
The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents


Abstract
1. Introduction
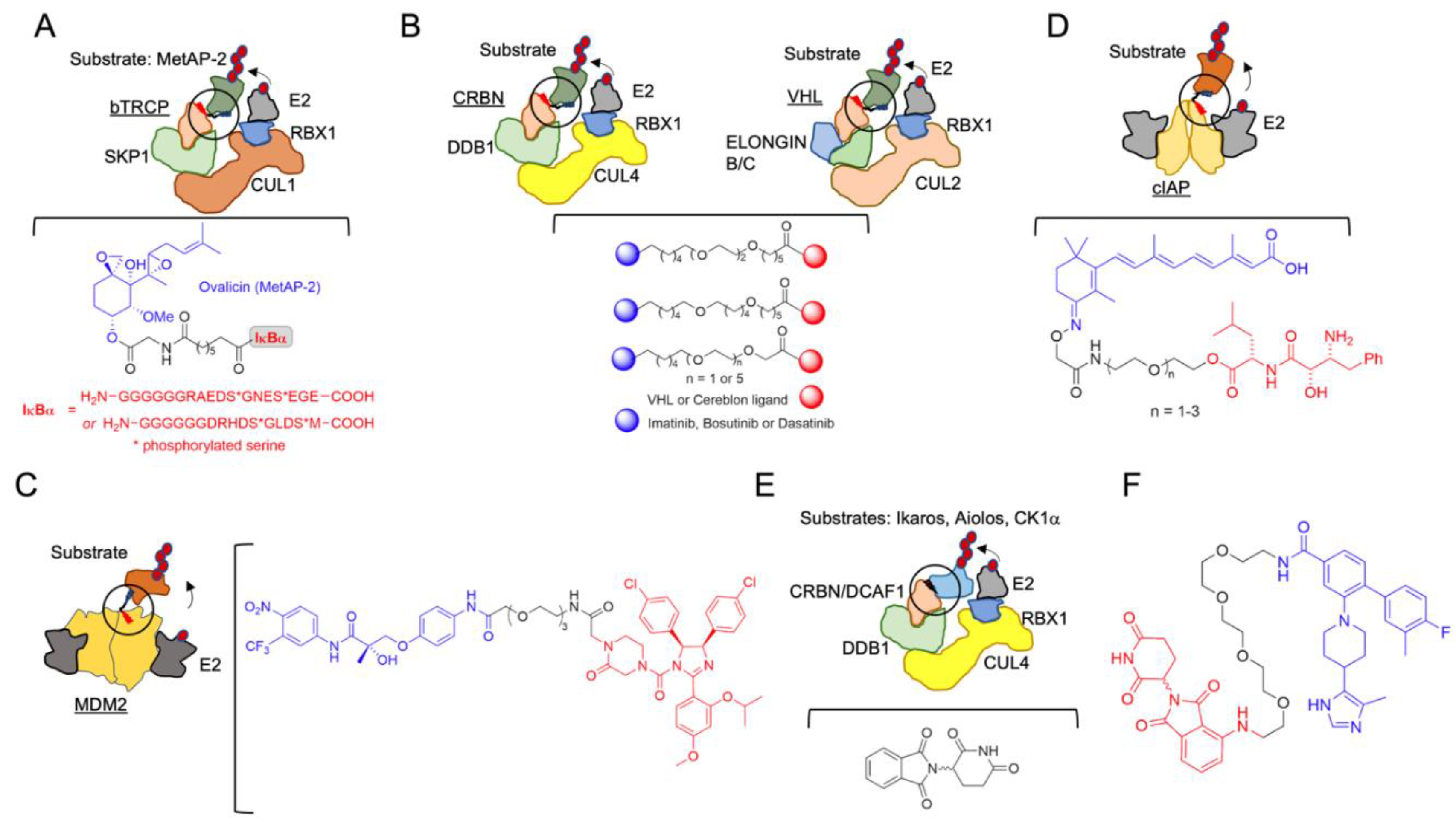
2. The Beginning of TPD
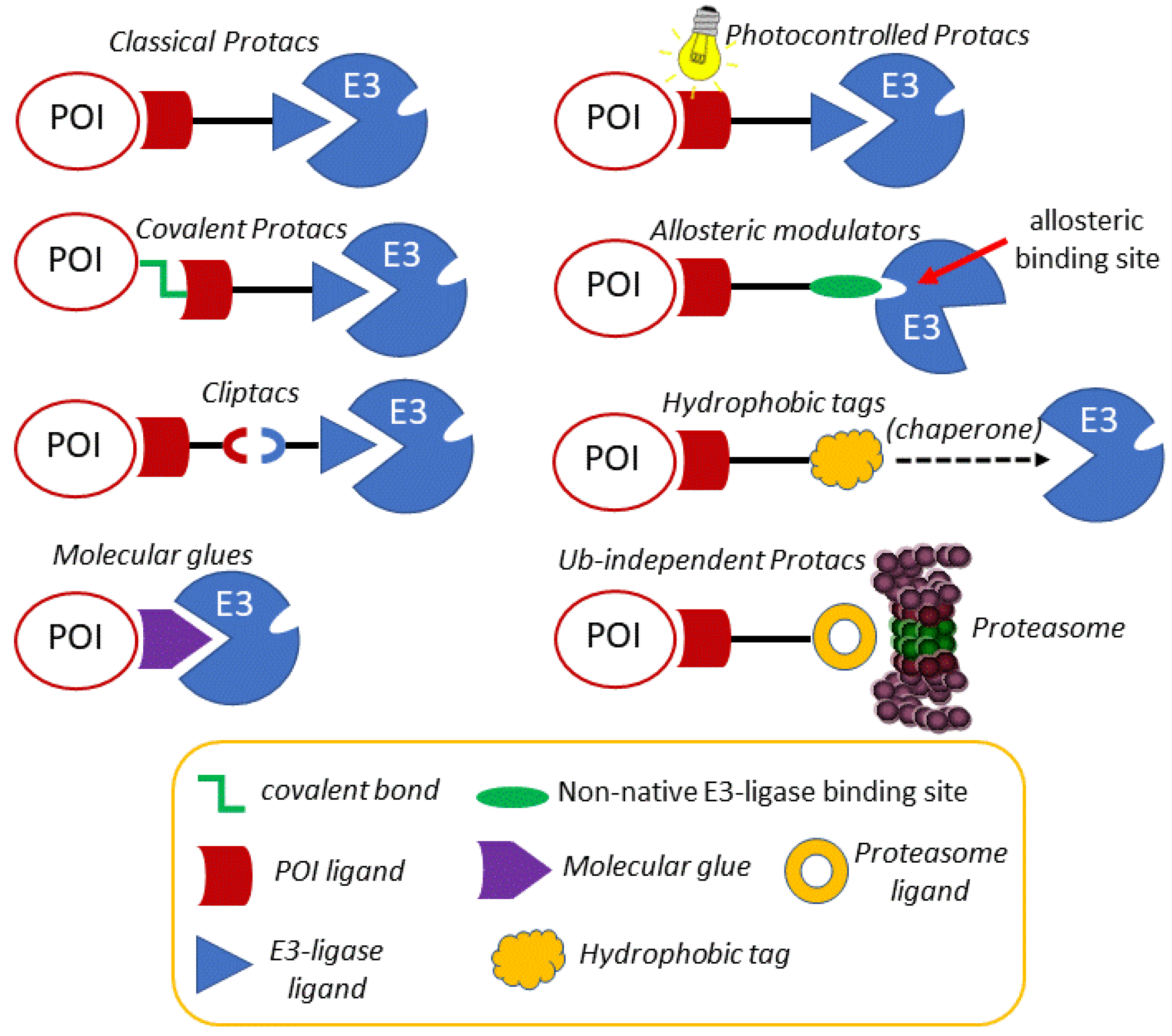
3. Modulating the Reactivity and Versatility of Proteolytic Chimeras
3.1. Photocontrolled Protacs
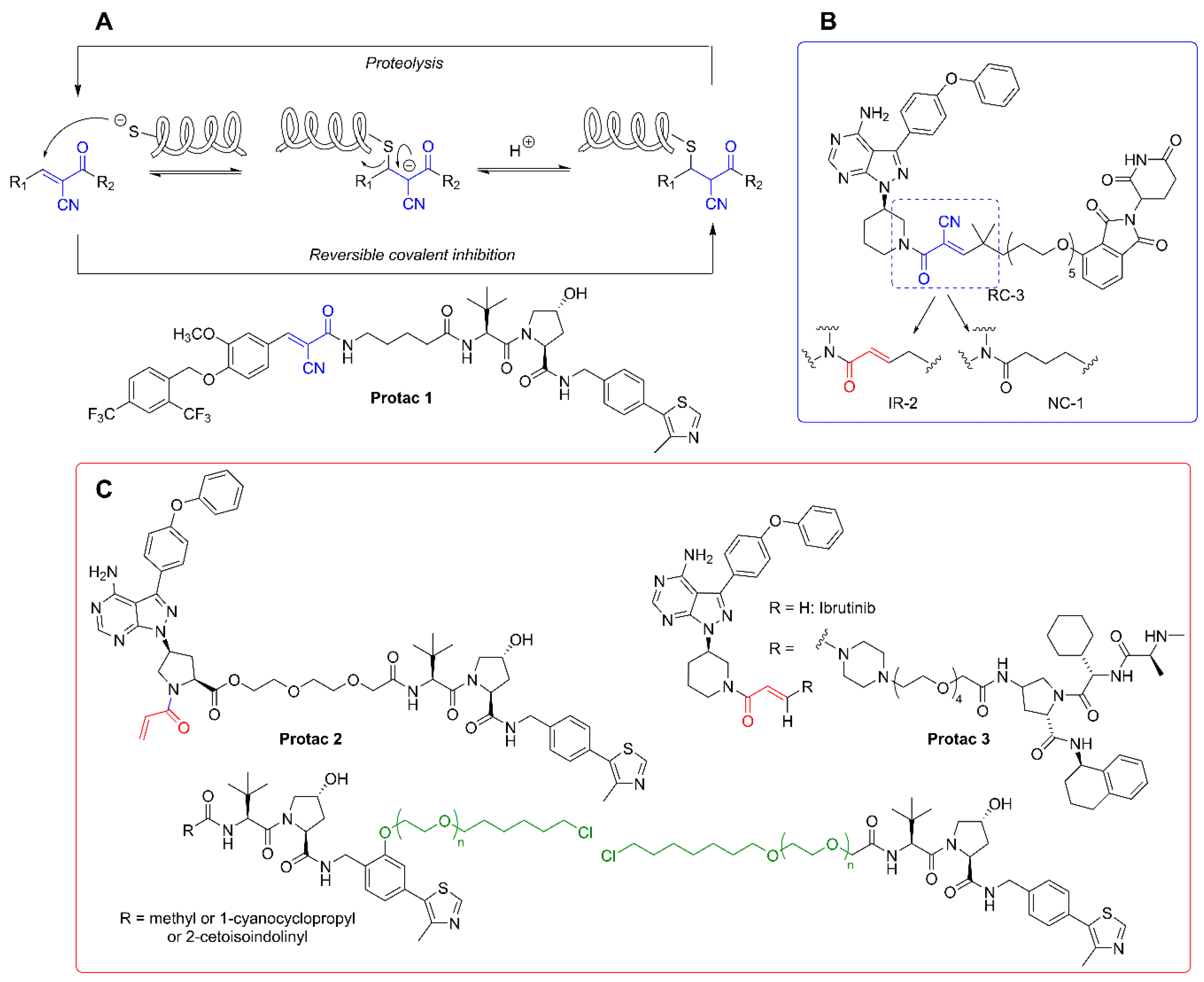
3.2. Covalent Protacs
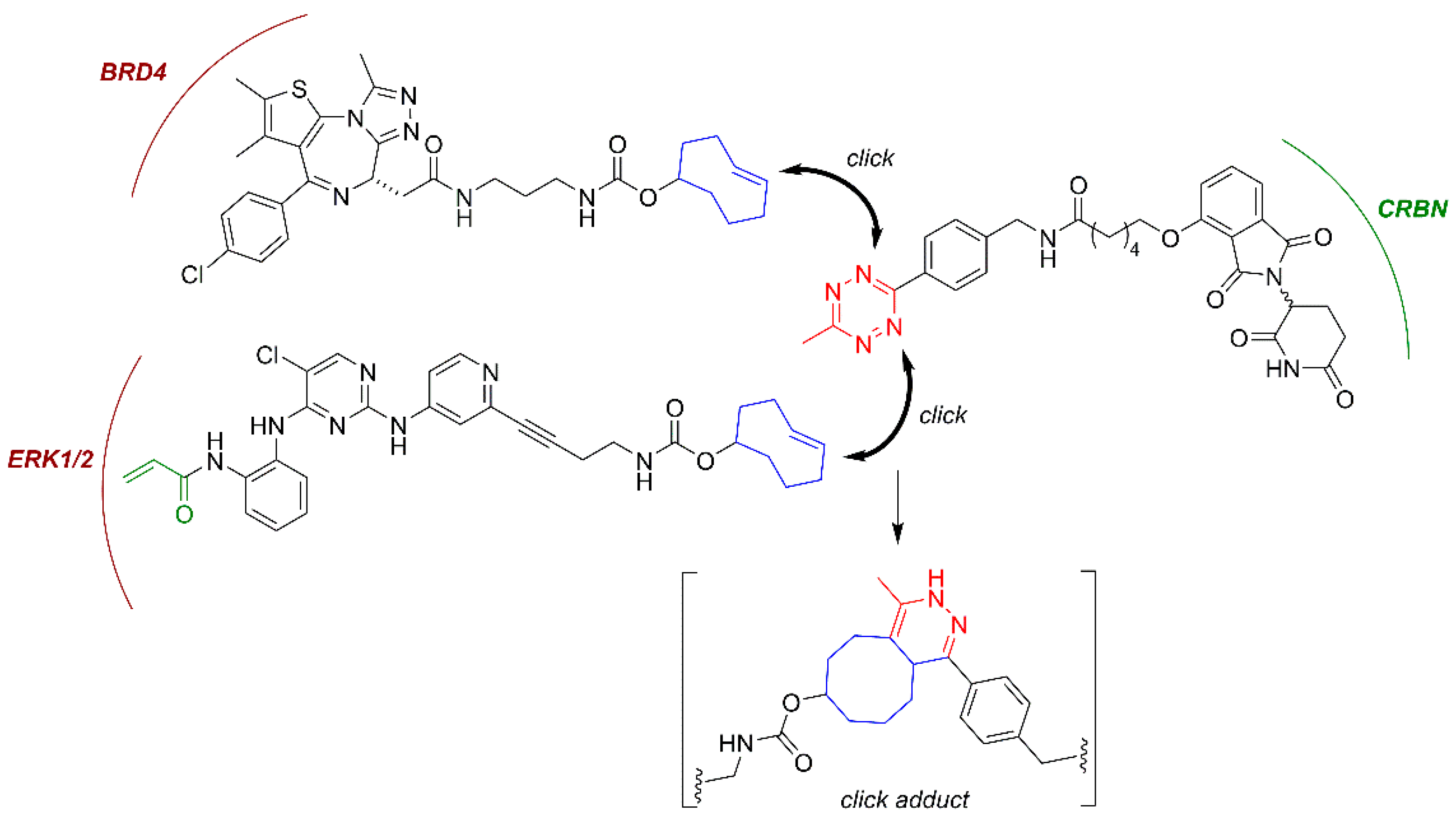
3.3. In Cell Click-Based Protacs (CLIPTACs)
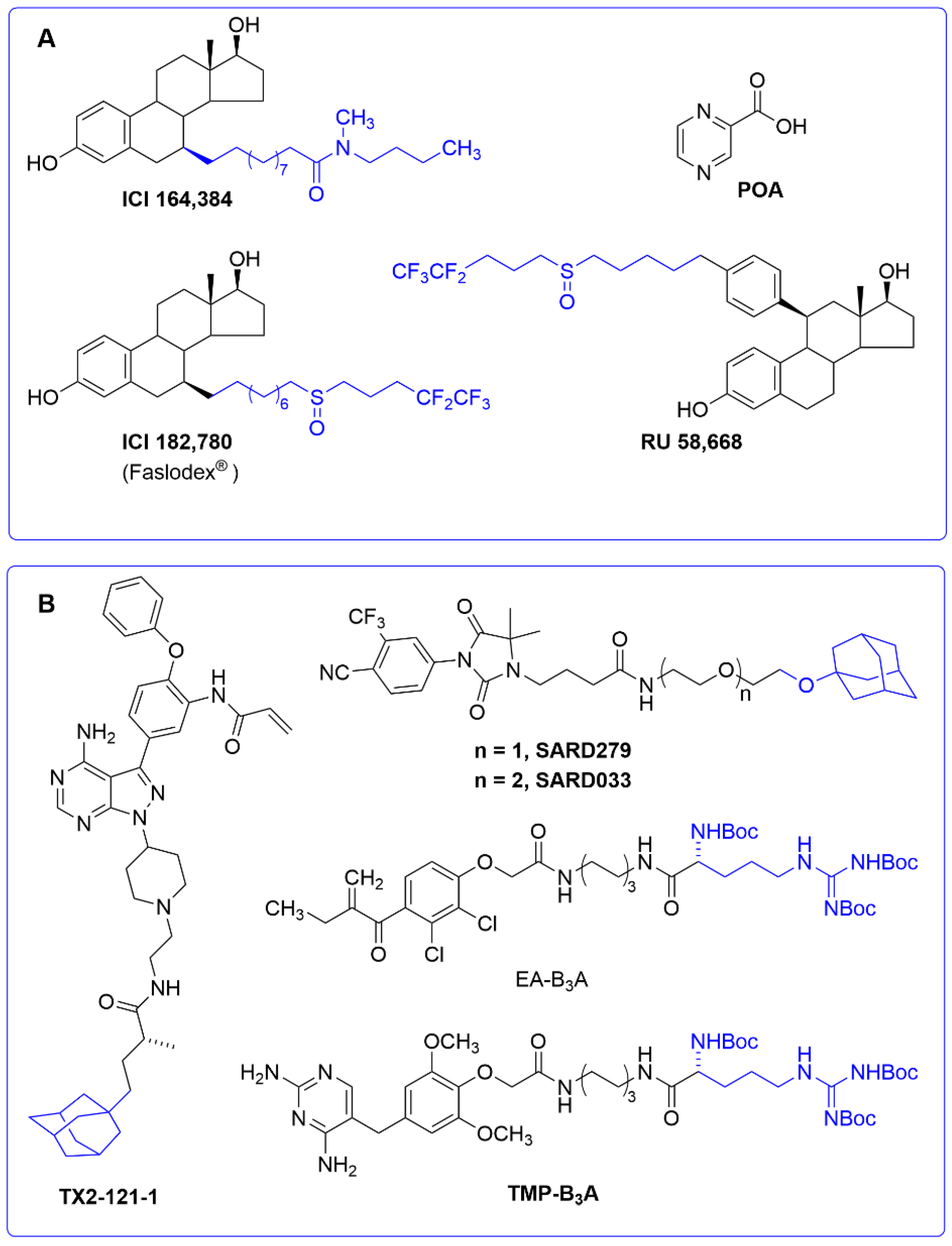
3.4. Molecular Glues, Allosteric Modulators and Hydrophobic Tags
3.5. Ubiquitin-Independent Protacs
3.6. Antibody-Chimeric Degrader Conjugates
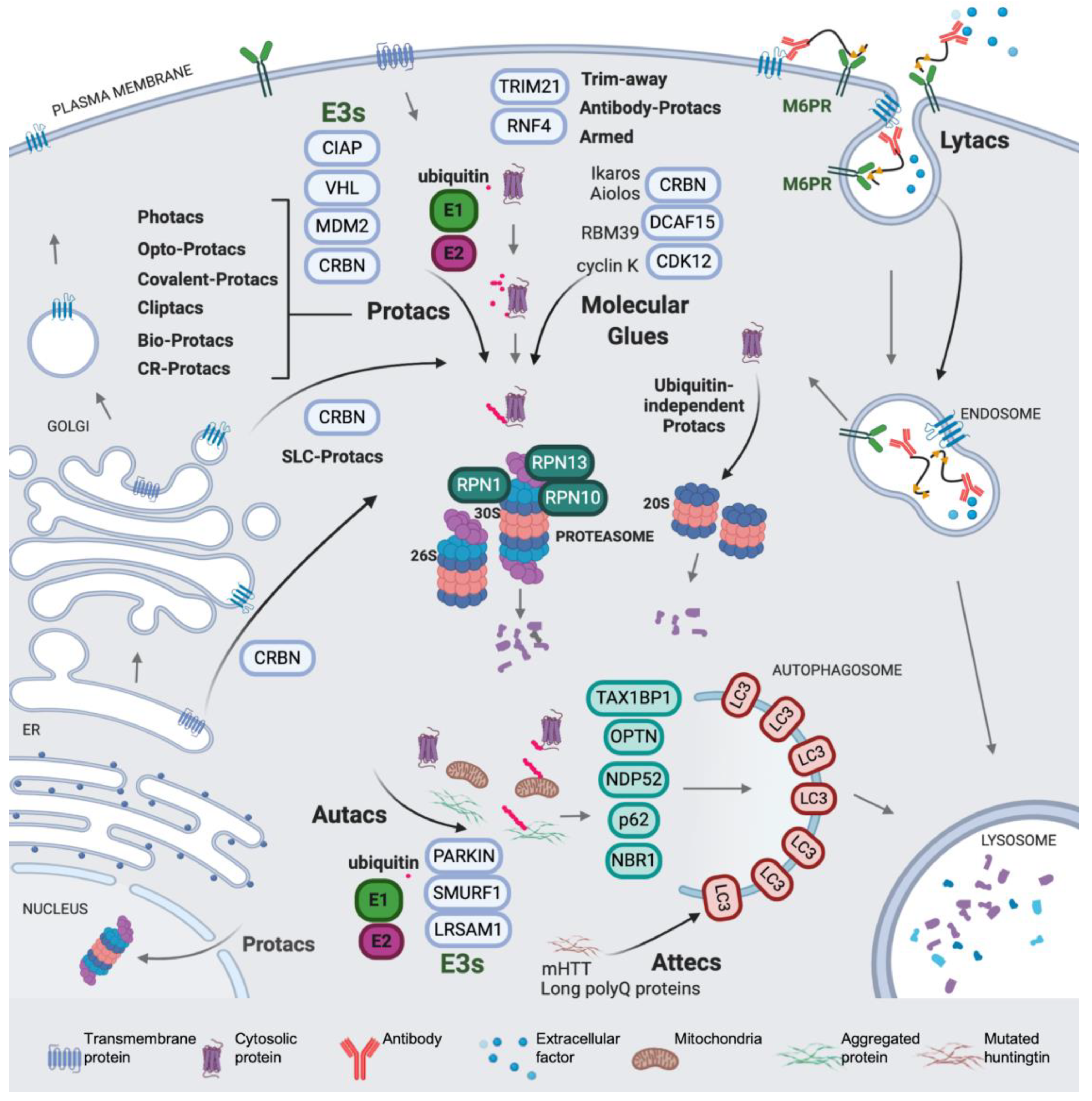
4. Expanding the (Sub)Cellular Landscape of Targetable Proteins (Factors)
4.1. Protacs for Solute Carrier Proteins (SLC-Protacs)
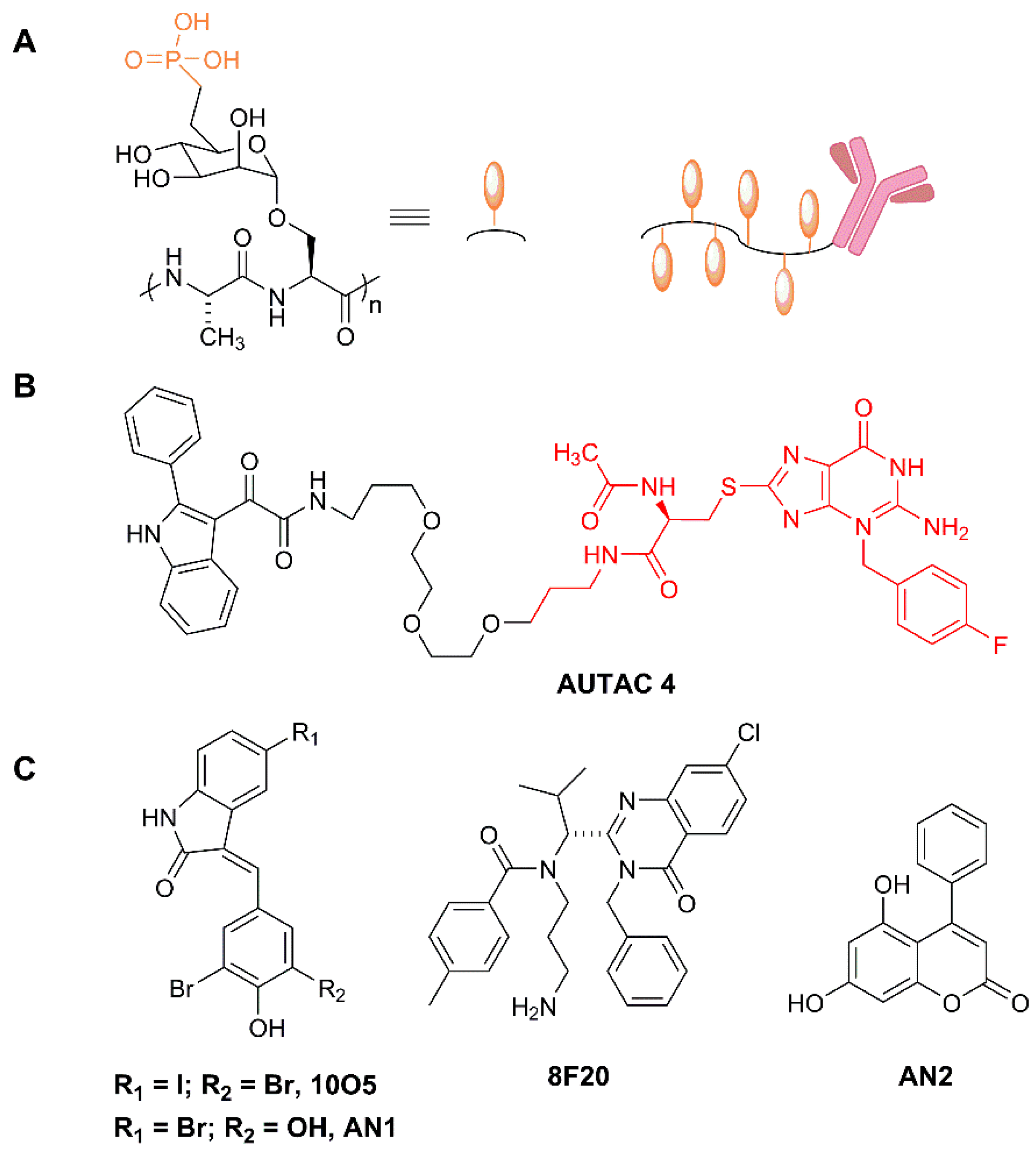
4.2. Lysosome-Targeting Chimeras (Lytacs) for Endocytically Internalized Targets
4.3. Autophagy Targeting Chimeras
4.3.1. Autacs
4.3.2. Autophagosome Tethering Compounds (Attecs)
5. Miscellaneous Protacs
5.1. “Bioprotacs”
5.2. Conformationally Restricted Protacs
5.3. N-Degron Pathway-Based Protacs
6. Concluding Remarks: An Exciting Third Generation of Protein-Degrading Chimeras

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Targeted Protein | Degradation Factor | Therapeutic Potential * | Reference |
|---|---|---|---|---|
| Protac | Multi-span transmembrane SLC proteins | CRBN | Oncology | [40] |
| Protac | ErbB1/HER1 (EGFR) | VHL | Oncology | [42] |
| Protac | MetAP-2 | SCF complex (SCFbTRCP) | Research use, first protac | [51] |
| Protac | Estrogen-related receptor alpha and RIPK2 | VHL | Immunology: Blau syndrome (RIPK2). Oncology: breast cancer(ERRa), early-onset sarcoidosis (RIPK2) | [63] |
| Protac | BCR-ABL | CRBN or VHL | Oncology: Chronic myelogenous leukemia | [64] |
| Protac | BET proteins | VHL | Oncology: castration-resistant prostate cancer. AR-related cancers | [65] |
| Protac | Androgen Receptor | MDM2 | Oncology: AR-related | [66] |
| Protac | Retinoic acid, estrogen and androgen receptors | cIAP1 | Oncology: CRABP-II-targeting; oncology: therapy for controlling tumor metastasis | [67] |
| Allosteric modulator | Ikaros and Aiolos and casein kinase 1α | CRBN | Oncology: Multiple myeloma, B cell malignancies | [88] |
| Photoactivated protac | BRD2-4 and FKBP12 | CRBN | Oncology, precision medicine | [94] |
| Photoactivated protac | BRD4, BTK | CRBN | Oncology | [100] |
| Photoactivated Protac | Bruton tyrosine kinase (BTK) | CRBN | Oncology: Precision medicine | [101] |
| Photoactivated protac | BRD2/3 and Anaplastic lymphoma kinase (ALK) | CRBN | Oncology, precision medicine | [102] |
| Photoactivated protac | BRD4 | VHL | Oncology, prescision medicine | [103] |
| Covalent protac | BTK and BLK | CRBN or VHL | Oncology | [106] |
| Covalent protac | ERRα | VHL | Metabolic disorders: Type II diabetes. Oncology: Her2+ and triple-negative breast tumors | [111] |
| Covalent protac | BTK and BLK | CRBN | Oncology: chronic lymphocytic leukemia | [112] |
| Covalent protac | Platform for Halo-tagged proteins. | VHL | Research use | [115] |
| Covalent protac | Platform for GFP-tagged proteins | VHL | Research use | [117] |
| Click protac | BRD4 and ERK1/2 | CRBN | Oncology | [119] |
| Molecular glue | RBM39 | E3 ligase receptor DCAF15 | Oncology | [122] |
| Molecular glue | RBM39, cyclinK | CRBN | Oncology | [123] |
