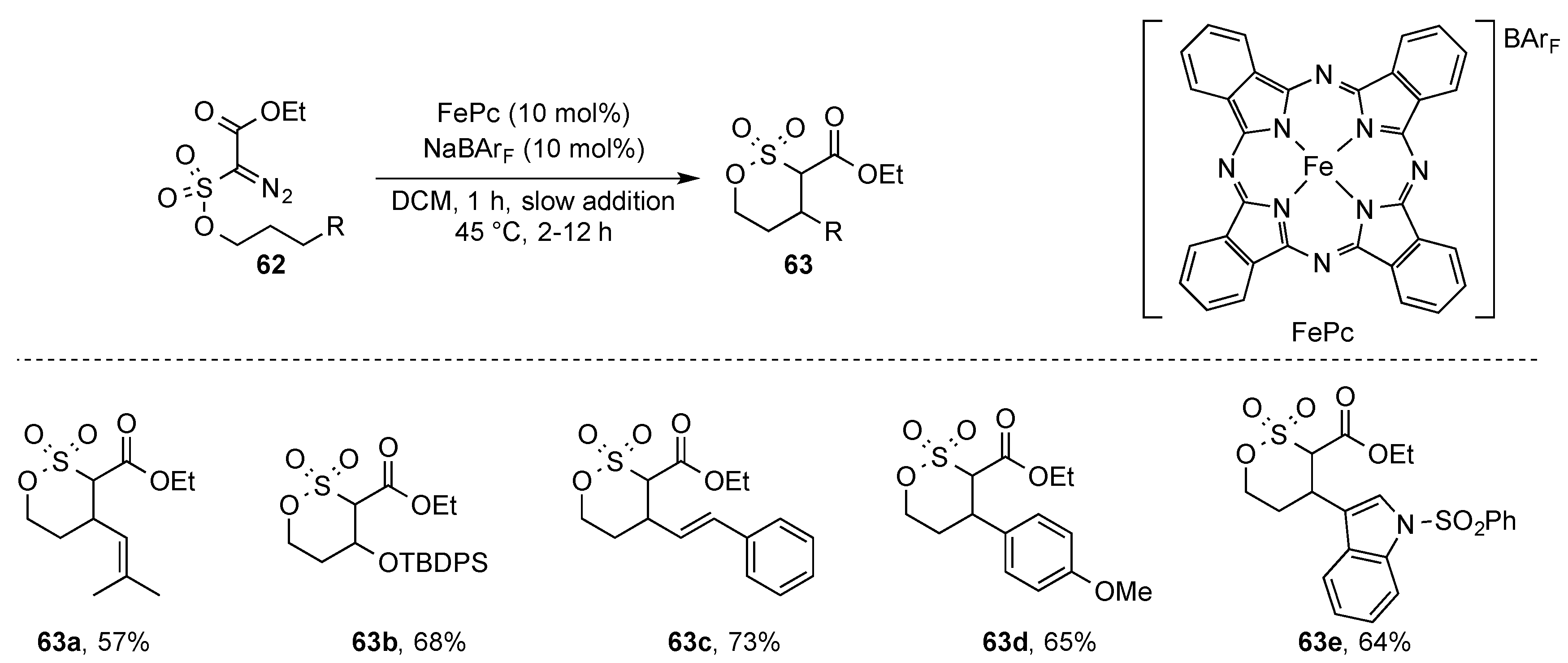
While intermolecular C(sp
3)-H functionalization reactions suffer from missing selectivity and reactivity, the development of intramolecular processes would enable important steps to be taken in the understanding of iron-catalyzed C(sp
3)-H functionalization. In this context, the White group explored an intramolecular cyclization via C-H functionalization using an iron phthalocyanin complex as the carbene-transfer catalyst; importantly, weakly coordinating counterions needed to be used to increase the electrophilicity of the iron complex. Under these conditions, the
bis-acceptor diazoalkanes (
62) underwent smooth intramolecular C-H functionalization reaction to yield product
63 in moderate to good yields with broad functional group tolerance (
Scheme 17). It is important to note that the C-H functionalization reaction selectivity occurred in the allylic, benzylic position or in the α position to a heteroatom [
43].
