
Generation of Bis(ferrocenyl)silylenes from Siliranes †


Abstract
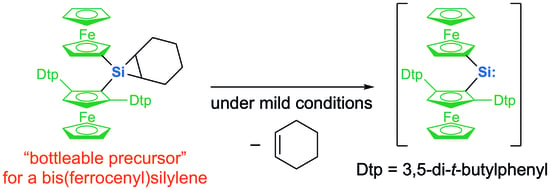
{kind=link}
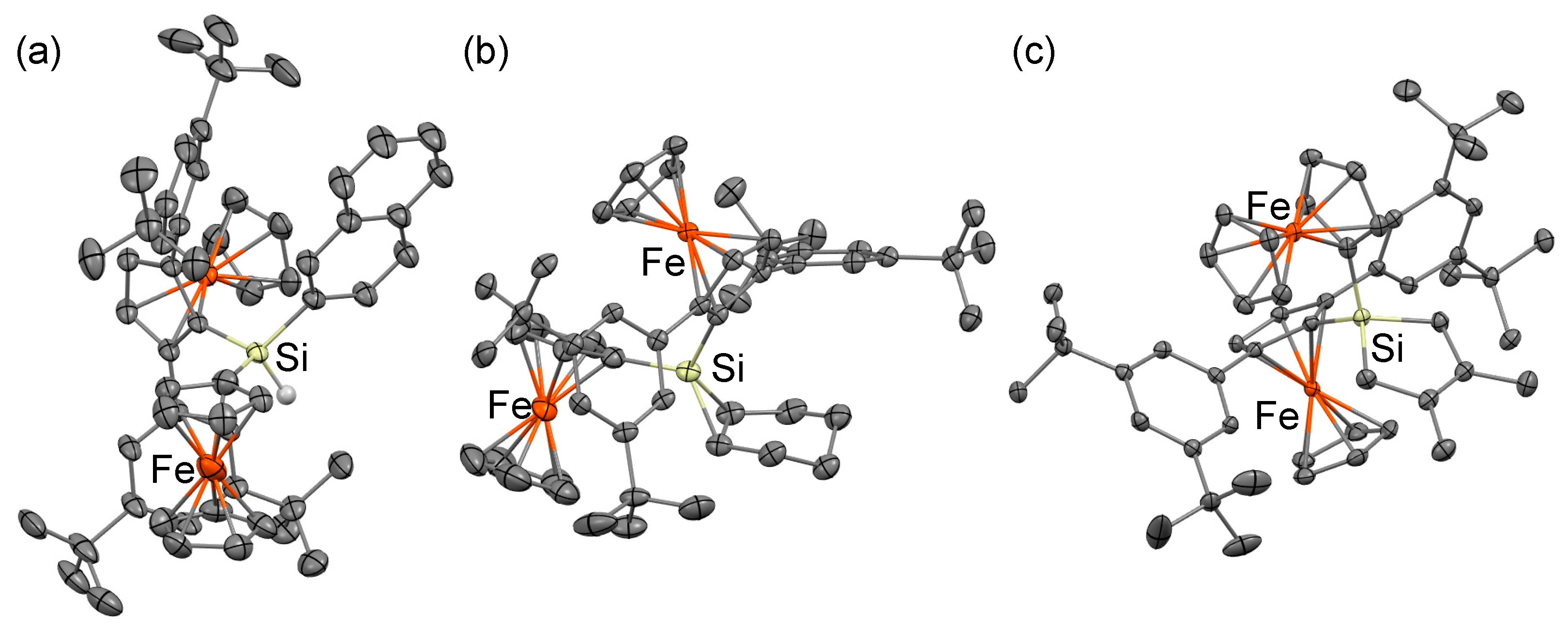{kind=link}
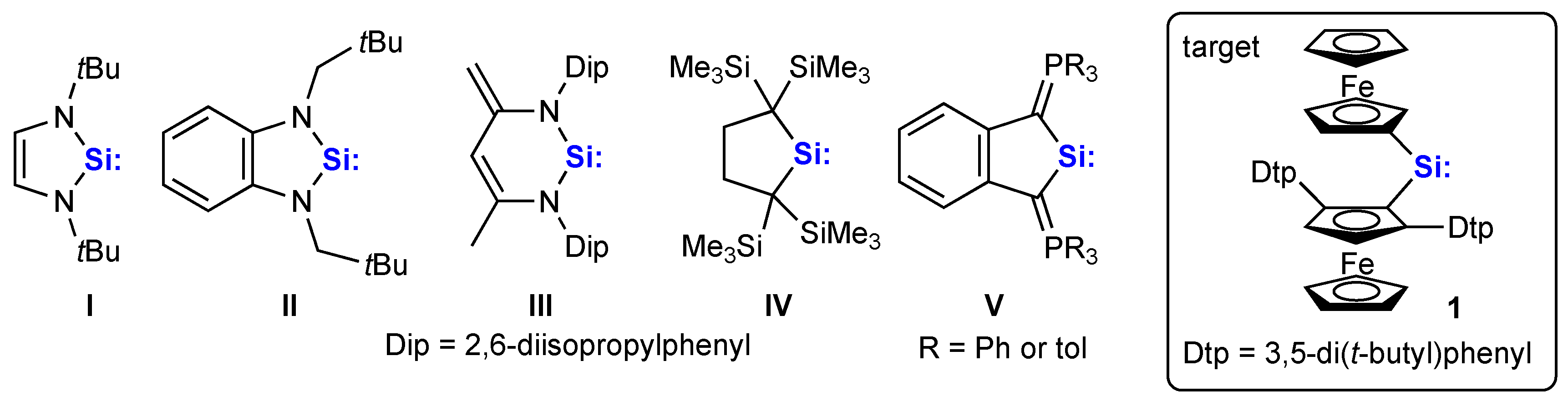{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
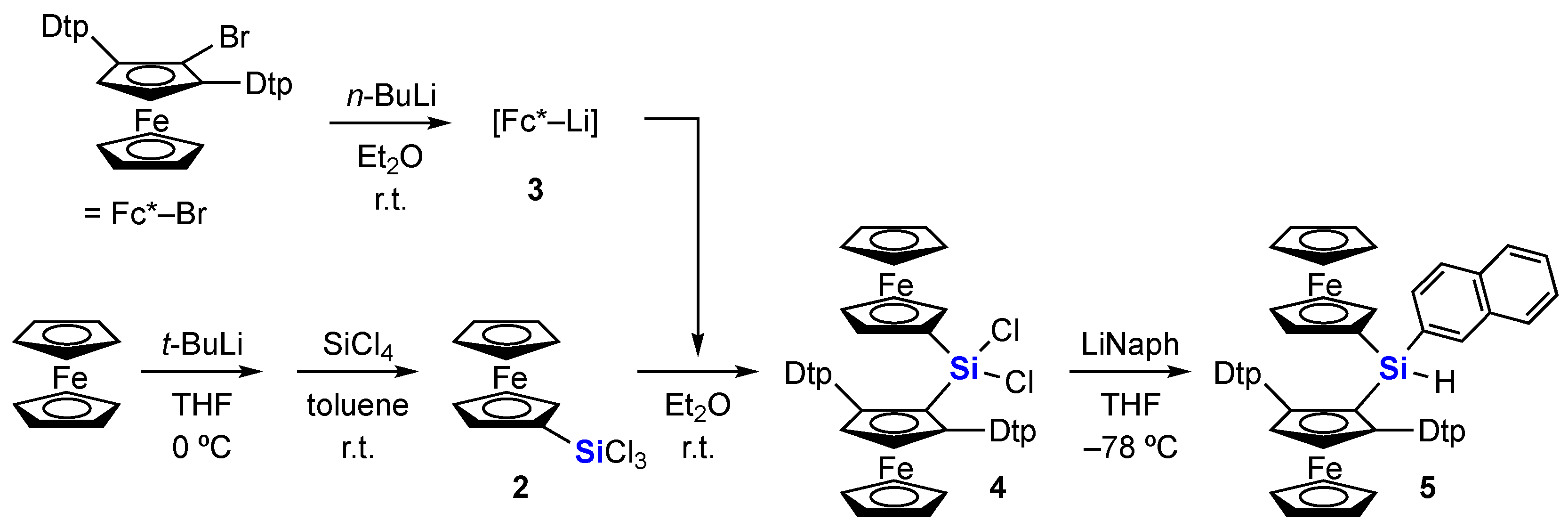
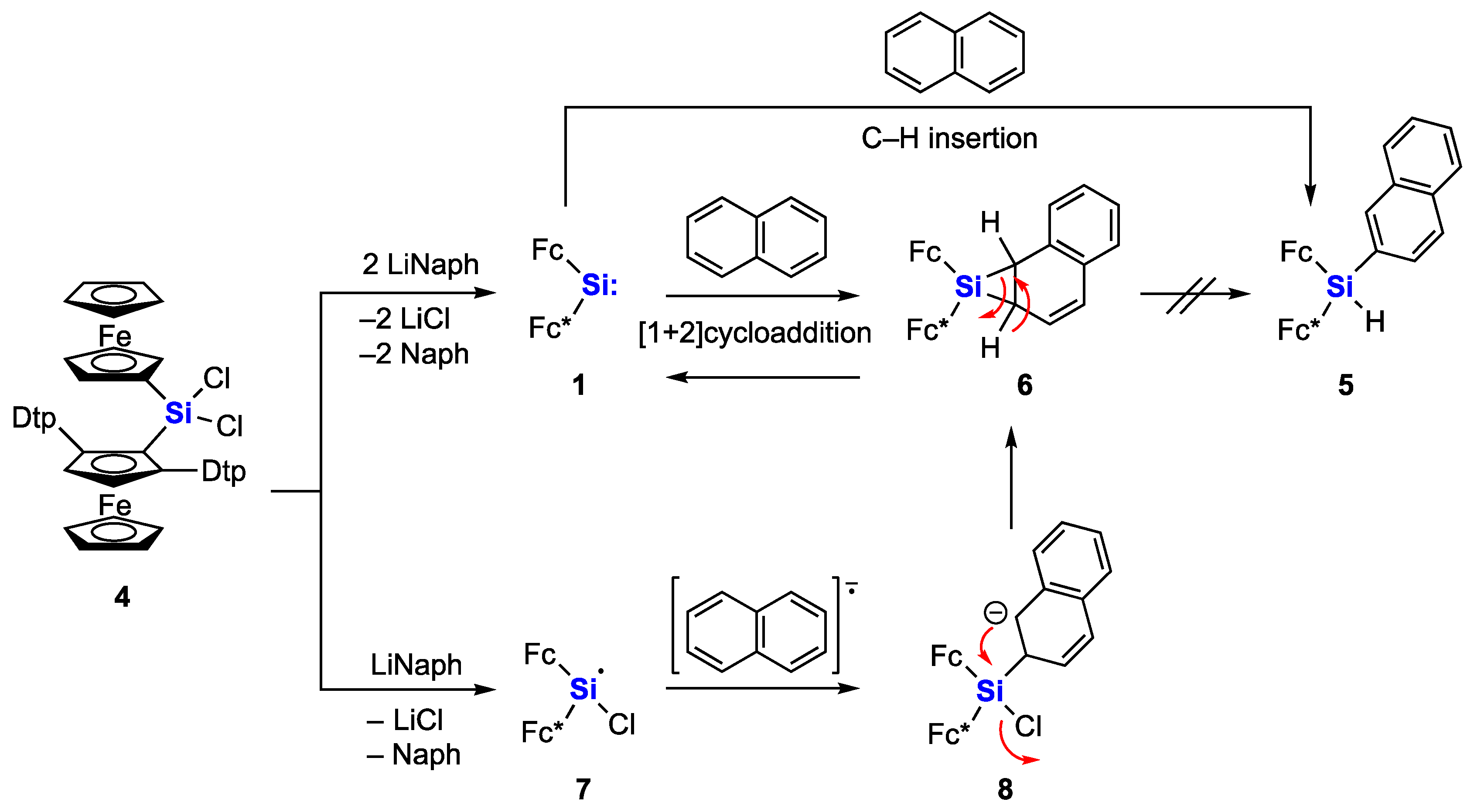
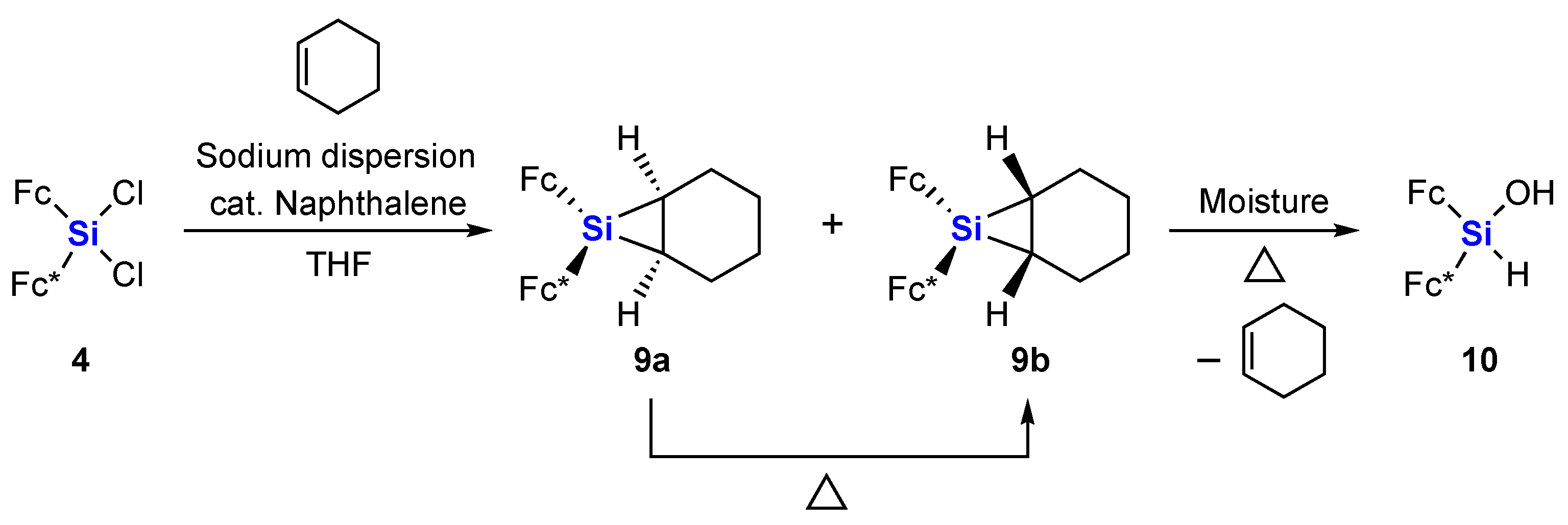
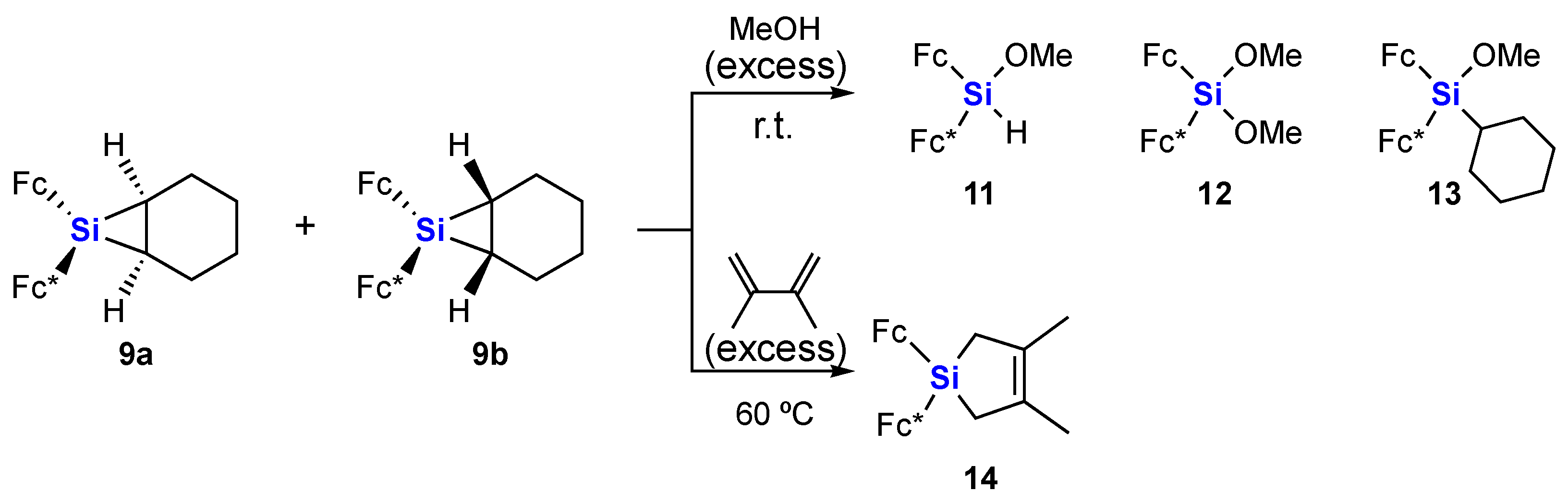
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Syntheses and Reactions
3.2.1. Bis(ferrocenyl)dichlorosilane 4
3.2.2. Bis(ferrocenyl)naphthylsilane 5
3.2.3. Siliranes 9a and 9b
3.2.4. Thermolysis of Siliranes 9a and 9b in Cyclohexene
3.2.5. Thermolysis of Siliranes 9a and 9b in the Solid State
3.2.6. Reaction of Siliranes 9a and 9b with Methanol
3.2.7. Reaction of Siliranes 9a and 9b with 2,3-Dimethyl-1,3-butadiene
3.2.8. Characterization of Fc*(Fc)Si(OH)2
3.3. Computational Methods
3.4. X-ray Crystallographic Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Raoufmoghaddam, S.; Zhou, Y.-P.; Wang, Y.; Driess, M. N-heterocyclic silylenes as powerful steering ligands in catalysis. J. Organomet. Chem. 2017, 829, 2–10. [Google Scholar] [CrossRef]
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato)silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: Synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [Google Scholar] [CrossRef]
