3. Materials and Methods
3.1. General Information
Commercial reagents were purified prior to use following the guidelines of Perrin and Armarego [
26]. All solvents were redistilled before using. Reactions under N
2 atmosphere were performed with a Schlenk apparatus or simple dry-box (Laboratory build, Hanoi, Vietnam). Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator using an ice-water bath for volatile compounds. Chromatographic purification of products was accomplished by flash chromatography on silica gel according to the method of Still [
27]. Thin-layer chromatography (TLC) was performed on Merk silica gel plates. Visualization of the developed chromatogram was performed by fluorescence quenching,
p–anisaldehyde, ceric ammonium molybdate, or potassium permanganate stains.
1H and
13C NMR spectra were recorded on a Bruker Advance–III 500 (500 and 125 MHz) instrument (Bruker Corp., Billerica, MA, USA) in the Faculty of Chemistry, VNU University of Science, Vietnam National University, Hanoi, Vietnam, and were internally referenced to residual protic solvent signals (note: CDCl
3 referenced at d 7.27 and 77.0 ppm respectively).
13C NMR spectra were recorded in J-MOD (J-modulated spin-echo) mode, which were the combination of
13C and DEPT without losing quaternary
13C signals. Data for
1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, h = heptet, m = multiplet, b = broad), integration, coupling constant (Hz), and assignment. Data for
13C NMR are reported in terms of chemical shift and multiplicity; no special nomenclature has been used for equivalent carbons. The IR spectrum (
Supplementary Materials) of alcohol
4 was recorded on the Shimadzu FTIR Affinity-1S (Shimadzu Corp., Kyoto, Japan) in the Department of Inorganic Chemistry, Faculty of Chemistry, VNU University of Science, Vietnam National University and has been reported in terms of frequency of absorption. High-resolution mass spectra were obtained via ESI method on Agilent 6530 Accurate-Mass Q-TOP LC/MS (Agilent Technonogy Inc., Santa Clara, CA, USA) in the Institute of Marine Biochemistry, Vietnam Academy of Science and Technology Hanoi, Vietnam.
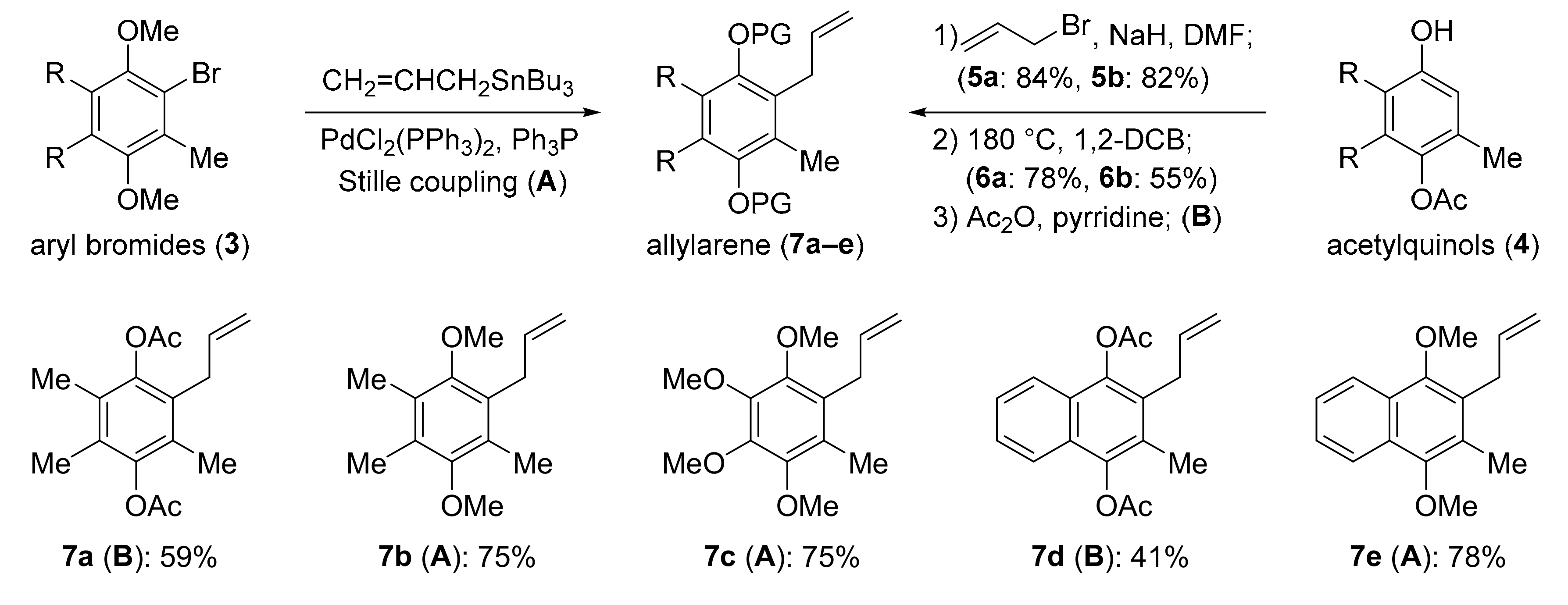
3.2. General Procedure for Allylation Reaction
NaH (0.78 g, 32.5 mmol) was slowly added to the solution of phenol (16 mmol) in DMF (60 mL) at 0 °C under an N2 atmosphere. The reaction mixture was stirred vigorously for 30 min to evaporate the gas in solution. After that, allyl bromide (2.18 mL, 25.2 mmol) was slowly added to the reaction flask and refluxed at 40 °C for 12 h. The mixture was poured into ice-water and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and evaporated under vacuum. The residue was purified via flash column chromatography with silica gel as the stationary phase (0%–2% ethyl acetate/n-hexane) to yield the titular allyl aryl ether.
