Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes


Abstract
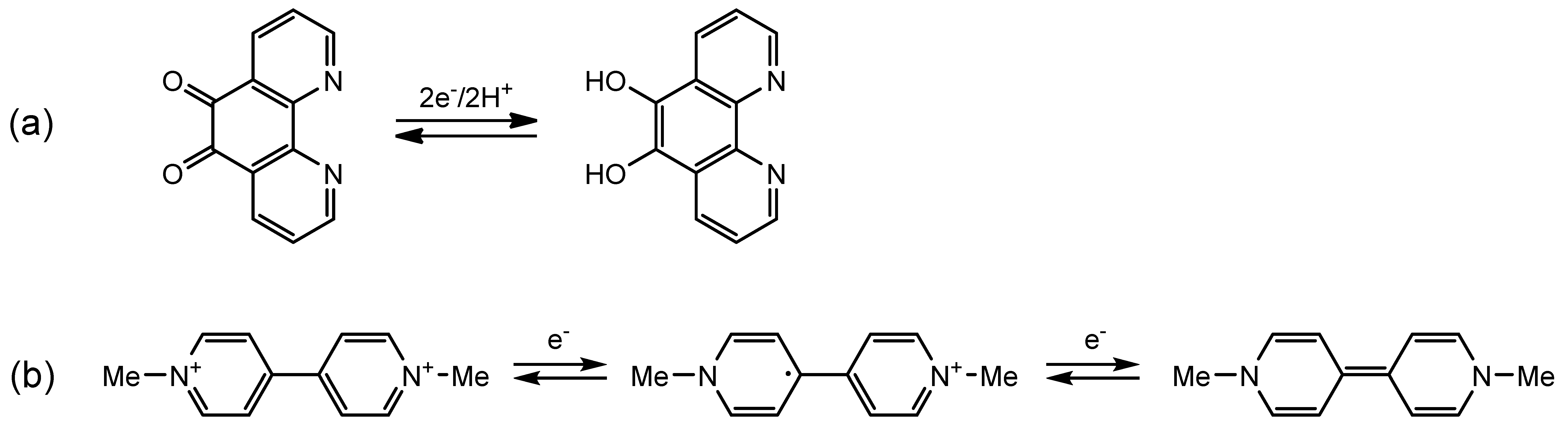
1. Introduction