- Zhou, Y.-P.; Driess, M. Isolable Silylene Ligands Can Boost Efficiencies and Selectivities in Metal-Mediated Catalysis. Angew. Chem. Int. Ed. 2018, 58, 3715–3728. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, S.; Inoue, S. Small Molecule Activation by Two-Coordinate Acyclic Silylenes. Eur. J. Inorg. Chem. 2020, 3131–3142. [Google Scholar] [CrossRef]
- Shan, C.; Yao, S.; Driess, M. Where silylene–silicon centres matter in the activation of small molecules. Chem. Soc. Rev. 2020, 49, 6733–6754. [Google Scholar] [CrossRef] [PubMed]
- Zark, P.; Schäfer, A.; Mitra, A.; Haase, D.; Saak, W.; West, R.; Müller, T. Synthesis and reactivity of N-aryl substituted N-heterocyclic silylenes. J. Organomet. Chem. 2010, 695, 398–408. [Google Scholar] [CrossRef]
- Reiter, D.; Holzner, R.; Porzelt, A.; Altmann, P.J.; Frisch, P.; Inoue, S. Disilene−Silylene Interconversion: A Synthetically Accessible Acyclic Bis(silyl)silylene. J. Am. Chem. Soc. 2019, 141, 13536–13546. [Google Scholar] [CrossRef]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Gehrhus, B.; Lappert, M.F.; Heinicke, J.; Boese, R.; Bläser, D. Synthesis, structures and reactions of new thermally stable silylenes. J. Chem. Soc. Chem. Commun. 1995, 1931–1932. [Google Scholar] [CrossRef]
- Schäfer, A.; Saak, W.; Weidenbruch, M.; Marsmann, H.; Henkel, G. Compounds of Germanium and Tin, 22—Reactions of a Silyene with a Germylene and a Stannylene: Formation of a Digermene with an Unusual Arrangement of the Substituents and of a Stannane. Chem. Ber. 1997, 130, 1733–1737. [Google Scholar] [CrossRef]