4-(Allyloxy)-2,3,6-trimethylphenyl acetate (5a): clear oil, 84% yield. IR (film) 2976, 2918, 2864, 1759, 1744, 1599, 1584, 1404, 1357, 1211, 1161, 1111, 1084, 937, 895, 839, 768, 745 cm–1; 1H NMR (500 MHz, CDCl3) δ 6.58 (s, 1H), 6.14–6.02 (m, 1H), 5.44 (dq, J = 17.3, 1.7 Hz,1H), 5.28 (dq, J = 10.5, 1.5 Hz, 1H), 4.50 (dt, J = 5.0, 1.6 Hz, 2H), 2.34 (s, 3H), 2.18 (s, 3H), 2.13 (s, 3H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.5, 154.2, 141.8, 133.8, 129.8, 127.1, 124.3, 116.9, 111.6, 69.4, 20.6, 16.7, 13.1, 12.1; HRMS (ESI) exact mass calculated for [M + H]+ (C14H19O3) requires m/z 235.1334, found m/z 235.1329.
4-(allyloxy)-2-methylnaphthalen-1-yl acetate (5d): clear oil, 82% yield. IR (film) 3073, 2918, 2862, 1759, 1744, 1632, 1599, 1506, 1462, 1404, 1356, 1269, 1202, 1159, 1111, 1084, 937, 895, 839, 768, 745 cm–1; 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J 8.3 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.52–7.48 (m, 1H), 7.45–7.42 (m, 1H), 6.65 (s, 1H), 6.20–6.14 (m, 1H), 5.53 (dd, J = 17.3, 1.5 Hz, 1H), 5.34 (dd, J = 6.8, 1 Hz, 1H), 4.70 (d, J 5.1 Hz, 2H), 2.46 (s, 3H), 2.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.6, 152.3, 137.8, 133.2, 127.9, 127.2, 126.3, 124.9, 122.6, 120.6, 117.6, 107.7, 69.3, 20.7, 16.9; HRMS (ESI) exact mass calculated for [M + H]+ (C16H17O3) requires m/z 257.1178, found m/z 257.1172.
3.3. General Procedure for Claisen Rearrangement
A solution of allyl aryl ether (11 mmol) in 1,2-dichlorobenzene (50 mL) was stirred for 24 h at 180 °C under an N2 atmosphere. The mixture was purified by column chromatography with silica gel as the stationary phase (0%–5% ethyl acetate/n-hexane) to yield the titular allylphenol.
3-allyl-4-hydroxy-2,5,6-trimethylphenyl acetate (6a): yellow solid, 78% yield, mp = 119–120 °C. IR 3485, 2924, 1728, 1639, 1574, 1458, 1365, 1300, 1225, 1192, 1070, 993, 905 cm–1; 1H NMR (500 MHz, CDCl3) δ 6.01–5.88 (m, 1H), 5.11–5.02 (m, 2H), 4.79 (s, 1H), 3.41 (dt, J = 10.0, 3 Hz, 2H), 2.34, (d, J = 0.5 Hz, 2H), 2.15 (s, 5H), 2.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.8, 150.1, 141.8, 135.5, 127.6, 126.4, 121.7, 121.4, 116.1, 31.5, 20.7, 13.3, 12.8, 12.3; HRMS (ESI) exact mass calculated for [M + H]+ (C14H19O3) requires m/z 235.1334, found m/z 235.1329.
3-allyl-4-hydroxy-2-methylnaphthalen-1-yl acetate (6d): light yellow crystal, 55% yield, mp = 117–118 °C. IR 3485, 3441, 3063, 2976, 2924, 1726, 1634, 1595, 1574, 1454, 1389, 1364, 1290, 1233, 1080, 1047, 1015, 982, 914, 768, 737 cm–1; 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 7.5 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.49–7.43 (m, 2H), 6.07–6.00 (m, 1H), 5.36 (s, 1H), 5.17 (dq, J = 10.1, 1.6 Hz, 1H), 5.11 (dq, J = 18.9, 1.7 Hz, 1H), 3.59 (dt, J = 5.6, 1.7 Hz, 2H), 2.47 (s, 3H), 2.25 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.7, 147.7, 138.4, 135.0, 126.7, 126.5, 126.5, 125.3, 124.0, 121.7, 120.8, 118.2, 116.7, 31.4, 20.8, 13.5; HRMS (ESI) exact mass calculated for [M + H]+ (C16H17O3) requires m/z 257.1178, found m/z 257.1172.