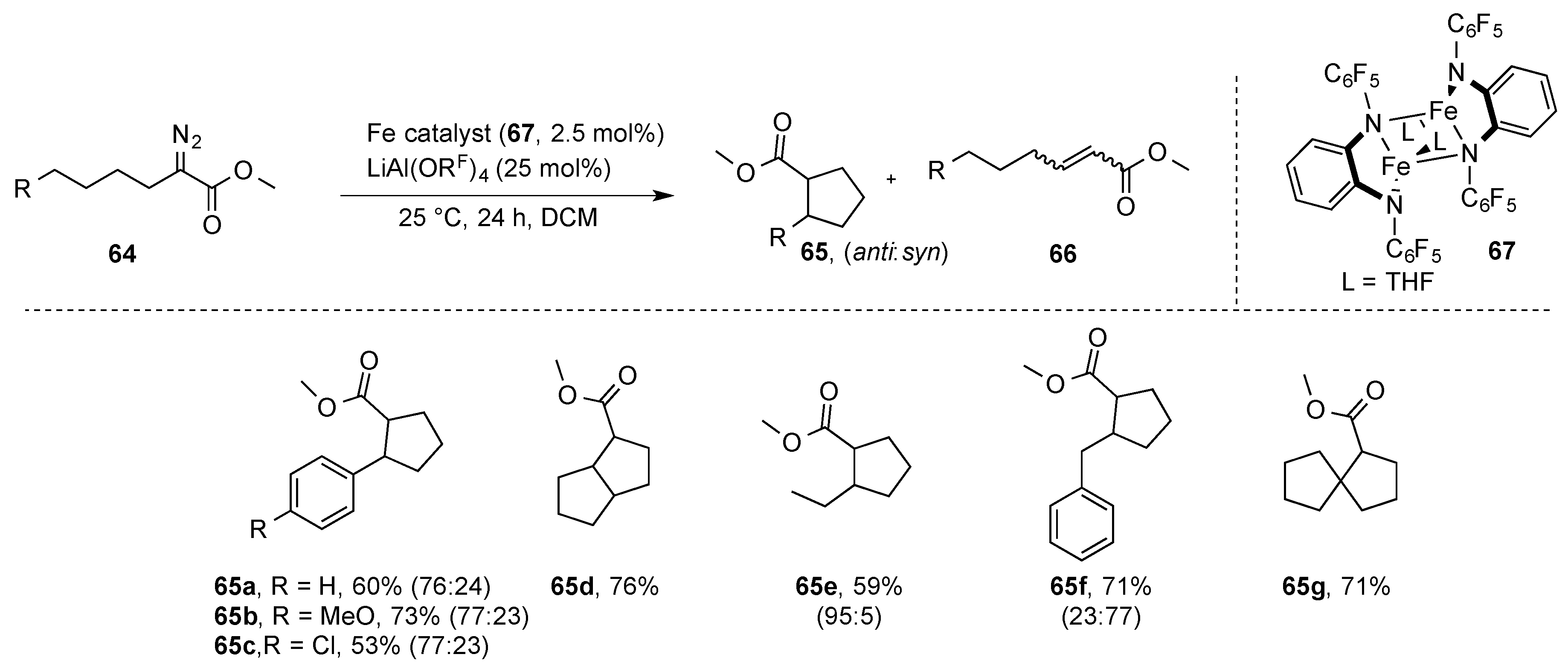
Recently, Costas and coworkers reported on the intramolecular functionalization of C(sp
3)-H bonds using the electrophilic [Fe(
Fpda)-(THF)]
2 complex (
67) as catalyst and a lithium salt with a weakly coordinating counterion as a co-catalyst. Importantly, the lithium salt is required to activate the diazoester under mild conditions, while the iron complex is needed for the actual carbene-transfer reaction. Following this strategy, intramolecular C-H functionalization reactions of C(sp
3)-H could be realized to selectively obtain the five membered ring products (
65). Following this strategy, bi- and spirocyclic systems were synthesized (
Scheme 18). Under these conditions, the five membered ring was is favored, e.g., over benzylic C-H functionalization (
65f). The formation of α,β-unsaturated esters (
66) occurred via a β-hydride elimination reaction [
44].
7. Biocatalytic C-H Functionalization Reactions of Aromatic C-H Bonds
Over the years, the development of non-natural activity of enzymes has developed as an important strategy by which to conduct highly efficient carbene-transfer reactions [
45,
46]. In this section, we have focused on the recent developments in this research area, with a focus on applications in C-H functionalization reactions.