- Moser, D.F.; Guzei, I.A. Robert West Crystal Structure of the Stable Silylene, N, N’-di-tert-butyl-1,3-diaza-2-silacyclopent-4-en-2-ylidene. Main Group Met. Chem. 2001, 24, 811–812. [Google Scholar] [CrossRef]
- Becker, J.S.; Staples, R.J.; Gordon, R.G. Analysis of the crystal structures of 1,3-di-tert-butyl-2,3-dihydro-1H-1,3,2-diazasilol-2-ylidene and 1,3-di-tert-butyl-2,2-dichloro-1,3-diaza-2-sila-4-cyclopentene. Cryst. Res. Technol. 2004, 39, 85–88. [Google Scholar] [CrossRef]
- Hill, N.J.; Moser, D.F.; Guzei, I.A.; West, R. Reactions of Stable Silylenes with Organic Azides. Organometallics 2005, 24, 3346–3349. [Google Scholar] [CrossRef]
- Driess, M.; Yao, S.; Brym, M.; van Wüllen, C.; Lentz, D. A New Type of N-Heterocyclic Silylene with Ambivalent Reactivity. J. Am. Chem. Soc. 2006, 128, 9628–9629. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, J.; Song, H.; Cui, C. N-Aryl substituted heterocyclic silylenes. Dalton Trans. 2009, 5444–5446. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Brodovitch, J.-C.; Krempner, C.; Percival, P.W.; Vyas, P.; West, R. A Silyl Radical formed by Muonium Addition to a Silylene. Angew. Chem. Int. Ed. 2010, 49, 2893–2895. [Google Scholar] [CrossRef]
