
Synthesis of Lactam-Bridged and Lipidated Cyclo-Peptides as Promising Anti-Phytopathogenic Agents

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
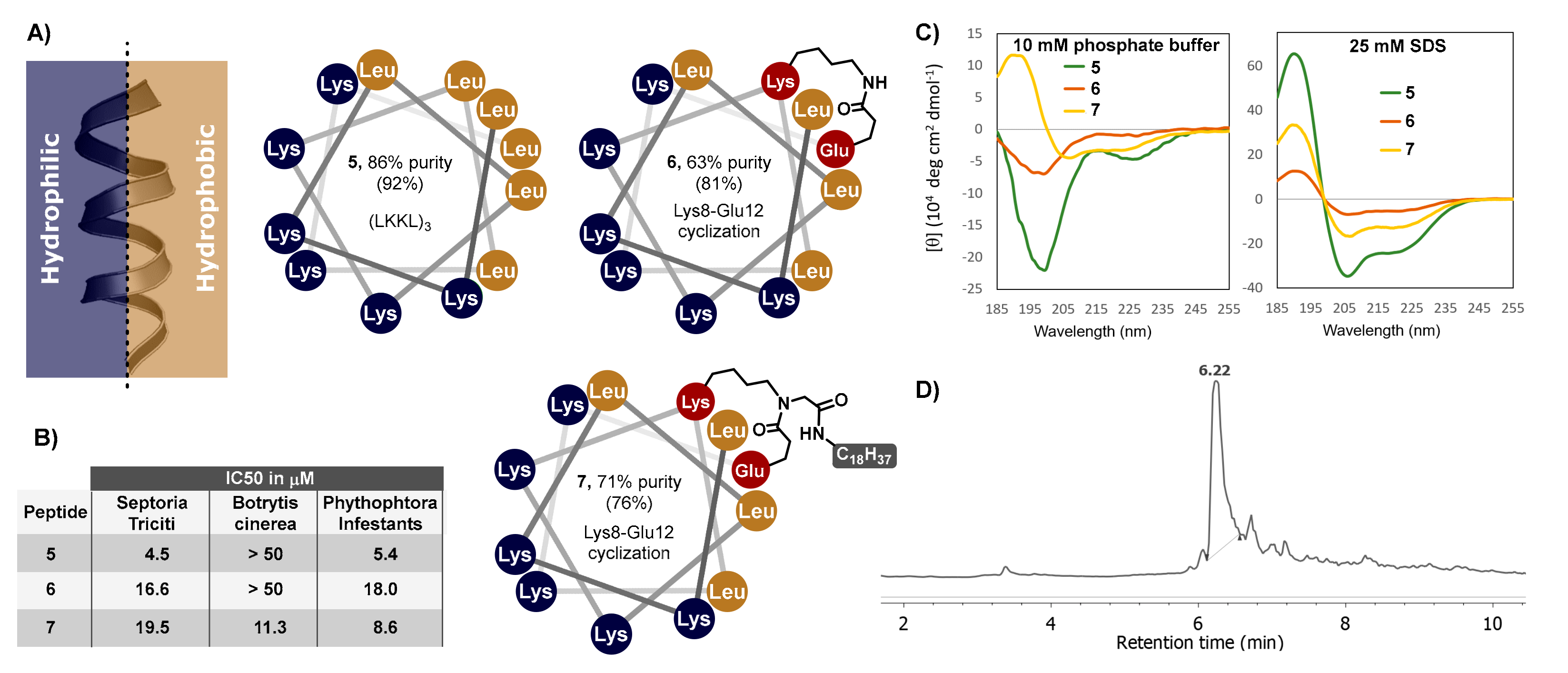
2.1. Synthesis of Peptides Based on the Ac-LAKLLKAKAKAD-NH2 Sequence
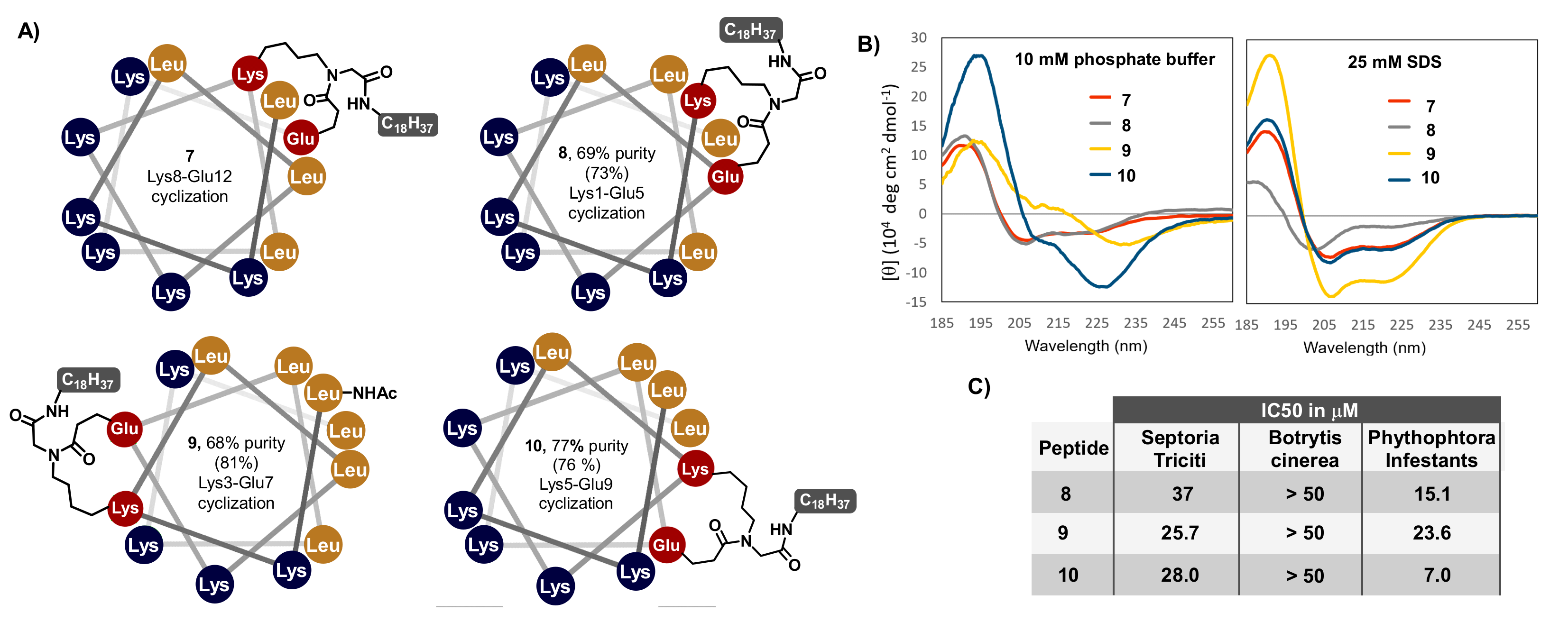
2.2. Secondary Structure and Bioactivity
2.3. Synthesis of Cyclic Lipopeptides Based on the (LKKL)3 Sequence
3. Materials and Methods
3.1. Materials and Equipment
3.2. General Procedures
3.2.1. Solid Phase Peptide Synthesis
3.2.2. N-Terminus Acetylation
3.2.3. Alloc/Allyl Ester Removal
3.2.4. Aminocatalysis-Mediated Ugi-4C Cyclization
3.2.5. Cyclization by Peptide Coupling
3.2.6. Test Cleavage (Mini-Cleavage)
3.2.7. Cleavage
3.3. Synthesis
3.4. Anti-Phytopathogenic Bioassays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chemie - Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R. Synthetic lipopeptides: A novel class of anti-infectives. Expert Opin. Investig. Drugs 2007, 16, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2009, 31, 71–82. [Google Scholar] [CrossRef]
- Martínez-de-Anda, A.; Valdivia, A.G.; Jaramillo-Juárez, F.; Reyes, J.L.; Ortiz, R.; Quezada, T.; de Luna, M.C.; Rodríguez, M.L. Effects of aflatoxin chronic intoxication in renal function of laying hens. Poult. Sci. 2010, 89, 1622–1628. [Google Scholar] [CrossRef]
- Khlangwiset, P.; Shephard, G.S.; Wu, F. Aflatoxins and growth impairment: A review. Crit. Rev. Toxicol. 2011, 41, 740–755. [Google Scholar] [CrossRef]
- Zeitler, B.; Herrera Diaz, A.; Dangel, A.; Thellmann, M.; Meyer, H.; Sattler, M.; Lindermayr, C. De-Novo Design of Antimicrobial Peptides for Plant Protection. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Makovitzki, A.; Shai, Y. pH-dependent antifungal lipopeptides and their plausible mode of action. Biochemistry 2005, 44, 9775–9784. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Pasupuleti, M.; Malmsten, M. Effect of hydrophobic modifications in antimicrobial peptides. Adv. Colloid Interface Sci. 2014, 205, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolym. - Pept. Sci. Sect. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, F.M.; Garro, A.D.; Masman, M.F.; Rodríguez, A.M.; Luiten, P.G.M.; Raimondi, M.; Zacchino, S.A.; Somlai, C.; Penke, B.; Enriz, R.D. New small-size peptides possessing antifungal activity. Bioorganic Med. Chem. 2010, 18, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Meena, K.R.; Kanwar, S.S. Lipopeptides as the antifungal and antibacterial agents: Applications in food safety and therapeutics. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Mahindra, A.; Bagra, N.; Wangoo, N.; Khan, S.I.; Jacob, M.R.; Jain, R. Discovery of short peptides exhibiting high potency against Cryptococcus neoformans. ACS Med. Chem. Lett. 2014, 5, 315–320. [Google Scholar] [CrossRef]
- Grimaldi, M.; De Rosa, M.; Di Marino, S.; Scrima, M.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Soriente, A.; D’ursi, A.M. Synthesis of new antifungal peptides selective against Cryptococcus neoformans. Bioorganic Med. Chem. 2010, 18, 7985–7990. [Google Scholar] [CrossRef]
- Johnson, E.T.; Evans, K.O.; Dowd, P.F. Antifungal activity of a synthetic cationic peptide against the plant pathogens Colletotrichum graminicola and three Fusarium species. Plant Pathol. J. 2015, 31, 316–321. [Google Scholar] [CrossRef]
- Balkovec, J.M. Section review: Anti-infectives: Lipopeptide antifungal agents. Expert Opin. Investig. Drugs 1994, 3, 65–82. [Google Scholar] [CrossRef]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef]
- Arima, K.; Kakinuma, A.; Tamura, G. Surfactin, a crystalline peptidelipid surfactant produced by Bacillus subtilis: Isolation, characterization and its inhibition of fibrin clot formation. Biochem. Biophys. Res. Commun. 1968, 31, 488–494. [Google Scholar] [CrossRef]
- Delcambe, L.; Peypoux, F.; Besson, F.; Guinand, M.; Michel, G. Structure of iturin and iturin like substances. Biochem. Soc. Trans. 1977, 5, 1122–1124. [Google Scholar] [CrossRef]
- Vanittanakom, N.; Loeffler, W.; Koch, U.; Jung, G. Fengycin-A Novel Antifungal Lipopeptide Antibiotic Produced by Bacillus Subtilis F-29-3. J. Antibiot. (Tokyo) 1986, 39, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Wessjohann, L.A.; Rivera, D.G.; Vercillo, O.E. Multiple Multicomponent Macrocyclization including Bifunctional Building Blocks (MiBs): A Strategic Development Towards Macrocycle Diversity. Chem. Rev. 2009, 109, 796–814. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Neves Filho, R.A.; Puentes, A.A.; Morejon, M.C. Macrocycles from Multicomponent Reactions. In Multicomponent Reactions in Organic Synthesis; Zhu, J., Wang, Q., Wang, M.-X., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 231–264. ISBN 9783527678174. [Google Scholar]