In the report of Koenigs, Weissenborn, and coworkers, the reaction of
N-methyl indole (
37) with diazoacetonitrile (
38) was also studied with the bacterial dye-decolorizing peroxidase YfeX from
E. coli. A library of YfeX variants and the wild-type protein were studied and, under the best conditions, a turnover number of 37 was achieved with the wild-type enzyme. This TON (= turnover number) could be improved to 80 by using the I230A variant of YfeX. This variant was also studied in the reaction of
N-methyl indole with ethyl diazoacetate
2, which led to an increased TON of 236 (
Scheme 17) [
39]. At the same time, the Fasan group reported on the application of an engineered myoglobin enzyme in the C3-functionalization reaction of unprotected indoles using ethyl diazoacetate
2 as a carbene precursor. The Mb(H64V,V68A) variant gave the highest conversion and a TON of 82 in a whole-cell setup, and
68a was obtained as the only product of C-H functionalization. In their report, Fasan and coworkers also investigated the functional group tolerance of this reaction, and different substitution patterns on the indole heterocycle were tolerated. Notably, in the case of
N-protected indoles, the Mb(L29F,H64V) variant was required to achieve a similar activity of the enzyme (
Scheme 19) [
47].
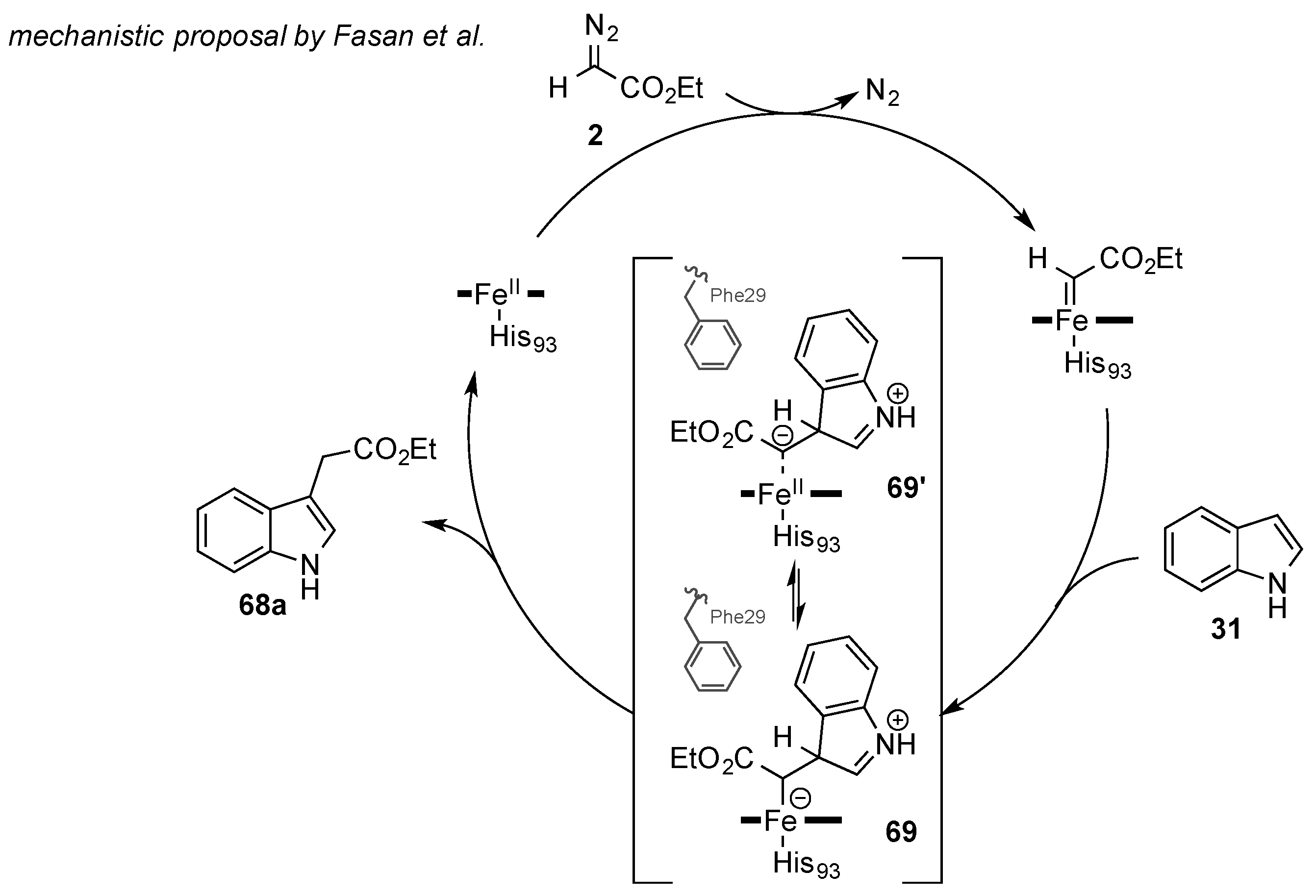
Fasan and coworkers studied the reaction mechanism of the enzymatic C-H functionalization reaction. For this purpose, the reaction of 3-deutero-indole
d-31 and ethyl diazoacetate
2 was investigated. The C-H functionalization product
68a was obtained as the protonated product exclusively, and no deuterium label was found in the reaction product. Moreover, in a competition experiment, no difference in reaction kinetics of the deuterated and non-deuterated starting material was observed. The absence of a kinetic isotope effect led the authors to the conclusion that no C-H insertion mechanism occurred. Based on this evidence, Fasan and coworkers concluded a nucleophilic attack by the indole substrate
31 to generate a zwitterionic intermediate
69/
69′, which underwent a solvent-assisted proton-transfer reaction to give the desired C-H functionalization product (
Scheme 20) [
47]. At this point, it is noteworthy that Koenigs and Weissenborn et al. were able to observe a retention of the deuterium label in the reaction of deuterated
N-methyl indole
d-
47 with diazoacetonitrile [
39].