- Kosai, T.; Ishida, S.; Iwamoto, T. A Two-Coordinate Cyclic (Alkyl)(amino)silylene: Balancing Thermal Stability and Reactivity. Angew. Chem. Int. Ed. 2016, 55, 15554–15558. [Google Scholar] [CrossRef]
- Takeda, N.; Suzuki, H.; Tokitoh, N.; Okazaki, R.; Nagase, S. Reaction of a Sterically Hindered Silylene with Isocyanides: The First Stable Silylene−Lewis Base Complexes. J. Am. Chem. Soc. 1997, 119, 1456–1457. [Google Scholar] [CrossRef]
- Gau, D.; Kato, T.; Saffon-Merceron, N.; Cossío, F.P.; Baceiredo, A. Stable Phosphonium Sila-ylide with Reactivity as a Sila-Wittig Reagent. J. Am. Chem. Soc. 2009, 131, 8762–8763. [Google Scholar] [CrossRef]
- Filippou, A.C.; Chernov, O.; Blom, B.; Stumpf, K.W.; Schnakenburg, G. Stable N-Heterocyclic Carbene Adducts of Arylchlorosilylenes and Their Germanium Homologues. Chem. Eur. J. 2010, 16, 2866–2872. [Google Scholar] [CrossRef]
- Rodriguez, R.; Gau, D.; Contie, Y.; Kato, T.; Saffon-Merceron, N.; Baceiredo, A. Synthesis of a Phosphine-Stabilized Silicon(II) Hydride and Its Addition to Olefins: A Catalyst-Free Hydrosilylation Reaction. Angew. Chem. Int. Ed. 2011, 50, 11492–11495. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsuo, T.; Hashizume, D.; Tamao, K. Room-Temperature Dissociation of 1,2-Dibromodisilenes to Bromosilylenes. J. Am. Chem. Soc. 2011, 133, 19710–19713. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Köhler, C.; Yang, Z.; Stalke, D.; Herbst-Irmer, R.; Roesky, H.W. Synthesis of Cyclic Alkyl(amino) Carbene Stabilized Silylenes with Small N-Donating Substituents. Chem. Eur. J. 2019, 25, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S. Theory and Calculations of Molecules Containing Heavier Main Group Elements and Fullerenes Encaging Transition Metals: Interplay with Experiment. Bull. Chem. Soc. Jpn. 2014, 87, 167–195. [Google Scholar] [CrossRef]
- Fischer, R.C.; Power, P.P. π-Bonding and the Lone Pair Effect in Multiple Bonds Involving Heavier Main Group Elements: Developments in the New Millennium. Chem. Rev. 2010, 110, 3877–3923. [Google Scholar] [CrossRef] [PubMed]
- Rivard, E.; Power, P.P. Multiple Bonding in Heavier Element Compounds Stabilized by Bulky Terphenyl Ligands. Inorg. Chem. 2007, 46, 10047–10064. [Google Scholar] [CrossRef] [PubMed]
- Power, P.P. Stable Two-Coordinate, Open-Shell (d1–d9) Transition Metal Complexes. Chem. Rev. 2012, 112, 3482–3507. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected organophosphorus compounds of unusual structures. Pure Appl. Chem. 2017, 89, 281–286. [Google Scholar] [CrossRef]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. The First Isolable Dialkylsilylene. J. Am. Chem. Soc. 1999, 121, 9722–9723. [Google Scholar] [CrossRef]
- Abe, T.; Tanaka, R.; Ishida, S.; Kira, M.; Iwamoto, T. New Isolable Dialkylsilylene and Its Isolable Dimer That Equilibrate in Solution. J. Am. Chem. Soc. 2012, 134, 20029–20032. [Google Scholar] [CrossRef]