3.4. General Procedure for Acetylation
Acetic anhydride (1 mL, 10 mmol) was added to the mixture of allylphenol (5 mmol) and pyridine (10 mL) at 0 °C under an N2 atmosphere. After that, 4-dimethyl pyridine (0.15 g, 1 mmol) was added as catalyst. The reaction mixture was stirred for 1 h at room temperature. After completion, the mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with water, saturate aqueous CuSO4 and brine, dried over Na2SO4, and evaporated under vacuum. The residue was purified by column chromatography with silica gel as the stationary phase (0%–5% ethyl acetate/n-hexane) to yield the titular allyl ester.
2-allyl-3,5,6-trimethyl-1,4-phenylene diacetate (7a): light yellow crystal, 90% yield, mp = 108–109 °C. IR 3458, 3074, 2976, 2927, 1728, 1639, 1573, 1460, 1366, 1225, 1192, 1070, 993, 905, 860, 827, 706, 683 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.81–5.76 (m, 1H), 5.00 (dq, J = 10.1, 1.6, 1H), 4.94 (dd, J = 17.1, 1H), 3.27 (bd, J = 39.5 Hz, 2H), 2.34 (s, 3H), 2.31 (s, 3H), 2.06 (b, 6H, 2CH3), 2.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.1 (b, 2 × C=O), 145.8, 145.6, 134.9, 128.4, 127.9, 127.7, 127.6, 127.4, 115.7, 31.8 (b, 2CH3CO), 20.5, 13.3, 12.7; HRMS (ESI) exact mass calculated for [M + Na]+ (C16H20NaO4) requires m/z 299.1259, found m/z 299.1257.
Our above NMR data for prepared compound (
7a) were comparable with the NMR data of 2-allyl-1,4-diacetoxy-3,5,6-trimethylbenzen in Reference [
28]:
1H NMR (90 MHz, CDCl
3) δ 5.7–5.9 (m, 1H), 5.0 (d,
J = 11, 1 H), 4.94 (d,
J = 18, 1H), 3.28 (br s, 2H), 2.32 and 2.34 (s, 6H), 2,04 and 2,06 (s, 9H);
13C NMR δ 168.90, 168.70, 145.97, 145.72, 134.97, 128.47, 127.92, 127.53, 127.36, 115.4, 31.78, 20.42, 20.29, 13.07, 12.47.
2-allyl-3-methylnaphthalene-1,4-diyl diacetate (7d): brown solid, 90% yield, mp = 107–108 °C. IR 3069, 2982, 2928, 2870, 1753, 1639, 1599, 1427, 1354, 1198, 1169, 1092, 1049, 1011, 895, 773, 742 cm–1; 1H NMR (500 MHz, CDCl3) δ 7.72–7.71 (m, 1H), 7.67–7.65 (m, 1H), 7.48–7.46 (m, 2H), 5.91–5.86 (m, 1H), 5.06 (dq, J = 10.2, 1.6, 1H), 4.99 (dq, J = 17.1, 1.7, 1H), 3.48 (b, 2H), 2.49 (s, 3H), 2.47 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.6 (2 × C=O) 142.9, 142.8, 134.7, 128.4, 127.1, 126.7, 126.6, 126.5, 126.3, 121.6, 121.4, 116.3, 32.1, 20.9, 20.8, 13.2. HRMS (ESI) exact mass calculated for [M + H]+ (C18H19O4) requires m/z 299.1283, found m/z 299.1282.
3.5. General Procedure for Stille Coupling
Bromoaryl (1.0 mmol), (PPh3)2PdCl2 (0.025 mmol) allylSnBu3 (1.1 mmol), and PPh3 (0.1 mmol) were dissolved in DMF (10 mL), and then the mixture was heated at 110 °C for 4 h under an N2 atmosphere. The reaction mixture was diluted with H2O, extracted five times with Et2O, and the combined extracts were dried over Na2SO4 and evaporated under vacuum. The residue was purified by column chromatography with silica gel as the stationary phase (10%–20% ethyl acetate/n-hexane) to yield the titular allylated arene.
1-allyl-2,5-dimethoxy-3,4,6-trimethylbenzene (7b): clear oil, 75% yield. IR (film) 2994, 2936, 2845, 1638, 1558, 1454, 1383, 1223, 1084, 1038, 1002, 937, 837, 758 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.97–5.92 (m, 1H), 5.00 (dq, J = 10.0, 1.9 Hz, 1H), 4.90 (dq, J = 17.5, 2.0 Hz, 1H), 3.66 (s, 3H), 3.64 (s, 3H), 3.43 (dt, J = 4.0 Hz, 1.7 Hz, 2H), 2.19 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 153.2, 153.0, 136.9, 129.3, 128.9, 128.2, 128.1, 114.9, 61.2, 60.3, 31.4, 12.9, 12.8, 12.0; HRMS (ESI) exact mass calculated for [M + H]+ (C14H21O2) requires m/z 221.1542, found m/z 221.1537.