- Morales, F.E.; Garay, H.E.; Muñoz, D.F.; Augusto, Y.E.; Otero-González, A.J.; Reyes Acosta, O.; Rivera, D.G. Aminocatalysis-mediated on-resin ugi reactions: Application in the solid-phase synthesis of n -substituted and tetrazolo lipopeptides and peptidosteroids. Org. Lett. 2015, 17, 2728–2731. [Google Scholar] [CrossRef]
- Rivera, D.G.; Vasco, A.V.; Echemendía, R.; Concepción, O.; Pérez, C.S.; Gavín, J.A.; Wessjohann, L.A. A Multicomponent Conjugation Strategy to Unique N-Steroidal Peptides: First Evidence of the Steroidal Nucleus as a β-Turn Inducer in Acyclic Peptides. Chem. A Eur. J. 2014, 20, 13150–13161. [Google Scholar] [CrossRef]
- Morejón, M.C.; Laub, A.; Westermann, B.; Rivera, D.G.; Wessjohann, L.A. Solution- and Solid-Phase Macrocyclization of Peptides by the Ugi–Smiles Multicomponent Reaction: Synthesis of N -Aryl-Bridged Cyclic Lipopeptides. Org. Lett. 2016, 18, 4096–4099. [Google Scholar] [CrossRef]
- Ricardo, M.G.; Llanes, D.; Wessjohann, L.A.; Rivera, D.G. Introducing the Petasis Reaction for Late-Stage Multicomponent Diversification, Labeling, and Stapling of Peptides. Angew. Chemie Int. Ed. 2019, 58, 2700–2704. [Google Scholar] [CrossRef]
- Vasco, A.V.; Méndez, Y.; Porzel, A.; Balbach, J.; Wessjohann, L.A.; Rivera, D.G. A Multicomponent Stapling Approach to Exocyclic Functionalized Helical Peptides: Adding Lipids, Sugars, PEGs, Labels, and Handles to the Lactam Bridge. Bioconjug. Chem. 2019, 30, 253–259. [Google Scholar] [CrossRef]
- Morejón, M.C.; Laub, A.; Kaluđerović, G.N.; Puentes, A.R.; Hmedat, A.N.; Otero-González, A.J.; Rivera, D.G.; Wessjohann, L.A. A multicomponent macrocyclization strategy to natural product-like cyclic lipopeptides: Synthesis and anticancer evaluation of surfactin and mycosubtilin analogues. Org. Biomol. Chem. 2017, 15, 3628–3637. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides [8]. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Kim, Y.W.; Grossmann, T.N.; Verdine, G.L. Synthesis of all-hydrocarbon stapled ±-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.M.; Heimer, E.P.; Wang, C.T.; Lambros, T.J.; Fournier, A.; Mowles, T.F.; Maines, S.; Campbell, R.M.; Wegrzynski, B.B.; Toome, V.; et al. Synthesis, biological activity and conformational analysis of cyclic GRF analogs. Int. J. Pept. Protein Res. 1988, 32, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.E.; Gannon, C.L.; Kay, C.M.; Hodges, R.S. Lactam bridge stabilization of α-helical peptides: Ring size, orientation and positional effects. J. Pept. Sci. 1995, 1, 274–282. [Google Scholar] [CrossRef]