- Kobayashi, R.; Ishida, S.; Iwamoto, T. An Isolable Silicon Analogue of a Ketone that Contains an Unperturbed Si= O Double Bond. Angew. Chem., Int. Ed. 2019, 58, 9425–9428. [Google Scholar] [CrossRef] [PubMed]
- Asay, M.; Inoue, S.; Driess, M. Aromatic Ylide-Stabilized Carbocyclic Silylene. Angew. Chem. Int. Ed. 2011, 50, 9589–9592. [Google Scholar] [CrossRef] [PubMed]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable Heavier Carbene Analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef] [PubMed]
- Sasamori, T.; Tokitoh, N. A New Family of Multiple-Bond Compounds between Heavier Group 14 Elements. Bull. Chem. Soc. Jpn. 2013, 86, 1005–1021. [Google Scholar] [CrossRef]
- Iwamoto, T.; Ishida, S. Multiple Bonds with Silicon: Recent Advances in Synthesis, Structure, and Functions of Stable Disilenes. In Functional Molecular Silicon Compounds II: Low Oxidation States; Scheschkewitz, D., Ed.; Structure and Bonding; Springer International Publishing: Cham, Switzerland, 2014; pp. 125–202. ISBN 978-3-319-03734-9. [Google Scholar]
- Ishida, S.; Iwamoto, T. Recent advances in η2-disilene and η2-disilyne mononuclear transition metal complexes and related compounds. Coord. Chem. Rev. 2016, 314, 34–63. [Google Scholar] [CrossRef]
- Matsuo, T.; Hayakawa, N. π-Electron systems containing Si= Si double bonds. Sci. Technol. Adv. Mater. 2018, 19, 108–129. [Google Scholar] [CrossRef]