2. Results and Discussion
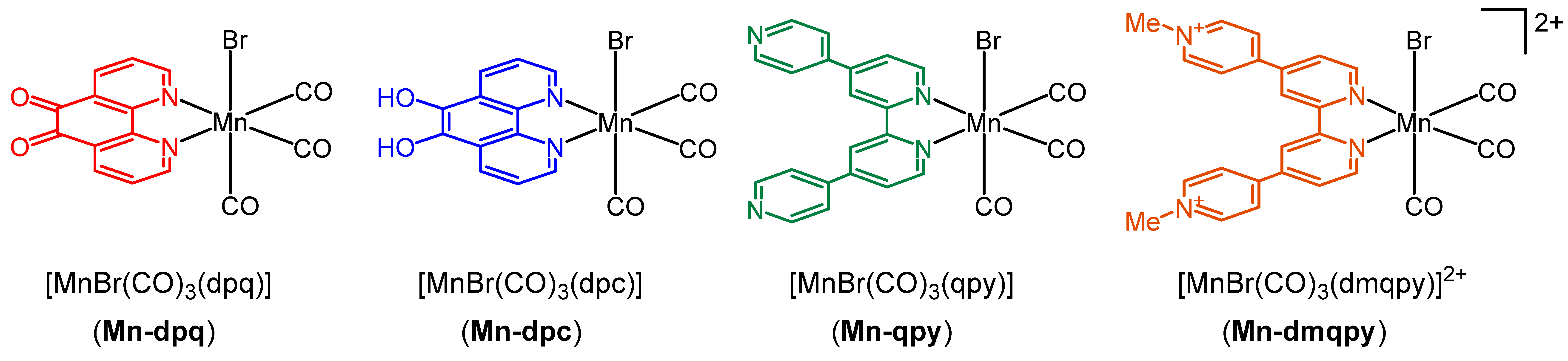
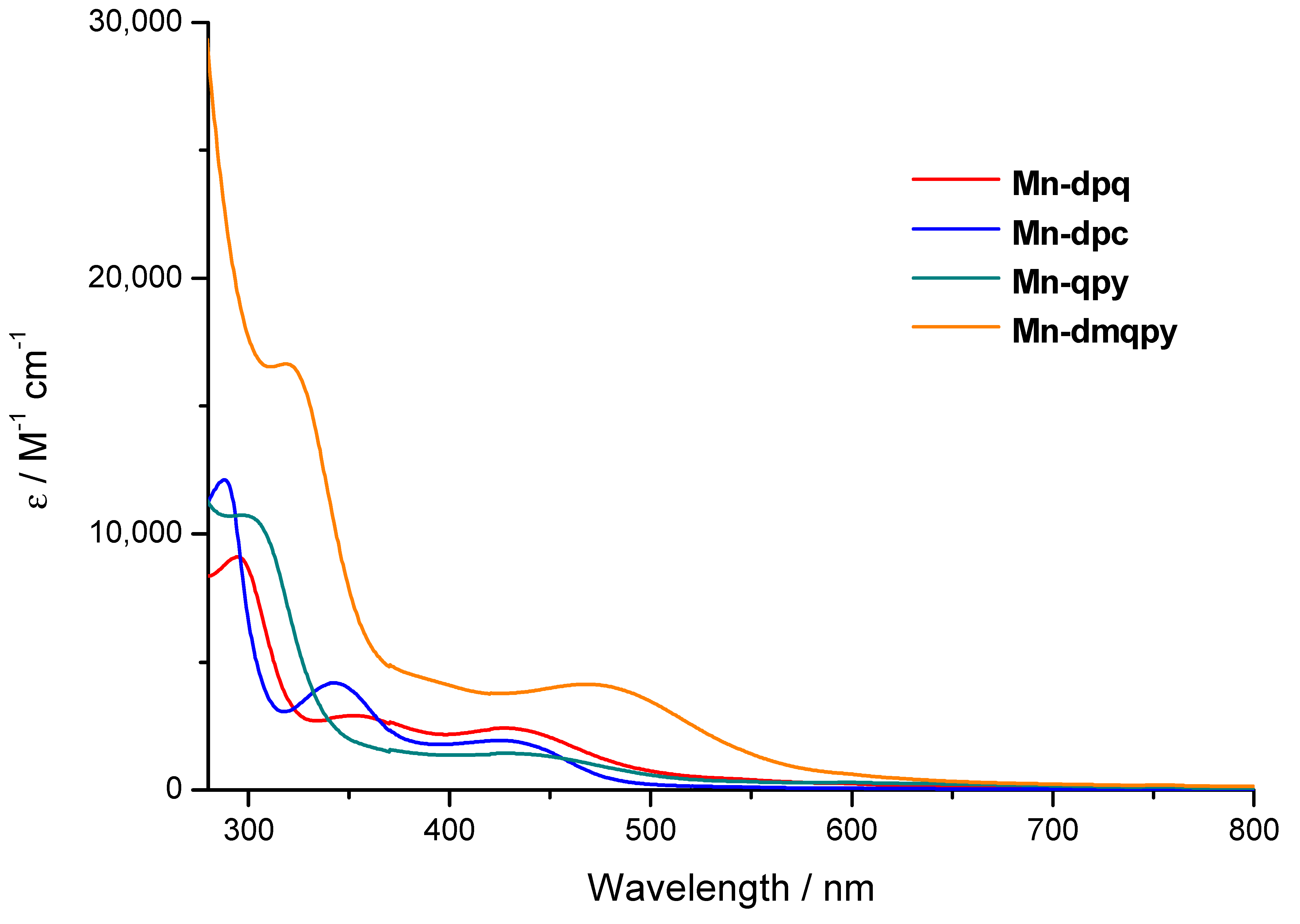