| Molecular glue | DDB1-CDK12 molecular glue | CRBN | Oncology | [124] |
| Molecular glue | CDK12-cyclin K | CRBN | Oncology | [125] |
| Allosteric modulator | Aspartate decarboxylase PanD | E3-independent | Research use, anti-microbial agents | [131] |
| Ubiquitin-independent degrader | Androgen receptor (AR) (F876L) | ND (indirectly, CHIP) | Oncology: prostate cancer | [137] |
| Ubiquitin-independent degrader | Her3 (ErbB3) | E3-independent (Hsp70 and Hsp90 chaperone mediated) | Oncology: breast cancer | [138] |
| Ubiquitin-independent degrader | eDHFR and GST- α1 GST-π | E3-independent | Research use | [140] |
| Ubiquitin-independent degrader | GST proteins, eDHFR | E3-independent | Oncology | [141] |
| Ubiquitin-independent degrader | Proprotein convertase substilisin-like/kexin type 9(PCSK9) | E3-independent (20S targeting) | Vascular disease | [142] |
| Ubiquitin-independent degrader | BRD2/4 | E3-independent | Oncology | [143] |
| Antibody-conjugated protac | BRD4 | VHL | Oncology, precision medicine | [148] |
| Antibody-conjugated protac | ERa | VHL | Oncology: breast cancer, precision medicine | [153] |
| Antibody-conjugated protac | BRD4 | VHL | Oncology, precision medicine | [154] |
| Nanobody-conjugated protac | Platform for GFP-tagged proteins or other nanobody-targetable proteins. | TRIM21 E3 | Research use | [156] |
| Nanobody-conjugated protac | Platform for GFP-tagged proteins or other nanobody-targetable proteins. | RNF4 | Research use | [159] |
| Protac for tagged proteins | Platform for FKBP12F36V-tagged proteins. | CRBN | Research use | [160] |
| Lytac | EGFR, CD71, and PD-L1 | E3-independent (M6PR) | Oncology, neurodegenerative disorders | [162] |
| Autac | MetAP2, FK506-binding protein (FKBP12), BRD4, fragmented mitochondria, | Autophagy E3s | Oncology, neurodegenerative disorders, diabetes | [164] |
| Attec | mHTT | E3-independent (LC3) | Neurodegenerative disorders | [166] |
| Nanobody-conjugated protac | Validated against GFP-fused substrates and proliferating cell nuclear antigen (PCNA) | Slmb, a Drosophila melanogaster E3. | Research tool | [167] |
| Protac | SRC-1 | UBR | Oncology: metastasis | [169] |
| Allosteric modulator | Estrogen receptor | ND (indirectly, CHIP) | Oncology: breast cancer | [134,136] |
Funding
Conflicts of Interest
Abbreviations
References
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- MacGurn, J.A.; Hsu, P.C.; Emr, S.D. Ubiquitin and membrane protein turnover: From cradle to grave. Annu. Rev. Biochem. 2012, 81, 231–259. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-binding domains from structures to functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef]
- Zuin, A.; Bichmann, A.; Isasa, M.; Puig-Sarries, P.; Diaz, L.M.; Crosas, B.; Puig-Sàrries, P.; Díaz, L.M.; Crosas, B. Rpn10 monoubiquitination orchestrates the association of the ubiquilin-type DSK2 receptor with the proteasome. Biochem. J. 2015, 472, 353–365. [Google Scholar] [CrossRef]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef]
- Finley, D.; Ulrich, H.D.; Sommer, T.; Kaiser, P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 2012, 192, 319–360. [Google Scholar] [CrossRef]
