
The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions




Abstract
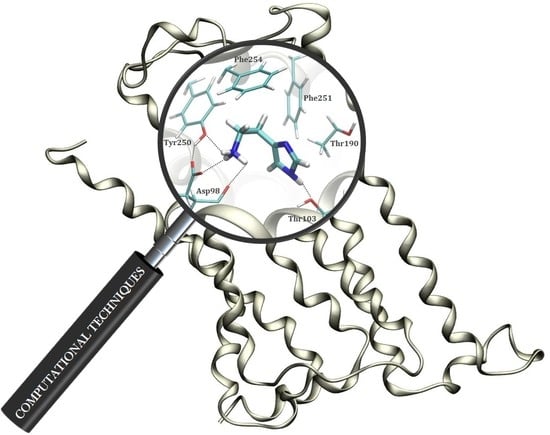
1. Introduction
2. Results and Discussion
2.1. Docking Simulation
2.2. Molecular Dynamics Simulation of Histamine in Aqueous Solution
2.3. Molecular Dynamics Simulation within the H2 Receptor
2.4. Quantum-Chemical Calculations
3. Computational Details
3.1. Docking Analysis
3.2. Molecular Dynamics Simulation
3.3. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walter, M.; Stark, H. Histamine receptor subtypes: A century of rational drug design. Front. Biosci. 2012, 4, 461–488. [Google Scholar] [CrossRef]
- Cong, X.; Topin, J.; Golebiowski, J. Class A GPCRs: Structure, function, modeling and structure-based ligand design. Curr. Pharm. Des. 2017, 23, 4390–4409. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Langmead, C.J.; Mason, J.S.; Marshall, F.H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011, 54, 4283–4311. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, A.; Solis, G.P.; Hersch, M.; Koval, A.; Kryuchkov, M.; Bergmann, S.; Katanaev, V.L. High capacity in G protein-coupled receptor signaling. Nat. Commun. 2018, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Ubbelohde, A.R.; Gallagher, K.J. Acid-base effects in hydrogen bonds in crystals. Acta Cryst. 1955, 8, 71–83. [Google Scholar] [CrossRef]
- De Souza, J.M.; Freire, P.T.C.; Bordallo, H.N.; Argyriou, D.N. Structural isotopic effects in the smallest chiral amino acid: Observation of a structural phase transition in fully deuterated alanine. J. Phys. Chem. B 2007, 111, 5034–5039. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, X.; Yu, C.-H.; Yao, Y.-F.; Zhang, W. Geometric isotope effect of deuteration in a hydrogen-bonded host-guest crystal. Nat. Commun. 2018, 9, 481. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Wang, Z.; McElmurry, B.A.; Willaert, F.F.; Lucchese, R.R.; Bevan, J.W.; Suenram, R.D.; Lovas, F.J. A ground state morphed intermolecular potential for the hydrogen bonded and van der Waals isomers in OC:HI and a prediction of an anomalous deuterium isotope effect. J. Chem. Phys. 2010, 133, 184305. [Google Scholar] [CrossRef]
- Mishra, A.K.; Murli, C.; Sharma, S.M. High pressure Raman spectroscopic study of deuterated γ-glycine. J. Phys. Chem. B 2008, 112, 15867–15874. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.O.; Freire, P.T.C.; Bordallo, H.N.; Lima, J.A., Jr.; Melo, F.E.A.; Mendes Filho, J.; Argyriou, D.N.; Lima, R.J.C. High-pressure Raman spectra of deuterated L-alanine crystal. J. Raman Spectrosc. 2009, 40, 958–963. [Google Scholar] [CrossRef]
- Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Benedict, H.; Schah-Mohammedi, P.; Limbach, H.-H. Hydrogen/deuterium isotope effects on the NMR chemical shifts and geometries of intermolecular low-barrier hydrogen-bonded complexes. J. Am. Chem. Soc. 1996, 118, 4094–4101. [Google Scholar] [CrossRef]
- Yarnell, A.T. Heavy-hydrogen drugs turn heads, again. Chem. Eng. News 2009, 87, 36–39. [Google Scholar] [CrossRef]
- Halford, B. Deuterium switcheroo breathes life into old drugs. Chem. Eng. News 2016, 94, 32–36. [Google Scholar]
- Belleau, B.; Burba, J.; Pindell, M.; Reiffenstein, J. Effect of Deuterium Substitution in Sympathomimetic Amines on Adrenergic Responses. Science 1961, 133, 102–104. [Google Scholar] [CrossRef]
- Falconnet, J.B.; Brazier, J.L.; Desage, M. Synthesis of seven deuteromethyl-caffeine analogues; observation of deuterium isotope effects on CMR analysis. J. Label. Compd. Radiopharm. 1986, 23, 267–276. [Google Scholar] [CrossRef]
- Brazier, J.L.; Ribon, B.; Falconnet, J.B.; Cherrah, Y.; Benchekroun, Y. Etude et utilisation des effets isotopiques en pharmacologie. Therapie 1987, 42, 445–450. [Google Scholar]
- Cherrah, Y.; Falconnet, J.B.; Desage, M.; Brazier, J.L.; Zini, R.; Tillement, J.P. Study of deuterium isotope effects on protein binding by gas chromatography/mass spectrometry. Caffeine and deuterated isotopomers. Biomed. Environ. Mass Spectrom. 1987, 14, 653–657. [Google Scholar] [CrossRef]