1-allyl-2,3,4,5-tetramethoxy-6-methylbenzene (7c): clear oil, 75% yield. IR (film) 2934, 2829, 1728, 1638, 1464, 1404, 1350, 1292, 1257, 1196, 1105, 1036, 1009, 978, 908, 871, 743 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.93–5.86 (m, 1H), 5.00 (dd, J = 10.1, 1.4 Hz, 1H), 4.91 (dd, J = 17.1, 1.6 Hz, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.38 (d, J = 5.8 Hz, 2H), 2.14 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 147.9 (2 × C-OMe), 145.4, 144.8, 136.7 (–CH=CH2), 127.0, 125.8, 114.9 (=CH2), 61.4, 61.3, 61.2, 60.8, 31.0, 11.7; HRMS (ESI) exact mass calculated for [M + H]+ (C14H21O4) requires m/z 253.1440, found m/z 253.1436.
2-allyl-1,4-dimethoxy-3-methylnaphthalene (7e): off-white crystal, 78% yield, mp = 83–84 °C. 1H NMR (500 MHz, CDCl3) δ 8.09–8.04 (m, 2H), 7.49–7.45 (m, 2H), 6.08–6.00 (m, 1H), 5.05 (dq, J = 10.2, 1.8 Hz, 1H), 4.91 (dq, J = 17.2, 1.9 Hz, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.64 (dt, J = 5.5, 1.9 Hz, 2H), 2.39 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.3, 150.2, 136.7, 128.6, 127.9, 127.3, 125.7, 125.4, 122.5, 122.2, 115.2, 62.5, 61.5, 31.4, 12.4; HRMS (ESI) exact mass calculated for [M + H]+ (C16H19O2) requires m/z 243.1385, found m/z 243.1380.
Our above NMR data for prepared compound (
7e) were comparable with the NMR data of compound
5 in Reference [
29]:
1H NMR (80 MHz, CDCl
3) δ 8.40–8.25 (m, 2H), 7.82–7.76 (m, 2H), 6.50–6.00 (m, 1H), 5.40–4.98 (m, 2H), 4.11 (s, 3H), 4.07 (s, 3H), 3.85 (d,
J = 6, 2H), 2.62 (s, 3H).
3.6. General Procedure for Key Cross Metathesis Reaction
Methyl methacrylate (200.0 mg, 2.0 mmol) and allylarene (0.1 mmol) were dissolvent in dichloromethane (100 mL), and Grubbs 2nd (42.1 mg, 0.05 mmol) was added. The reaction mixture was refluxed for 24 h at 40 °C under an N2 atmosphere. The mixture was then concentrated under vacuum and the residue was purified by column chromatography with silica gel as the stationary phase (5%–10% ethyl acetate/n-hexane) to yield the titular key ester intermediate. The E- configuration of the cross-coupling products was determined by NOESY NMR spectroscopy analysis.
(E)-2-(4-methoxy-3-methyl-4-oxobut-2-en-1-yl)-3,5,6-trimethyl-1,4-phenylene diacetate (8a): clear oil, 56% yield. IR (film) 2927, 2926, 1740, 1639, 1495, 1445, 1373, 1238, 1045, 935, 878, 841, 731 cm–1; 1H NMR (500 MHz, CDCl3) δ 6.58 (td, J = 6.8, 1.4 Hz, 1H), 3.69 (s, 3H), 3.36 (b, 2H), 2.35 (s, 3H), 2.31 (s, 3H), 2.06 (s, 3H), 2.04 (s, 6H), 1.96 (d, J = 1.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.4, 168.2, 146.1, 145.7, 139.5, 128.7, 128.3, 127.9, 127.4, 112.9, 51.9, 27.6, 20.7, 20.6, 13.4 (d), 13.0, 12.8; HRMS (ESI) exact mass calculated for [M + H]+ (C19H25O6) requires m/z 349.1651, found m/z 349.1646.
Methyl (E)-4-(2,5-dimethoxy-3,4,6-trimethylphenyl)-2-methylbut-2-enoate (8b): clear oil, 61% yield. IR (film) 2916, 2849, 1719, 1649, 1458, 1402, 1300, 1259, 1244, 1206, 1132, 1080, 1059, 997, 802, 743 cm–1; 1H NMR (500 MHz, CDCl3) δ 6.69 (td, J = 7.0, 1.0 Hz, 1H), 3.69 (s, 3H), 3.64 (s, 3H), 3.63 (s, 3H), 3.53 (d, J = 6.9 Hz, 2H), 2.19 (s, 6H), 2.17 (s, 3H), 2.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.7, 153.3, 152.9, 141.3, 129.3, 129.1, 128.3, 127.9, 127.5, 61.0, 60.3, 51.8 27.1, 12.9, 12.8, 12.7, 12.5; HRMS (ESI) exact mass calculated for [M + H]+ (C17H25O4) requires m/z 293.1753, found m/z 295.1750.