- Mulder, M.P.C.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.P.; Chang, J.G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.O.; et al. A cascading activity-based probe sequentially targets E1-E2-E3 ubiquitin enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The ubiquitin system, an Immense Realm. Annu. Rev. Biochem. 2012, 81, 167–176. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 5249. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Baboshina, O.V.; Crinelli, R.; Siepmann, T.J.; Haas, A.L. N-end Rule Specificity within the Ubiquitin/Proteasome Pathway Is Not an Affinity Effect. J. Biol. Chem. 2001, 276, 39428–39437. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Linke, K.; Mace, P.D.; Smith, C.A.; Vaux, D.L.; Silke, J.; Day, C.L. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008, 15, 841–848. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kleffmann, T.; Linke, K.; Condon, S.M.; Hinds, M.G.; Day, C.L. Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem. J. 2013, 450, 629–638. [Google Scholar] [CrossRef]
- Trempe, J.F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Meńade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Chen, Z.; Sui, J.; Zhang, F.; Zhang, C. Cullin family proteins and tumorigenesis: Genetic association and molecular mechanisms. J. Cancer 2015, 6, 233–242. [Google Scholar] [CrossRef]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Kile, B.T.; Schulman, B.A.; Alexander, W.S.; Nicola, N.A.; Martin, H.M.E.; Hilton, D.J. The SOCS box: A tale of destruction and degradation. Trends Biochem. Sci. 2002, 27, 235–241. [Google Scholar] [CrossRef]
- Lipkowitz, S.; Weissman, A.M. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 2011, 11, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Matsuzaki, M.; Nakatsukasa, K.; Kamura, T. The role of Elongin BC-containing ubiquitin ligases. Front. Oncol. 2012, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Glickman, M.H.; Rubin, D.M.; Fried, V.A.; Finley, D. The Regulatory Particle of the Saccharomyces cerevisiae Proteasome. Mol. Cell. Biol. 1998, 18, 3149–3162. [Google Scholar] [CrossRef]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Bashore, C.; Dong, K.C.; Martin, A. The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 2019, 177, 286–298.e15. [Google Scholar] [CrossRef]
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147.e5. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Huett, A.; Heath, R.J.; Begun, J.; Sassi, S.O.; Baxt, L.A.; Vyas, J.M.; Goldberg, M.B.; Xavier, R.J. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular salmonella typhimurium. Cell Host Microbe 2012, 12, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yasuda, S.; Fujita, T.; Hamasaki, M.; Murakami, A.; Kawawaki, J.; Iwai, K.; Saeki, Y.; Yoshimori, T.; Matsuda, N.; et al. Ubiquitination of exposed glycoproteins by SCFFBXO27 directs damaged lysosomes for autophagy. Proc. Natl. Acad. Sci. USA 2017, 114, 8574–8579. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ebelle, D.L.; Wright, B.J.; Sridharan, V.; Hooper, E.; Walters, K.J. Structure of hRpn10 Bound to UBQLN2 UBL Illustrates Basis for Complementarity between Shuttle Factors and Substrates at the Proteasome. J. Mol. Biol. 2019, 431, 939–955. [Google Scholar] [CrossRef]
- Adjei, A.A. “Targeted” therapies for non-small-cell lung cancer. Clin. Lung Cancer 2002, 4, 124. [Google Scholar] [CrossRef]
- Rudmann, D.G. On-target and off-target-based toxicologic effects. Toxicol. Pathol. 2013, 41, 310–314. [Google Scholar] [CrossRef]