- Rammo, A.; Scheschkewitz, D. Functional Disilenes in Synthesis. Chem. Eur. J. 2018, 24, 6866–6885. [Google Scholar] [CrossRef]
- Sasamori, T. Ferrocenyl-substituted low-coordinated heavier group 14 elements. Dalton Trans. 2020, 49, 8029–8035. [Google Scholar] [CrossRef]
- Sasamori, T.; Suzuki, Y.; Sakagami, M.; Miyake, H.; Tokitoh, N. Structure of stable telluradiphosphirane bearing bulky ferrocenyl ligands. Chem. Lett. 2014, 43, 1464–1466. [Google Scholar] [CrossRef]
- Sakagami, M.; Sasamori, T.; Sakai, H.; Furukawa, Y.; Tokitoh, N. 1,2-Bis(ferrocenyl)dipnictenes: Bimetallic Systems with a Pn= Pn Heavy π-Spacer (Pn: P, Sb, and Bi). Bull. Chem. Soc. Jpn. 2013, 86, 1132–1143. [Google Scholar] [CrossRef]
- Sasamori, T.; Suzuki, Y.; Tokitoh, N. Isolation and Structural Characterization of a Lewis Base-Free Monolithioferrocene. Organometallics 2014, 33, 6696–6699. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sasamori, T.; Guo, J.-D.; Tokitoh, N. A Redox-Active Bis(ferrocenyl)germylene and Its Reactivity. Chem. Eur. J. 2018, 24, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Herberhold, M.; Ayazi, A.; Milius, W.; Wrackmeyer, B. Silyl derivatives of ferrocene with pending indenyl or fluorenyl substituents at silicon. J. Organomet. Chem. 2002, 656, 71–80. [Google Scholar] [CrossRef]
- Fc*(Fc)Si(OH)2 was characterized based on the spectroscopic and XRD analyses. See the Material and Methods section.