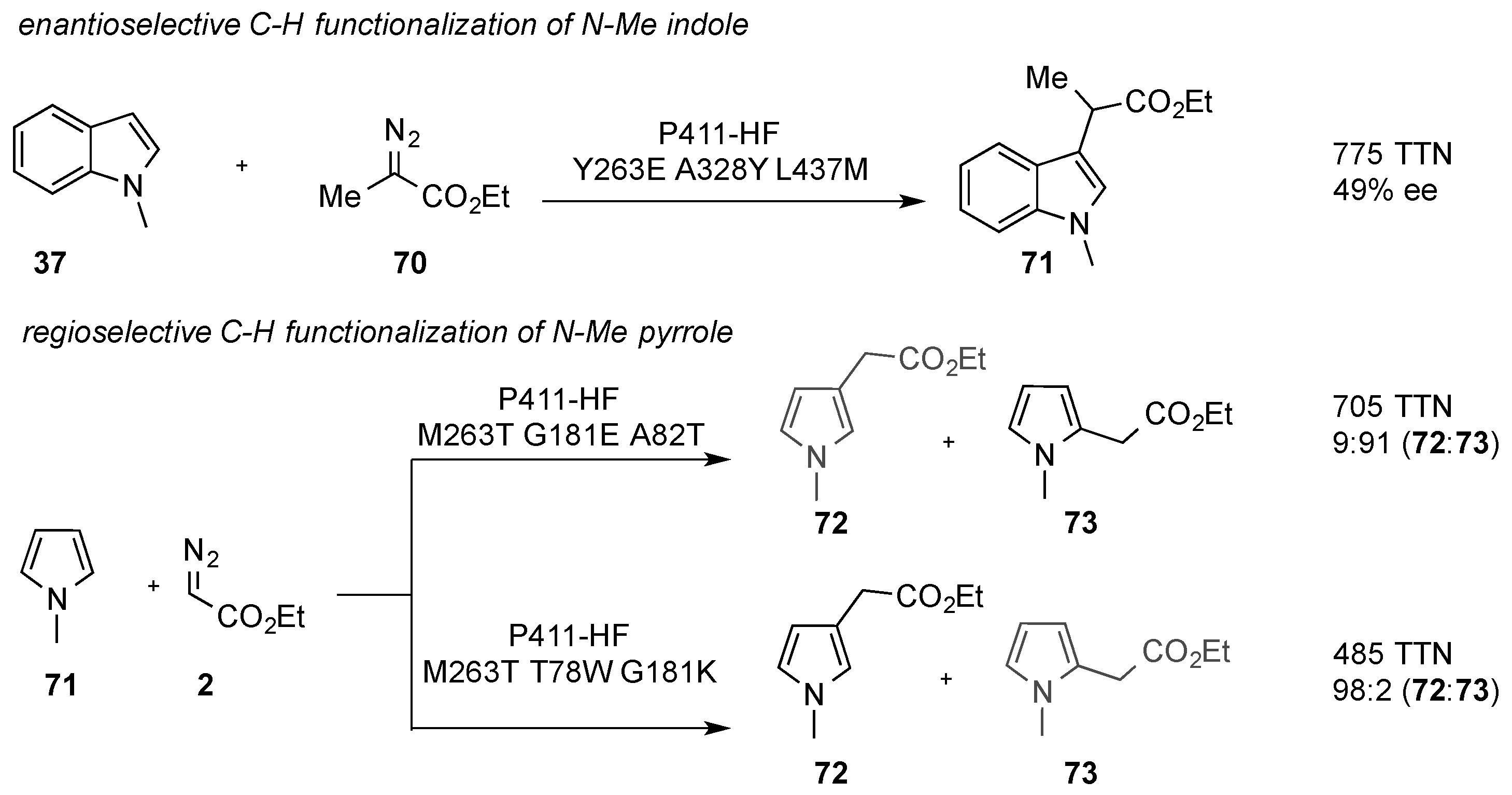
Shortly after the reports by Koenigs, Weissenborn. and Fasan, the enzymatic C-H functionalization of indole and other heterocycles with an engineered cytochrome P411 enzyme was studied by Arnold and coworkers. They reported on the directed evolution of a “carbene transferase” enzyme that enables the highly efficient, chemoselective, and biocatalytic C-H functionalization of indole and pyrrole heterocycles. Using a UV-Vis spectrophotometry-based high-throughput screening, the authors were able study several thousand mutants to engineer enzymes to conduct chemoselective C-H functionalization reactions. Cytochrome P411 variants were tested in reactions with
N-Me pyrrole (
71), and two different variants allowed the regioselective C-H functionalization in the C3 position or the C2 position as well as enantioselective C-H functionalization of indole heterocycles with diazopropionate (