Methyl (E)-2-methyl-4-(2,3,4,5-tetramethoxy-6-methylphenyl)but-2-enoate (8c): clear oil, 58% yield. IR (film) 2918, 2849, 1719, 1649, 1458, 1400, 1300, 1259, 1244, 1206, 1132, 1080, 1059, 999, 802, 741 cm–1; 1H NMR (500 MHz, CDCl3) δ 6.64 (t, J = 6.9 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.79 (s, 3H), 3.78 (s, 3H), 3.70 (s, 3H), 3.48 (d, J = 7.0 Hz, 2H), 2.13 (s, 2H), 2.00 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.7, 148.1, 147.9, 145.8, 144.9, 140.8, 127.5, 126.5, 125.6, 61.3, 61.2, 61.1, 60.9, 51.9, 29.8, 12.7, 12.0; HRMS (ESI) exact mass calculated for [M + H]+ (C17H25O6) requires m/z 325.1651, found m/z 325.1547.
(E)-2-(4-methoxy-3-methyl-4-oxobut-2-en-1-yl)-3-methylnaphthalene-1,4-diyl diacetate (8d): clear oil, 64% yield. IR (film) 2981, 2907, 1736, 1699, 1597, 1446, 1371, 1300, 1234, 1157, 1097, 1043, 937, 846, 785 cm–1; 1H NMR (500 MHz, CDCl3) δ 7.73–7.71 (m, 1H), 7.69–7.67 (m, 1H), 7.51–7.47 (m, 2H), 6.67 (td, J = 6.8, 1.4 Hz, 1H), 3.69 (s, 3H), 3.58 (b, 2H), 2.49 (s, 3H), 2.47 (s, 3H), 2.24 (s, 3H), 2.02 (d, J = 1.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 169.4, 168.3 (2 × C=OCH3), 142.9, 142.8, 138.9, 128.6, 128.1, 126.9, 126.8, 126.7, 126.5, 126.3, 121.6, 121.4, 51.9, 27.8, 20.8, 20.7, 13.4, 12.9; HRMS (ESI) exact mass calculated for [M + H]+ (C21H23O6) requires m/z 371.1495, found m/z 371.1489.
Methyl (E)-4-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methylbut-2-enoate (8e): clear oil, 64% yield. IR (film) 2982, 2939, 1736, 1699, 1597, 1447, 1371, 1300, 1234, 1098, 1043, 937, 847, 785 cm–1; 1H NMR (500 MHz, CDCl3) δ 8.07–8.03 (m, 2H), 7.50–7.46 (m, 2H), 6.74 (t, J = 6.8 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.73 (d, J = 6.9 Hz, 2H), 3.70 (s, 3H), 2.36 (s, 3H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.6, 150.4, 150.3, 140.7, 128.4, 128.1, 128.0, 127.3, 126.5, 125.9, 125.7, 122.4, 122.3, 62.4, 61.6, 51.9, 27.7, 12.9, 12.8; HRMS (ESI) exact mass calculated for [M + H]+ (C19H23O4) requires m/z 315.1596, found m/z 315.1592.
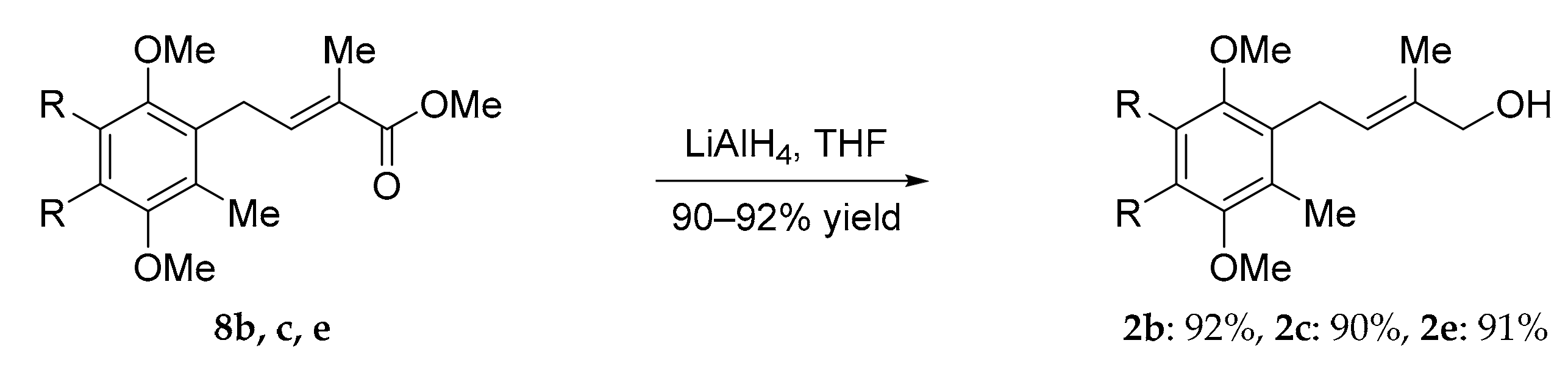
3.7. General Procedure for the Reduction of Cross Metathesis Products
To a stirred solution of ester 6c (325.1 mg, 6.0 mmol) in anhydrous diethyl ether (10 mL) was added dropwise a suspension of LiAlH4 in dry THF at 0 °C. The reaction was allowed to warm to room temperature until completion as indicated by TLC (4 h). The reaction mixture was quenched at 0 °C by addition of water (2 mL) and aq sodium hydroxide (20%, 1 mL). The workup was continued by the further addition of water (10 mL) and aq sodium hydroxide (20%, 10 mL). The mixture was stirred for 30 min, after which the mineral solid precipitate was filtrated and washed with diethyl ether (40 mL). The residue was purified by column chromatography with silica gel as the stationary phase (5–10% ethyl acetate/n-hexane) to yield alcohol 4 (266.6 mg, 90% yield) as a clear and colorless viscous oil.