- Bensimon, A.; Pizzagalli, M.D.; Kartnig, F.; Dvorak, V.; Essletzbichler, P.; Winter, G.E.; Superti-Furga, G. Targeted Degradation of SLC Transporters Reveals Amenability of Multi-Pass Transmembrane Proteins to Ligand-Induced Proteolysis. Cell Chem. Biol. 2020, 27, 728–739.e9. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharmacol. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Luh, L.M.; Scheib, U.; Juenemann, K.; Wortmann, L.; Brands, M.; Cromm, P.M. Prey for the Proteasome: Targeted Protein Degradation—A Medicinal Chemist’s Perspective. Angew. Chem. Int. Ed. 2020, 59, 15448–15466. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Tomaselli, D.; Mautone, N.; Mai, A.; Rotili, D. Recent advances in epigenetic proteolysis targeting chimeras (Epi-PROTACs). Eur. J. Med. Chem. 2020, 207, 112750. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, X.; Feng, F.; Liu, W.; Sun, H. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B 2020, 10, 207–238. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Baek, S.H.; Lee, H.; Kim, K.; Mohan, R. Use of PROTACS as molecular probes of angiogenesis. Bioorg. Med. Chem. Lett. 2005, 15, 2724–2727. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.Y.; Lee, H.; Swanson, H.; Kim, K.B. Jostling for position: Optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem 2010, 5, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.M.; Deshaies, R.J.; Sakamoto, K.M. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Baek, S.-H.; Ho, A.; Kim, K. Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorg. Med. Chem. Lett. 2004, 14, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Puppala, D.; Choi, E.Y.; Swanson, H.; Kim, K.B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. ChemBioChem 2007, 8, 2058–2062. [Google Scholar] [CrossRef]
- Puppala, D.; Lee, H.; Kyung, B.K.; Swanson, H.I. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: A potential tool for chemoprevention. Mol. Pharmacol. 2008, 73, 1064–1071. [Google Scholar] [CrossRef]
- Montrose, K.; Krissansen, G.W. Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740. [Google Scholar] [CrossRef]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef]
- Henning, R.K.; Varghese, J.O.; Das, S.; Nag, A.; Tang, G.; Tang, K.; Sutherland, A.M.; Heath, J.R. Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci. 2016, 22, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Okuhira, K.; Naito, M.; Hashimoto, Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg. Med. Chem. Lett. 2012, 22, 4453–4457. [Google Scholar] [CrossRef]
- Okuhira, K.; Shoda, T.; Omura, R.; Ohoka, N.; Hattori, T.; Shibata, N.; Demizu, Y.; Sugihara, R.; Ichino, A.; Kawahara, H.; et al. Targeted degradation of proteins localized in subcellular compartments by hybrid small molecules. Mol. Pharmacol. 2017, 91, 159–166. [Google Scholar] [CrossRef]
- Demizu, Y.; Okuhira, K.; Motoi, H.; Ohno, A.; Shoda, T.; Fukuhara, K.; Okuda, H.; Naito, M.; Kurihara, M. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 2012, 22, 1793–1796. [Google Scholar] [CrossRef]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef] [PubMed]
- Okuhira, K.; Demizu, Y.; Hattori, T.; Ohoka, N.; Shibata, N.; Nishimaki-Mogami, T.; Okuda, H.; Kurihara, M.; Naito, M. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013, 104, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Nagai, K.; Hattori, T.; Okuhira, K.; Shibata, N.; Cho, N.; Naito, M. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 2014, 5, e1513. [Google Scholar] [CrossRef]
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. [Google Scholar] [CrossRef]