- Cherrah, Y.; Zini, R.; Falconnet, J.B.; Desage, M.; Tillement, J.P.; Brazier, J.L. Study of deutero-isotopomer-induced inhibition of caffeine and phenobarbitone binding to human serum albumin. Biochem. Pharmacol. 1988, 37, 1311–1315. [Google Scholar] [CrossRef]
- Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 2017, 35, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Bowers, S.G.; Truong, A.P.; Probst, G. The Role and Significance of Unconventional Hydrogen Bonds in Small Molecule Recognition by Biological Receptors of Pharmaceutical Relevance. Curr. Pharm. Des. 2007, 13, 3476–3493. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Mavri, J. Microsolvation of the histamine monocation in aqueous solution: The effect on structure, hydrogen bonding ability and vibrational spectrum. New J. Chem. 2012, 36, 954–962. [Google Scholar] [CrossRef]
- Stare, J.; Mavri, J.; Grdadolnik, J.; Zidar, J.; Maksić, Z.B.; Vianello, R. Hydrogen bond dynamics of histamine monocation in aqueous solution: Car–Parrinello molecular dynamics and vibrational spectroscopy study. J. Phys. Chem. B 2011, 115, 5999–6010. [Google Scholar] [CrossRef] [PubMed]
- Kržan, M.; Vianello, R.; Maršavelski, A.; Repič, M.; Zakšek, M.; Kotnik, K.; Fijan, E.; Mavri, J. The quantum nature of drug-receptor interactions: Deuteration changes binding affinities for histamine receptor ligands. PLoS ONE 2016, 11, e0154002. [Google Scholar] [CrossRef]
- Kržan, M.; Keuschler, J.; Mavri, J.; Vianello, R. Relevance of Hydrogen Bonds for the Histamine H2 Receptor-Ligand Interactions: A Lesson from Deuteration. Biomolecules 2020, 10, 196. [Google Scholar] [CrossRef]
- Cheng, H.C. The power issue: Determination of KB or Ki from IC50—A closer look at the Cheng–Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 2001, 46, 61–71. [Google Scholar] [CrossRef]
- Singh, V.; Gohil, N.; Ramírez-García, R. New insight into the control of peptic ulcer by targeting the histamine H2 receptor. J. Cell Biochem. 2018, 119, 2003–2011. [Google Scholar] [CrossRef]
- Strasser, A.; Wittmann, H.J. Molecular Modelling Approaches for the Analysis of Histamine Receptors and Their Interaction with Ligands. In Histamine and Histamine Receptors in Health and Disease. Handbook of Experimental Pharmacology; Hattori, Y., Seifert, R., Eds.; Springer: Cham, Switzerland, 2017; Volume 241, pp. 31–61. [Google Scholar]
- Sun, X.; Li, Y.; Li, W.; Xu, Z.; Tang, Y. Computational investigation of interactions between human H2 receptor and its agonists. J. Mol. Graph. Model. 2011, 29, 693–701. [Google Scholar] [CrossRef]
- Schreeb, A.; Łażewska, D.; Dove, S.; Buschauer, A.; Kieć-Kononiwcz, K.; Stark, H. Histamine H4 receptor ligands. In Histamine H4 Receptor: A Novel Drug Target for Immunoregulation and Inflammation; Stark, H., Ed.; Versita: London, UK, 2013; pp. 21–61. [Google Scholar]
- Collado, J.A.; Tuñón, I.; Silla, E.; Ramírez, F.J. Vibrational Dynamics of Histamine Monocation in Solution: An Experimental (FT-IR, FT-Raman) and Theoretical (SCRF-DFT) Study. J. Phys. Chem. A 2000, 104, 2120–2131. [Google Scholar] [CrossRef]
- Collado, J.A.; Ramírez, F.J. Vibrational spectra and assignments of histamine dication in the solid state and in solution. J. Raman Spectrosc. 2000, 31, 925–931. [Google Scholar] [CrossRef]
- Drozdzewski, P.; Kordon, E. Isotope effects in the far-infrared spectra of histamine complexes with palladium(II). Vib. Spectrosc. 2000, 24, 243–248. [Google Scholar] [CrossRef]
- Xerri, B.; Flament, J.-P.; Petitjean, H.; Berthomieu, C.; Berthomieu, D. Vibrational modeling of copper-histamine complexes: Metal-ligand IR modes investigation. J. Phys. Chem. B 2009, 113, 15119–15127. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Birdsall, N.J. Cloning and structure-function of the H2 histamine receptor. Trends Pharmacol. Sci. 1991, 12, 9–10. [Google Scholar] [CrossRef]
- Gantz, I.; DelValle, J.; Wang, L.D.; Tashiro, T.; Munzert, G.; Guo, Y.J.; Konda, Y.; Yamada, T. Molecular basis for the interaction of histamine with the histamine H2 receptor. J. Biol. Chem. 1992, 267, 20840–20843. [Google Scholar]