- Shepherd, N.E.; Hoang, H.N.; Abbenante, G.; Fairlie, D.P. Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127, 2974–2983. [Google Scholar] [CrossRef]
- Sharma, K.; Kunciw, D.L.; Xu, W.; Wiedmann, M.M.; Wu, Y.; Sore, H.F.; Galloway, W.R.J.D.; Lau, Y.H.; Itzhaki, L.S.; Spring, D.R. CHAPTER 8. Double-click Stapled Peptides for Inhibiting Protein–Protein Interactions. In Cyclic Peptides: From Bioorganic Synthesis to Applications; The Royal Society of Chemistry: London, UK, 2017; pp. 164–187. ISBN 978-1-78262-528-5. [Google Scholar]
- Lautrette, G.; Touti, F.; Lee, H.G.; Dai, P.; Pentelute, B.L. Nitrogen Arylation for Macrocyclization of Unprotected Peptides. J. Am. Chem. Soc. 2016, 138, 8340–8343. [Google Scholar] [CrossRef]
- Brown, S.P.; Smith, A.B. Peptide/protein stapling and unstapling: Introduction of s-tetrazine, photochemical release, and regeneration of the peptide/protein. J. Am. Chem. Soc. 2015, 137, 4034–4037. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.S.; Pentelute, B.L. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef]
- Jo, H.; Meinhardt, N.; Wu, Y.; Kulkarni, S.; Hu, X.; Low, K.E.; Davies, P.L.; Degrado, W.F.; Greenbaum, D.C. Development of α-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. [Google Scholar] [CrossRef]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. ChemBioChem 2005, 6, 821–824. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.A.; Shepherd, N.E.; Diness, F.; Fairlie, D.P. Constraining cyclic peptides to mimic protein structure motifs. Angew. Chemie - Int. Ed. 2014, 53, 13020–13041. [Google Scholar] [CrossRef] [PubMed]
- Khara, J.S.; Obuobi, S.; Wang, Y.; Hamilton, M.S.; Robertson, B.D.; Newton, S.M.; Yang, Y.Y.; Langford, P.R.; Ee, P.L.R. Disruption of drug-resistant biofilms using de novo designed short α-helical antimicrobial peptides with idealized facial amphiphilicity. Acta Biomater. 2017, 57, 103–114. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Fundamentals of Modern Peptide Synthesis. In Peptide Synthesis and Applications; Howl, J., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 3–24. ISBN 978-1-58829-317-6. [Google Scholar]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Gomez-Martinez, P.; Dessolin, M.; Guibé, F.; Albericio, F. N -Alloc temporary protection in solid-phase peptide synthesis. The use of amine–borane complexes as allyl group scavengers. J. Chem. Soc. Perkin Trans. 1 1999, 2871–2874. [Google Scholar] [CrossRef]