- Crystallographic data for 5, 9a, 10, 11, 13, 14, and Fc*FcSi(OH)2 were deposited at the Cambridge Crystallographic Data Centre (CCDC) under the numbers CCDC-2044338-2044344. These data can be obtained free of charge via www.ccdc.cam.ac.uk/structures.
- Suzuki, H.; Tokitoh, N.; Okazaki, R. A Novel Reactivity of a Silylene: The First Examples of [1 + 2] Cycloaddition with Aromatic Compounds. J. Am. Chem. Soc. 1994, 116, 11572–11573. [Google Scholar] [CrossRef]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. Excited-State Reactions of an Isolable Silylene with Aromatic Compounds. J. Am. Chem. Soc. 2002, 124, 3830–3831. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, T.; Uchida, K.; Sung, Y.M.; Shin, J.-Y.; Ishida, S.; Lim, J.M.; Hiroto, S.; Furukawa, K.; Kim, D.; Iwamoto, T.; et al. Near-IR Absorbing Nickel(II) Porphyrinoids Prepared by Regioselective Insertion of Silylenes into Antiaromatic Nickel(II) Norcorrole. Angew. Chem. Int. Ed. 2014, 53, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Kosai, T.; Ishida, S.; Iwamoto, T. Transformation of azulenes to bicyclic [4]dendralene and heptafulvene derivatives via photochemical cycloaddition of dialkylsilylene. Chem. Commun. 2015, 51, 10707–10709. [Google Scholar] [CrossRef]
- Xiong, Y.; Yao, S.; Driess, M. Versatile Reactivity of a Zwitterionic Isolable Silylene toward Ketones: Silicon-Mediated, Regio- and Stereoselective C–H Activation. Chem. Eur. J. 2009, 15, 5545–5551. [Google Scholar] [CrossRef]
- Azhakar, R.; Ghadwal, R.S.; Roesky, H.W.; Mata, R.A.; Wolf, H.; Herbst-Irmer, R.; Stalke, D. Reaction of N-Heterocyclic Silylenes with Thioketone: Formation of Silicon–Sulfur Three (Si–C–S)- and Five (Si–C–C–C–S)-Membered Ring Systems. Chem. Eur. J. 2013, 19, 3715–3720. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]







Sample Availability: Sample of compound 4 is available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Morisako, S.; Aoyagi, S.; Sasamori, T. Generation of Bis(ferrocenyl)silylenes from Siliranes. Molecules 2020, 25, 5917. https://doi.org/10.3390/molecules25245917
Pan Y, Morisako S, Aoyagi S, Sasamori T. Generation of Bis(ferrocenyl)silylenes from Siliranes. Molecules. 2020; 25(24):5917. https://doi.org/10.3390/molecules25245917
Chicago/Turabian StylePan, Yang, Shogo Morisako, Shinobu Aoyagi, and Takahiro Sasamori. 2020. "Generation of Bis(ferrocenyl)silylenes from Siliranes" Molecules 25, no. 24: 5917. https://doi.org/10.3390/molecules25245917
APA StylePan, Y., Morisako, S., Aoyagi, S., & Sasamori, T. (2020). Generation of Bis(ferrocenyl)silylenes from Siliranes. Molecules, 25(24), 5917. https://doi.org/10.3390/molecules25245917