- Tomoshige, S.; Hashimoto, Y.; Ishikawa, M. Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg. Med. Chem. 2016, 24, 3144–3148. [Google Scholar] [CrossRef] [PubMed]
- Orning, L.; Fitzpatrick, F.A. Albumins Activate Peptide Hydrolysis by the Bifunctional Enzyme LTA4 Hydrolase/Aminopeptidase. Biochemistry 1992, 31, 4218–4223. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Takubo, K.; Kikuchi, R.; Nishimoto, M.; Kitagawa, M.; Abe, F.; Nishikawa, K.; Tsuruo, T.; Naito, M. Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J. Biol. Chem. 2008, 283, 8961–8968. [Google Scholar] [CrossRef]
- McBride, W.G. the Teratogenic Action of Drugs. Med. J. Aust. 1963, 2, 689–692. [Google Scholar] [CrossRef]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor α production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Corral, L.G.; Haslett, P.A.J.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. Int. J. Lepr. Other Mycobact. Dis. 1999, 67, 501. [Google Scholar]
- Haslett, P.A.J.; Corral, L.G.; Albert, M.; Kaplan, G. Thalidomide costimulates primary human t lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J. Exp. Med. 1998, 187, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Scialli, A.R. Thalidomide: The tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 2011, 122, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.V.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Verma, S.; Manna, D. Controlling PROTACs with Light. ChemMedChem 2020, 15, 1258–1261. [Google Scholar] [CrossRef]
- Wu, P.; Manna, D. Optochemical Control of Protein Degradation. ChemBioChem 2020, 21, 2250–2252. [Google Scholar] [CrossRef]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef] [PubMed]
- Sekkat, Z. Photoisomerization Effects in Organic Nonlinear Optics: Photo-Assisted Poling and Depoling and Polarizability Switching. In Photoreactive Organic Thin Films; Academic Press: New York, NY, USA, 2002. [Google Scholar]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef]
- Dong, M.; Babalhavaeji, A.; Samanta, S.; Beharry, A.A.; Woolley, G.A. Red-Shifting Azobenzene Photoswitches for in Vivo Use. Acc. Chem. Res. 2015, 48, 2662–2670. [Google Scholar] [CrossRef]
- Kienzler, M.A.; Reiner, A.; Trautman, E.; Yoo, S.; Trauner, D.; Isacoff, E.Y. A red-shifted, fast-relaxing azobenzene photoswitch for visible light control of an ionotropic glutamate receptor. J. Am. Chem. Soc. 2013, 135, 17683–17686. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. O -fluoroazobenzenes as readily synthesized photoswitches offering nearly quantitative two-way isomerization with visible light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef]
- Castellanos, S.; Goulet-Hanssens, A.; Zhao, F.; Dikhtiarenko, A.; Pustovarenko, A.; Hecht, S.; Gascon, J.; Kapteijn, F.; Bléger, D. Structural Effects in Visible-Light-Responsive Metal-Organic Frameworks Incorporating ortho-Fluoroazobenzenes. Chem. A Eur. J. 2016, 22, 746–752. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [PubMed]