(E)-4-(2,5-dimethoxy-3,4,6-trimethylcyclohexa-1,5-dienyl)-2-methylbut-2-en-1-ol (2b): clear oil, 92%. IR (film) 3435, 2954, 2927, 2870, 1724, 1641, 1456, 1400, 1375, 1248, 1148, 1082, 1006, 804, 765 cm–1; 1H NMR Spectrum (500 MHz, CDCl3): 5.36 (t, J = 6 Hz, 1H), 4.00 (s, 1H), 3.65 (s, 3H), 3.64 (s, 3H), 3.41 (d, J = 6 Hz, 2H), 2.19 (s, 3H), 2.18 (s, 6H); 13C NMR Spectrum (125 MHz, CDCl3): 153.29, 152.92, 134.92, 130.93, 128.79, 128.21, 127.79, 125.22, 69.10, 61.01, 60.30, 26,08, 14.10, 12.98, 12.89, 12.43; HRMS (ESI) exact mass calculated for [M + H]+ (C16H25O3) requires m/z 265.1804, found m/z 265.1802.
(E)-2-methyl-4-(2,3,4,5-tetramethoxy-6-methylphenyl)but-2-en-1-ol (2c): clear oil, 90% yield. IR (film) 3414, 2931, 2860, 1463, 1406, 1350, 1257, 1193, 1103, 1050, 1005, 970, 871 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.34 (t, J 6.7 Hz, 1H), 4.00 (s, 2H), 3.90 (s, 3H), 3.90 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.37 (d, J = 6.7 Hz, 2H); 2.14 (s, 2H), 1.83 (d, J = 1.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 148.0, 147.8, 145.3, 144.8, 134.8, 128.4, 125.2, 124.6, 69.0, 61.3, 61.2 (b, 2CH3O), 60.7, 25.6, 14.0, 11.9; HRMS (ESI) exact mass calculated for [M + H]+ (C16H25O5) requires m/z 297.1702, found m/z 297.1701.
Our above IR and NMR data for prepared alcohol (
2c) were comparable with the IR and NMR data of alcohol
6 in Reference [
25]: IR (CHCl
3) 3432, 2935, 2862, 1466, 1407, 1351, 1219, 1105, 1067, 1039, 1013 cm
–1;
1H NMR (400 MHz, CDCl
3) δ 5.26 (1H, m), 3.93 (2H, s), 3.83 (3H, s), 3.82 (3H, s), 3.72 (3H, s), 3.70 (3H, s), 3.29 (2H, dd,
J = 6.8, 0.8 Hz), 2.07 (3H, s), 1.76 (3H, s),
13C NMR (100 MHz, CDCl
3) δ 147.93, 147.72, 145.19, 144.75, 134.68, 128.29, 125.23, 124.52, 68.83, 61.02, 60.99, 60.63, 25.51, 13.81, 11.76.
(E)-4-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methylbut-2-en-1-ol (2e): clear oil, 91%. IR 3397, 1659, 1597, 1454, 1379, 1366, 1263, 1159, 1066, 1037, 1007, 899, 758, 739 cm–1; 1H NMR Spectrum (500 MHz, CDCl3): 8.05 (m, 2H), 7.59–7.36 (m, 2H), 5.54–5.31 (m, 1H), 4.02 (d, J = 5.1 Hz, 2H), 3.89 (s, 3H), 3.87 (s, 3H), 3.61 (dd, J = 6.6, 2 Hz), 2.38 (s, 3H), 1.89 (d, J = 0.9 Hz, 3H); 13C NMR Spectrum (125 MHz, CDCl3): 150.42, 150.10, 135.55, 130.21, 127.83, 127.45, 126.82, 125.73, 125.56, 124.61, 122.47, 122.36, 68.98, 62.35, 61.52, 26.22, 14.19, 12.69; HRMS (ESI) exact mass calculated for [M + H]+ (C18H23O3) requires m/z 287.1647, found m/z 287.1645.