Scheme 21) [
48].
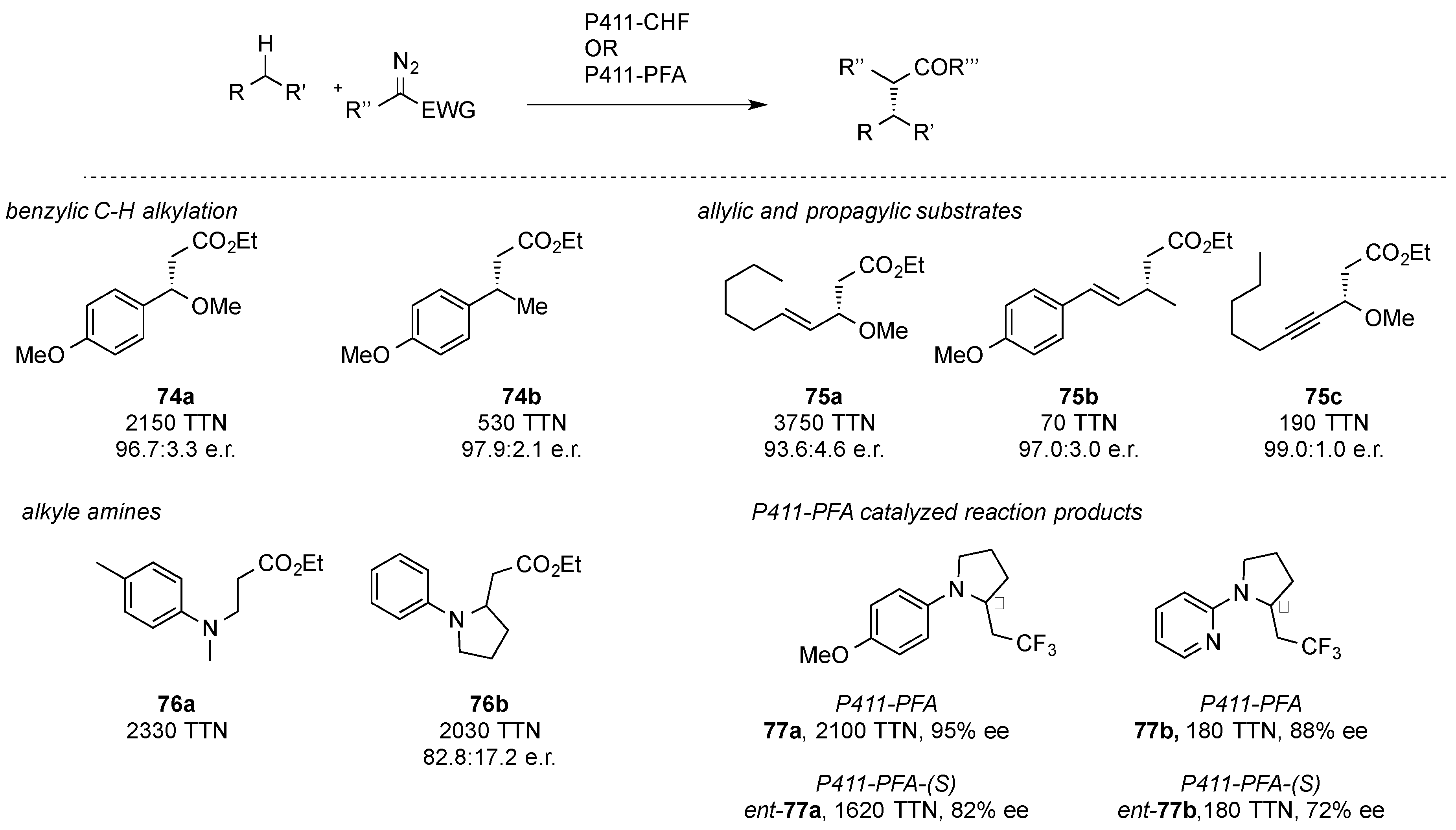
In further reports, the Arnold group studied engineered cytochrome P411 enzymes in benzylic C-H functionalization reactions of alkyl arenes with ethyl diazoacetate (
2). A cytochrome P411 variant with an axial serine ligand served as the starting point for the directed evolution, which led to the P411-CHF variant that gave a TTN (= total turnover number) of 2020 and an enantiomeric ratio of 96.7:3.3 (
74a,
Scheme 22). In further studies, the C-H functionalization of allylic and propargylic substrates as well as alkyl amines was studied (
75,
76,
Scheme 22) [
49]. Only recently, Arnold et al. were able to further extend the substrate scope of carbene precursors to trifluoro diazoethane, which was studied in the α-C(sp