- Lau, Y.H.; Wu, Y.; De Andrade, P.; Galloway, W.R.J.D.; Spring, D.R. A two-component “double-click” approach to peptide stapling. Nat. Protoc. 2015, 10, 585–594. [Google Scholar] [CrossRef]
- Stammler, G. Phytophthora infestans microtiter method with sporangia. Available online: https://www.frac.info/docs/default-source/monitoring-methods/approved-methods/phytin-microtiter-method-sporangia-basf-2006-v1.pdf?sfvrsn=4&sfvrsn=4. (accessed on 24 October 2005).
- Stammler, G.; Semar, M. Sensitivity of Mycosphaerella graminicola (anamorph: Septoria tritici) to DMI fungicides across Europe and impact on field performance. EPPO Bull. 2011, 41, 149–155. [Google Scholar] [CrossRef]
- Stammler, G.; Speakman, J. Microtiter method to test the sensitivity of Botrytis cinerea to boscalid. J. Phytopathol. 2006, 154, 508–510. [Google Scholar] [CrossRef]
- Otto, A.; Laub, A.; Porzel, A.; Schmidt, J.; Wessjohann, L.; Westermann, B.; Arnold, N. Isolation and Total Synthesis of Albupeptins A-D: 11-Residue Peptaibols from the Fungus Gliocladium album. European J. Org. Chem. 2015, 2015, 7449–7459. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors (limited). |




{kind=link}
{kind=link}
{kind=link}
| Peptide | Septoria Tritici | Botrytis Cinerea | Phytophthora Infestans |
|---|---|---|---|
| 1 | >50 | >50 | >50 |
| 2 | >50 | >50 | 26.2 |
| 3 | 5.9 | 9.8 | 14.3 |
| 4 | >50 | >50 | 20.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasco, A.V.; Brode, M.; Méndez, Y.; Valdés, O.; Rivera, D.G.; Wessjohann, L.A. Synthesis of Lactam-Bridged and Lipidated Cyclo-Peptides as Promising Anti-Phytopathogenic Agents. Molecules 2020, 25, 811. https://doi.org/10.3390/molecules25040811
Vasco AV, Brode M, Méndez Y, Valdés O, Rivera DG, Wessjohann LA. Synthesis of Lactam-Bridged and Lipidated Cyclo-Peptides as Promising Anti-Phytopathogenic Agents. Molecules. 2020; 25(4):811. https://doi.org/10.3390/molecules25040811
Chicago/Turabian StyleVasco, Aldrin V., Martina Brode, Yanira Méndez, Oscar Valdés, Daniel G. Rivera, and Ludger A. Wessjohann. 2020. "Synthesis of Lactam-Bridged and Lipidated Cyclo-Peptides as Promising Anti-Phytopathogenic Agents" Molecules 25, no. 4: 811. https://doi.org/10.3390/molecules25040811
APA StyleVasco, A. V., Brode, M., Méndez, Y., Valdés, O., Rivera, D. G., & Wessjohann, L. A. (2020). Synthesis of Lactam-Bridged and Lipidated Cyclo-Peptides as Promising Anti-Phytopathogenic Agents. Molecules, 25(4), 811. https://doi.org/10.3390/molecules25040811