Our above NMR data for prepared ancol (
2e) were comparable with the NMR data of compound
21 in Reference [
30]:
1H NMR (400 MHz, CDCl
3) δ 8.07–8.02 (m, 2H), 7.48–7.44 (m, 2H), 5.43–5.41 (m, 1H), 4.02 (s, 2H), 3.89 (s, 3H), 3.87 (s, 3H), 3.61 (d,
J = 6.5, 2H), 2.38 (s, 3H), 1.90 (d,
J = 1.0, 3H)
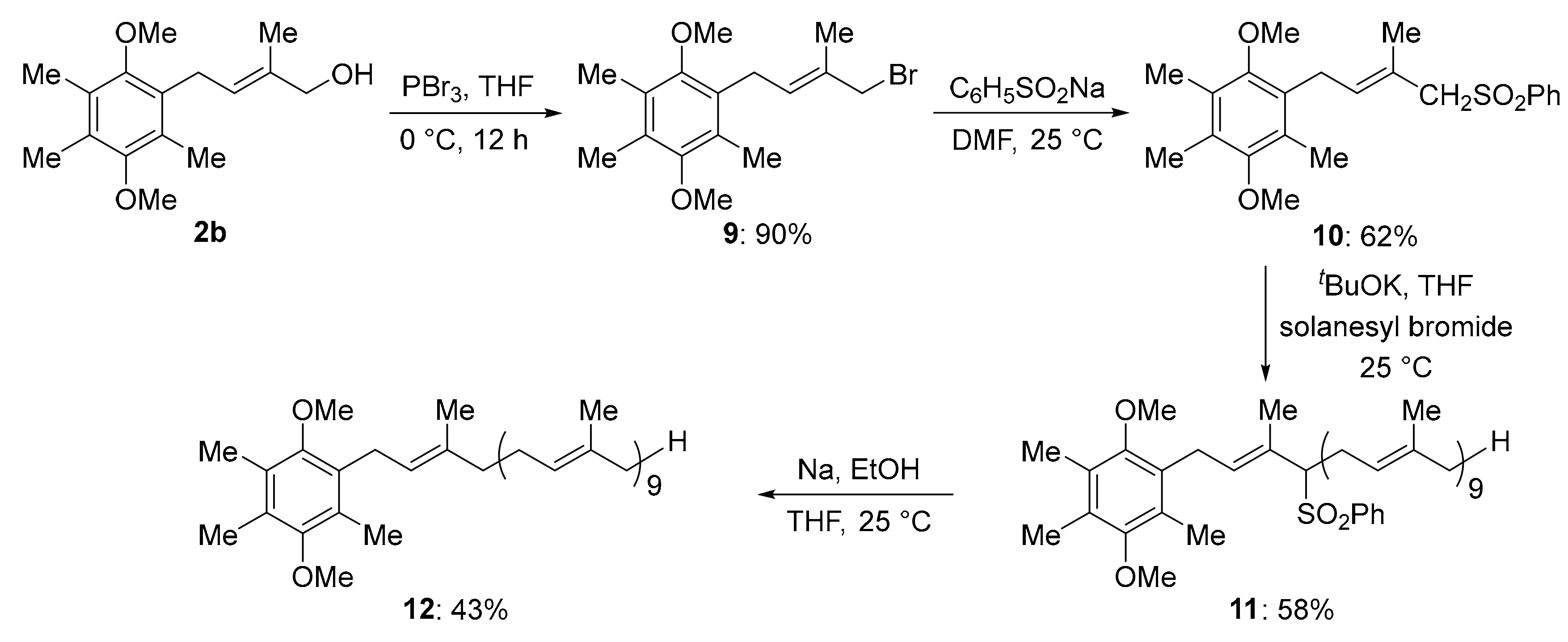
3.8. Bromination of 2b
Alcohol 2b (5 mmol) was dissolved in 20 mL of anhydrous tetrahydrofuran and PBr3 (2.5 mmol) was slowly added to the reaction at −5 °C for 15 min under an N2 atmosphere. After 2 h, the reaction mixture was poured into cold saturated sodium bicarbonate solution and then extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and concentrated under vacuum. The crude bromide 9 was used for the next reaction without purification.
3.9. General Procedure for the Sulfination
Bromide (9) (4 mmol) and sodium benzene sulphinate were dissolved in 25 mL dimethyl formamide. The reaction mixture was stirred at room temperature for 12 h under an N2 atmosphere. The mixture was then poured into ice-cold water, acidified to pH ~4 with 2N HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography with silica gel as the stationary phase (0%–10% ethyl acetate/n-hexane) to yield the titular sulfonyl compound (10).
(E)-1,4-dimethoxy-2,3,5-trimethyl-6-(3-methyl-4-(phenylsulfonyl)but-2-enyl)benzene (10): white solid, 90% yield, mp = 186–187 °C. IR 3065, 2938, 2847, 1587, 1447, 1400, 1350, 1302, 1292, 1248, 1165, 1132, 1082, 1055, 1009, 955, 901, 768, 743 cm–1; 1H NMR Spectrum (500 MHz, CDCl3): 7.83–7.75 (m, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 4.97 (t, J = 6.4 Hz, 1H), 3.71 (s, 2H), 3.61 (s, 3H), 3.54 (s, 3H), 3.27 (d, J = 6.6 Hz, 1H), 2.17 (s, 3H), 2.15 (s, 3H), 2.01 (s, 3H), 1.95 (s, 3H); 13C NMR Spectrum (125 MHz, CDCl3): 138.49, 138.46, 136.71, 135.36, 135.12, 133.50, 129.00, 128.54, 128.19, 127.85, 127.57, 123.44, 66.23, 60.93, 60.29, 26.78, 17.16, 12.96, 12.87, 12.37; HRMS (ESI) exact mass calculated for [M + H]+ (C22H29O4S) requires m/z 389.1787, found m/z 389.1784.