3)-H functionalization of N,N-dialkyl aniline derivatives. By directed evolution of the wild-type cytochrome P411 enzyme the C-H functionalization reaction with trifluoro diazoethane was realized. Further studies focused on the access of the opposite stereoisomer. Starting from a P411 variant that provided the opposite stereochemistry, direct evolution led to a variant providing the inverse stereochemistry. This example showcases the power of biocatalytic transformations and the fact that both enantiomers of a desired product can be obtained by means of directed evolution (
77,
Scheme 22) [
50].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
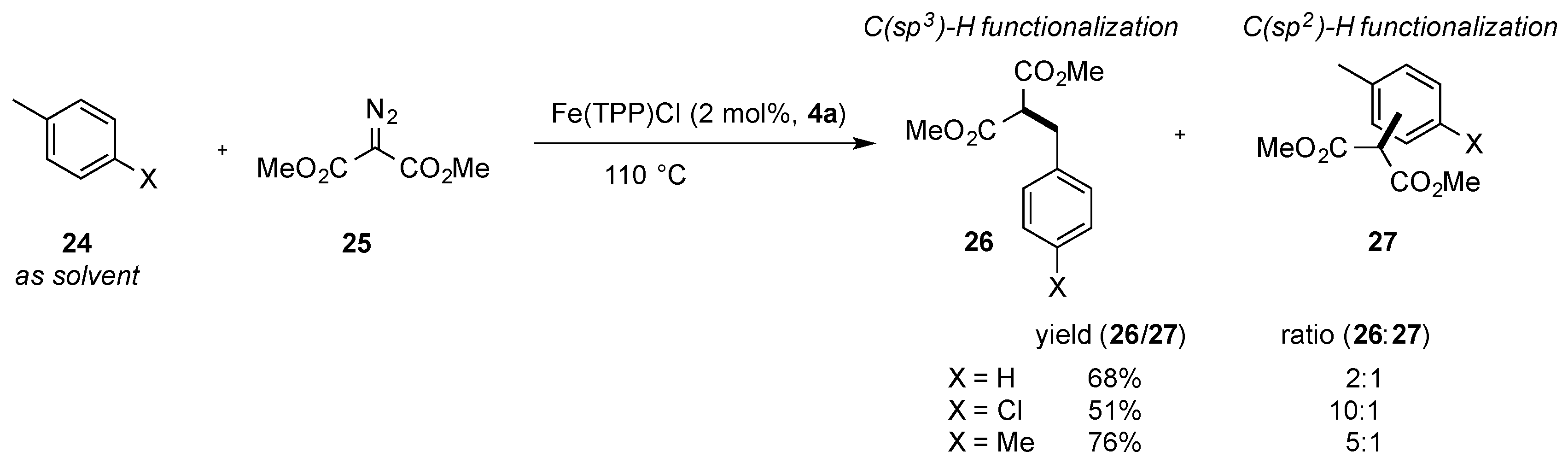{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