- Kounde, C.S.; Shchepinova, M.M.; Saunders, C.N.; Muelbaier, M.; Rackham, M.D.; Harling, J.D.; Tate, E.W. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun. 2020, 56, 5532–5535. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Xue, G.; Chen, J.; Liu, L.; Zhou, D.; Zuo, Y.; Fu, T.; Pan, Z. Protein degradation through covalent inhibitor-based PROTACs. Chem. Commun. 2020, 56, 1521–1524. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Lithgow, H.; Dittus, L.; Bassi, Z.I.; Hughes, S.E.; Muelbaier, M.; Dai, H.; Smith, I.E.D.; Kerr, W.J.; Burley, G.A.; et al. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem. Biol. 2019, 14, 342–347. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116. [Google Scholar] [CrossRef]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Lee, C.U.; Grossmann, T.N. Reversible covalent inhibition of a protein target. Angew. Chem. Int. Ed. 2012, 51, 8699–8700. [Google Scholar] [CrossRef]
- Peng, L.; Zhang, Z.; Lei, C.; Li, S.; Zhang, Z.; Ren, X.; Chang, Y.; Zhang, Y.; Xu, Y.; Ding, K. Identification of New Small-Molecule Inducers of Estrogen-related Receptor α (ERRα) Degradation. ACS Med. Chem. Lett. 2019, 10, 767–772. [Google Scholar] [CrossRef]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Tovell, H.; Testa, A.; Maniaci, C.; Zhou, H.; Prescott, A.R.; Macartney, T.; Ciulli, A.; Alessi, D.R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14, 882–892. [Google Scholar] [CrossRef]
- CRISPR Genome Editing, cDNA Clone, shRNA Knockdown, miRNA Expression, 3’UTR|GeneCopoeia. Available online: https://www.genecopoeia.com/ (accessed on 24 October 2020).
- Simpson, L.M.; Macartney, T.J.; Nardin, A.; Fulcher, L.J.; Röth, S.; Testa, A.; Maniaci, C.; Ciulli, A.; Ganley, I.G.; Sapkota, G.P. Inducible Degradation of Target Proteins through a Tractable Affinity-Directed Protein Missile System. Cell Chem. Biol. 2020, 27, 1164–1180.e5. [Google Scholar] [CrossRef]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- Baek, K.; Schulman, B.A. Molecular glue concept solidifies. Nat. Chem. Biol. 2020, 16, 2–3. [Google Scholar] [CrossRef]
- Tan, X.; Calderon-Villalobos, L.I.A.; Sharon, M.; Zheng, C.; Robinson, C.V.; Estelle, M.; Zheng, N. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 2007, 446, 640–645. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Chen, P.; Cao, L.; Li, Y.; Zeng, Z.; Cui, Y.; Wu, Q.; Li, J.; Wang, J.H.; Dong, M.Q.; et al. Discovery of a molecular glue promoting cdk12-ddb1 interaction to trigger cyclin k degradation. Elife 2020, 9, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Der Li, Y.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Baunbæk, D.; Delehouze, C.; Loaëc, N.; Hole, A.J.; Baumli, S.; Endicott, J.A.; Douc-Rasy, S.; Bénard, J.; Oumata, N.; et al. CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer 2010, 1, 369–380. [Google Scholar] [CrossRef]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thomä, N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4 CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Gopal, P.; Nartey, W.; Ragunathan, P.; Sarathy, J.; Kaya, F.; Yee, M.; Setzer, C.; Manimekalai, M.S.S.; Dartois, V.; Grüber, G.; et al. Pyrazinoic Acid Inhibits Mycobacterial Coenzyme A Biosynthesis by Binding to Aspartate Decarboxylase PanD. ACS Infect. Dis. 2017, 3, 807–819. [Google Scholar] [CrossRef]
- Shi, W.; Chen, J.; Feng, J.; Cui, P.; Zhang, S.; Weng, X.; Zhang, W.; Zhang, Y. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2014, 3, 1–8. [Google Scholar] [CrossRef]