- Ramesh Kumara, G.; Gokul Raj, S.; Saxena, A.; Karnal, A.K.; Raghavalu, T.; Mohan, R. Deuteration effects on structural, thermal, linear and nonlinear properties of L-threonine single crystals. Mater. Chem. Phys. 2008, 108, 359–363. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, T.; Wei, J. Homology Modeling and Antagonist Binding Site Study of the Human Histamine H2 Receptor. Med. Chem. 2012, 8, 1084–1092. [Google Scholar]
- Strasser, A. Molecular modeling and QSAR-based design of histamine receptor ligands. Expert Opin. Drug Discov. 2009, 4, 1061–1075. [Google Scholar] [CrossRef]
- Catalan, J.; Abboud, J.L.M.; Elguero, J. Basicity and Acidity of Azoles. Adv. Heterocycl. Chem. 1897, 41, 187–274. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N∙log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational Insight into the Mechanism of the Irreversible Inhibition of Monoamine Oxidase Enzymes by the Antiparkinsonian Propargylamine Inhibitors Rasagiline and Selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines—Harmine and Cinnamic Acid Hybrids as Novel Antiplasmodial Hits. Eur. J. Med. Chem. 2020, 187, 111927. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein-Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras-Raf and Ras-RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Georgieva, P.; Yu, J.G.; Himo, F. Mechanism of mycolic acid cyclopropane synthase: A theoretical study. Biochemistry 2011, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 36, 7057–7065. [Google Scholar] [CrossRef]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef]
- Himo, F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modeling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Brás, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wires Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Quesne, M.G.; Silveri, F.; de Leeuw, N.H.; Catlow, C.R.A. Advances in sustainable catalysis: A computational perspective. Front. Chem. 2019, 7, 182. [Google Scholar] [CrossRef]
- Kaur, S.; Gupta, M. Deuteration as a tool for optimization of metabolic stability and toxicity of drugs. Glob. J. Pharm. Sci. 2017, 1, 555566. [Google Scholar]
- Tung, R.D. Deuterium medicinal chemistry comes of age. Future Med. Chem. 2016, 8, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Don, C.G.; Riniker, S. Scents and sense: In silico perspectives on olfactory receptors. J. Comput. Chem. 2014, 35, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histamine | |||
|---|---|---|---|
| ΔGBIND = −14.2 kcal mol−1 | |||
| Favorable Contributions | Unfavorable Contributions | ||
| Asp98 | −5.45 | Leu193 | +0.02 |
| Val99 | −1.78 | Gly157 | +0.02 |
| Phe254 | −1.60 | Lys166 | +0.02 |
| Thr103 | −1.29 | Lys83 | +0.02 |
| Phe251 | −0.97 | Val273 | +0.02 |
| Thr190 | −0.84 | Asn108 | +0.02 |
| Tyr250 | −0.83 | Lys88 | +0.02 |
| Gly187 | −0.64 | Tyr192 | +0.03 |
| Asp186 | −0.64 | Arg260 | +0.03 |
| Met100 | −0.20 | Ala253 | +0.05 |
| Glu270 | −0.16 | Ser153 | +0.05 |
| Cys102 | −0.14 | Arg161 | +0.06 |
| Glu267 | −0.11 | Lys173 | +0.11 |
| Val178 | −0.10 | Gln177 | +0.11 |
| Tyr94 | −0.08 | Gly183 | +0.13 |
| Val189 | −0.08 | Arg257 | +0.15 |
| Val185 | −0.07 | Leu97 | +0.19 |
| Val255 | −0.06 | Lys175 | +0.29 |
| Ligand | In H2O | In D2O | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEHYDR | ΔEINTER | ΔEBIND | ΔEHYDR | ΔEINTER | ΔEBIND | ΔΔEBIND,calc | ΔΔEBIND,exp | |
| Histamine | −71.63 | −82.31 | −10.69 | −71.20 | −82.73 | −11.54 | −0.85 | −0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hok, L.; Mavri, J.; Vianello, R. The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules 2020, 25, 6017. https://doi.org/10.3390/molecules25246017
Hok L, Mavri J, Vianello R. The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules. 2020; 25(24):6017. https://doi.org/10.3390/molecules25246017
Chicago/Turabian StyleHok, Lucija, Janez Mavri, and Robert Vianello. 2020. "The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions" Molecules 25, no. 24: 6017. https://doi.org/10.3390/molecules25246017
APA StyleHok, L., Mavri, J., & Vianello, R. (2020). The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules, 25(24), 6017. https://doi.org/10.3390/molecules25246017