3.10. General Procedure for the Solanesylation:
Sulfonyl (10) (0.50 mmol) and solanesol bromide (0.75 mmol) were dissolved in 10 mL of anhydrous tetrahydrofuran. Solution of potassium tert-butoxide (0.75 mmol) in 5 mL anhydrous tetrahydrofuran was added dropwise to the above solution containing sulfonyl and solanesol bromide at −25 °C. After 2 h, the reaction was allowed to reach room temperature and the stirring was continued for 12 h under an N2 atmosphere. After completion, the mixture was poured into ice-water, acidified to pH ~4 with 2N HCl, and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography with silica gel as the stationary phase (0%–5% ethyl acetate/n-hexane) to yield the titular compound.
1-((2E,6E,10E,14E,18E,22E,26E,30E,34E)-3,7,11,15,19,23,27,31,35,39-decamethyl-4-(phenylsulfonyl)tetraconta-2,6,10,14,18,22,26,30,34,38-decaenyl)-2,5-dimethoxy-3,4,6-trimethylbenzene (11): clear oil, 43%. IR (film) 3296, 3233, 2914, 2849, 1728, 1468, 1418, 1290, 1217, 1194, 1177, 1101, 1047, 991, 943, 719 cm–1; 1H NMR Spectrum (500 MHz, CDCl3): 7.77 (m, 2H), 7.52 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.7 Hz, 2H), 5.13–5.10 (m, 7H), 5.05 (t, J = 6.6 Hz, 1H), 4.97 (t, J = 5.9 Hz, 1H), 4.87 (t, J = 6.8 Hz, 1H), 3.60 (s, 3H), 3.48 (m, 4H), 3.30 (dd, J = 15.7, 6.9 Hz, 1H), 3.17 (dd, J = 15.4, 5.3 Hz, 1H), 2.86–2.77 (m, 1H), 2.68–2.56 (m, 1H), 2.16 (s, 3H), 2.14 (s, 3H), 2.10–2.04 (m, 16H), 2.02–1.92 (m, 19H), 1.84 (s, 3H), 1.68 (s, 3H), 1.64–1.50 (s, 32H), 1.26 (s, 6H); 13C NMR Spectrum (125 MHz, CDCl3): 153.16, 152.68, 135.53, 135.09, 131.41, 130.04, 128.93, 128.86, 128.86,128.13, 127.68, 126.97, 124.63, 124.50, 124.36, 123.96, 118.84, 74.07, 60.89, 60.26, 39.91, 29.89, 26.77, 26.59, 25.87, 17.84, 16.50, 16.22, 12.93, 12.87, 12.31; HRMS (ESI) exact mass calculated for [M + H]+ (C27H37O4S) requires m/z 457.2413, found m/z 457.2413.
3.11. General Procedure for the De-Sulfination
To a solution of 11 (0.1 mmol) in 5 mL anhydrous tetrahydrofuran was added ethanol (0.005 mol). Sodium (1.5 mmol) pieces ware slowly added to the reaction mixture under vigorous stirring at room temperature for 12 h under an N2 atmosphere. The reaction was then poured into ice-water and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and evaporated under vacuum. The residue was purified by column chromatography with silica gel as the stationary phase (0%–2% ethyl acetate/n-hexane) to yield the titular compound.
1-((2E,6E,10E,14E,18E,22E,26E,30E,34E)-3,7,11,15,19,23,27,31,35,39-decamethyltetraconta-2,6,10,14,18,22,26,30,34,38-decaenyl)-2,5-dimethoxy-3,4,6-trimethylbenzene (12): clear oil, 41%. IR (film) 3103, 2989, 2936, 2626, 1587, 1510, 1450, 1400, 1300, 1246, 1169, 1130, 1246, 1169, 1130, 1082, 1053, 1002, 903, 822, 741 cm–1; 1H NMR Spectrum (500 MHz, CDCl3): 5.28–5.00 (m, 10H), 3.64 (s, 6H), 3.36 (d, J = 6.3 Hz, 2H), 2.69 (m, 3H), 2.18 (s, 12H), 2.08(m, 22H), 2.06(m, 24H), 1.78 (s, 3H), 1.68 (s, 6H), 2.63–1.56 (m, 38H); 13C NMR Spectrum (125 MHz, CDCl3): 153.25, 152.90, 135.27, 135.22, 135.13, 135.08, 131.84, 128.43, 128.07, 127.94, 124.62, 124.46, 124.33, 123.46, 123.36, 61.06, 60.29, 40.38, 39.94, 39.89, 27.21, 26.97, 26.92, 26.82, 26.37, 25.88, 16.50, 16.21, 12.96, 12.89, 12.34, 12.21; HRMS (ESI) exact mass calculated for [M + H]+ (C21H33O2) requires m/z 317.2481, found m/z 317.2478.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