- Gopal, P.; Sarathy, J.P.; Yee, M.; Ragunathan, P.; Shin, J.; Bhushan, S.; Zhu, J.; Akopian, T.; Kandror, O.; Lim, T.K.; et al. Pyrazinamide triggers degradation of its target aspartate decarboxylase. Nat. Commun. 2020, 11, 1661. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van Der Veken, P.; De Winter, H.; Augustyns, K. ClpP protease, a promising antimicrobial target. Int. J. Mol. Sci. 2019, 20, 2232. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164, 384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, B.M.; Sherk, A.; McDonnell, D.P. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007, 67, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Xie, T.; Lim, S.M.; Westover, K.D.; Dodge, M.E.; Ercan, D.; Ficarro, S.B.; Udayakumar, D.; Gurbani, D.; Tae, H.S.; Riddle, S.M.; et al. Pharmacological targeting of the pseudokinase Her3. Nat. Chem. Biol. 2014, 10, 1006–1012. [Google Scholar] [CrossRef]
- Gopal, P.; Dick, T. Targeted protein degradation in antibacterial drug discovery? Prog. Biophys. Mol. Biol. 2020, 152, 10–14. [Google Scholar] [CrossRef]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor mediated protein degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef]
- Petrilli, W.L.; Adam, G.C.; Erdmann, R.S.; Abeywickrema, P.; Agnani, V.; Ai, X.; Baysarowich, J.; Byrne, N.; Caldwell, J.P.; Chang, W.; et al. From Screening to Targeted Degradation: Strategies for the Discovery and Optimization of Small Molecule Ligands for PCSK9. Cell Chem. Biol. 2020, 27, 32–40.e3. [Google Scholar] [CrossRef]
- Testa, A.; Hughes, S.; Butcher, S.P.; Ciulli, A. Preparation of Bifunctional Molecules for Targeting Usp14; PCT International Application: Washington, DC, USA, 2019. [Google Scholar]
- Boselli, M.; Lee, B.H.; Robert, J.; Prado, M.A.; Min, S.W.; Cheng, C.; Catarina Silva, M.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.H.; Lee, M.J.; Park, S.; Oh, D.-C.C.; Elsasser, S.; Chen, P.-C.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Crosas, B.; Hanna, J.; Kirkpatrick, D.S.; Zhang, D.P.; Tone, Y.; Hathaway, N.A.A.; Buecker, C.; Leggett, D.S.; Schmidt, M.; King, R.W.; et al. Ubiquitin Chains Are Remodeled at the Proteasome by Opposing Ubiquitin Ligase and Deubiquitinating Activities. Cell 2006, 127, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Goldberg, A.L. Ubiquitinated proteins promote the association of proteasomes with the deubiquitinating enzyme Usp14 and the ubiquitin ligase Ube3c. Proc. Natl. Acad. Sci. USA 2017, 114, E3404–E3413. [Google Scholar] [CrossRef]
- Pillow, T.H.; Adhikari, P.; Blake, R.A.; Chen, J.; Del Rosario, G.; Deshmukh, G.; Figueroa, I.; Gascoigne, K.E.; Kamath, A.V.; Kaufman, S.; et al. Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. ChemMedChem 2020, 15, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.Q.A.B.H.; Van Den Oudenrijn, S.; Bakker, A.Q.A.B.H.; Feller, N.; Van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.S.; Tice, D.A.; Soria, J.C. Antibody-drug conjugates: Future directions in clinical and translational strategies to improve the therapeutic index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D.F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: A drug metabolism and pharmacokinetics perspective. Drug Discov. Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Adhikari, P.; Blake, R.A.; Blaquiere, N.; Chen, J.; Cheng, Y.X.; den Besten, W.; Han, J.; Hartman, S.J.; He, J.; et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorg. Med. Chem. Lett. 2020, 30, 126907. [Google Scholar] [CrossRef]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Clift, D.; McEwan, W.A.; Labzin, L.I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 2017, 171, 1692–1706.e18. [Google Scholar] [CrossRef] [PubMed]
- Clift, D.; So, C.; McEwan, W.A.; James, L.C.; Schuh, M. Acute and rapid degradation of endogenous proteins by Trim-Away. Nat. Protoc. 2018, 13, 2149–2175. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; Falcon, B.; Vaysburd, M.; Clift, D.; Oblak, A.L.; Ghetti, B.; Goedert, M.; James, L.C. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 574–579. [Google Scholar] [CrossRef]
- Ibrahim, A.F.M.; Shen, L.; Tatham, M.H.; Dickerson, D.; Prescott, A.R.; Abidi, N.; Xirodimas, D.P.; Hay, R.T. Antibody RING-Mediated Destruction of Endogenous Proteins. Mol. Cell 2020, 79, 155–166.e9. [Google Scholar] [CrossRef]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Salyer, S.A.; Parks, J.; Barati, M.T.; Lederer, E.D.; Clark, B.J.; Klein, J.D.; Khundmiri, S.J. Aldosterone regulates Na+, K+ ATPase activity in human renal proximal tubule cells through mineralocorticoid receptor. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 2143–2152. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Olson, L.J.; Castonguay, A.C.; Lasanajak, Y.; Peterson, F.C.; Cummings, R.D.; Smith, D.F.; Dahms, N.M. Identification of a fourth mannose 6-phosphate binding site in the cation-independent mannose 6-phosphate receptor. Glycobiology 2014, 25, 591–606. [Google Scholar] [CrossRef]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef]
- Zamponi, E.; Helguera, P.R. The Shape of Mitochondrial Dysfunction in Down Syndrome. Dev. Neurobiol. 2019, 79, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J.; et al. BioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. USA 2020, 117, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.; Hughes, S.J.; Lucas, X.; Wright, J.E.; Ciulli, A. Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem. Int. Ed. 2020, 59, 1727–1734. [Google Scholar] [CrossRef]
- Lee, Y.; Heo, J.; Jeong, H.; Hong, K.T.; Kwon, D.H.; Shin, M.H.; Oh, M.; Sable, G.A.; Ahn, G.O.; Lee, J.S.; et al. Targeted Degradation of Transcription Coactivator SRC-1 through the N-Degron Pathway. Angew. Chem. Int. Ed. 2020, 59, 17548–17555. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Z.; Chen, H.; Xu, J. The steroid receptor coactivator-1 regulates Twist expression and promotes breast cancer metastasis. Cancer Res. 2009, 69, 3819–3827. [Google Scholar] [CrossRef]
- Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019, 18, 237–239. [Google Scholar] [CrossRef]
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744. [Google Scholar] [CrossRef]
- Yamazoe, S.; Tom, J.; Fu, Y.; Wu, W.; Zeng, L.; Sun, C.; Liu, Q.; Lin, J.; Lin, K.; Fairbrother, W.J.; et al. Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study. J. Med. Chem. 2020, 63, 2807–2813. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coll-Martínez, B.; Delgado, A.; Crosas, B. The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents. Molecules 2020, 25, 5956. https://doi.org/10.3390/molecules25245956
Coll-Martínez B, Delgado A, Crosas B. The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents. Molecules. 2020; 25(24):5956. https://doi.org/10.3390/molecules25245956
Chicago/Turabian StyleColl-Martínez, Bernat, Antonio Delgado, and Bernat Crosas. 2020. "The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents" Molecules 25, no. 24: 5956. https://doi.org/10.3390/molecules25245956
APA StyleColl-Martínez, B., Delgado, A., & Crosas, B. (2020). The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents. Molecules, 25(24), 5956. https://doi.org/10.3390/molecules25245956