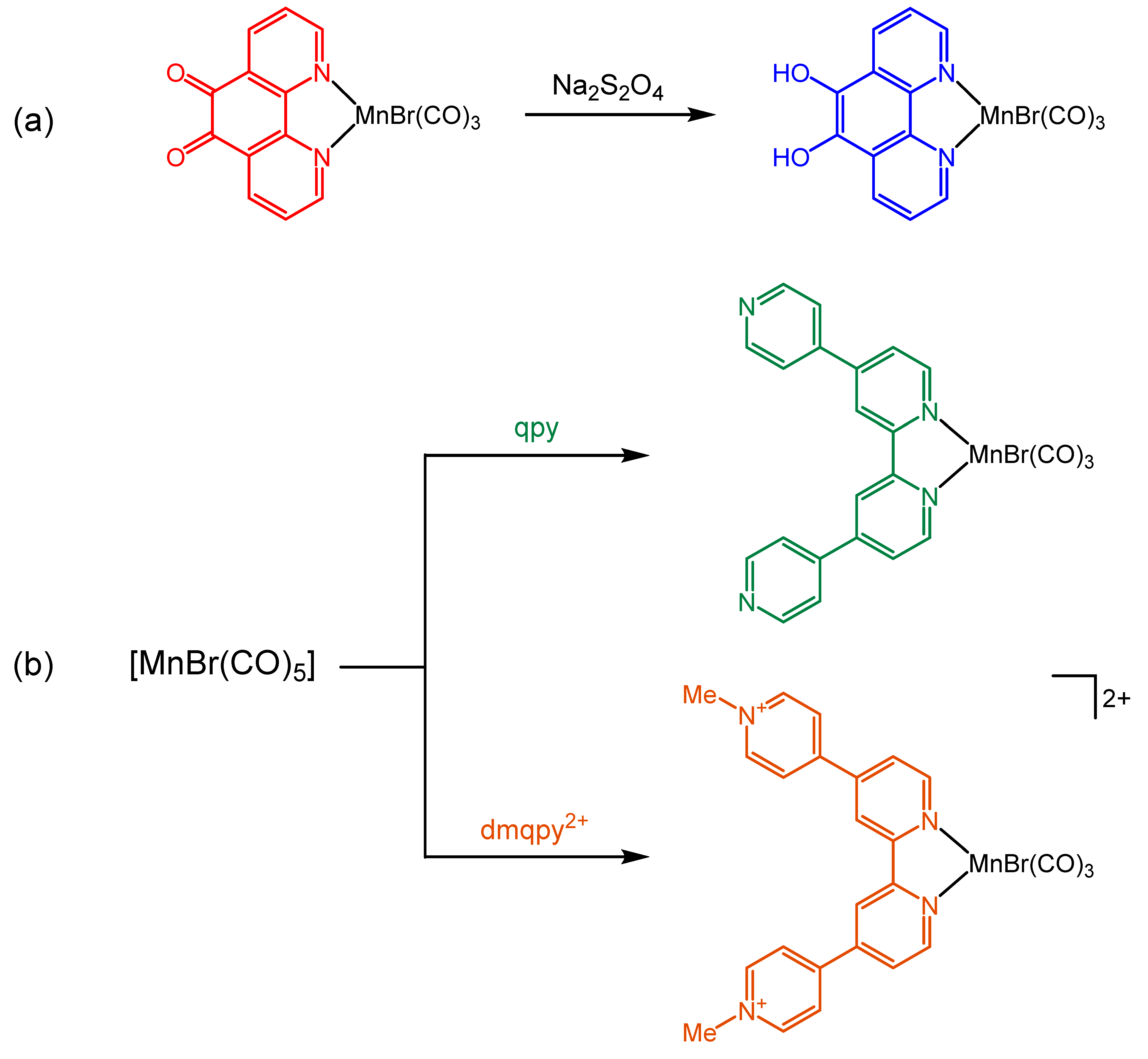
2.1. Synthesis and Spectroscopic Characterization of the Tricarbonylmanganese Complexes
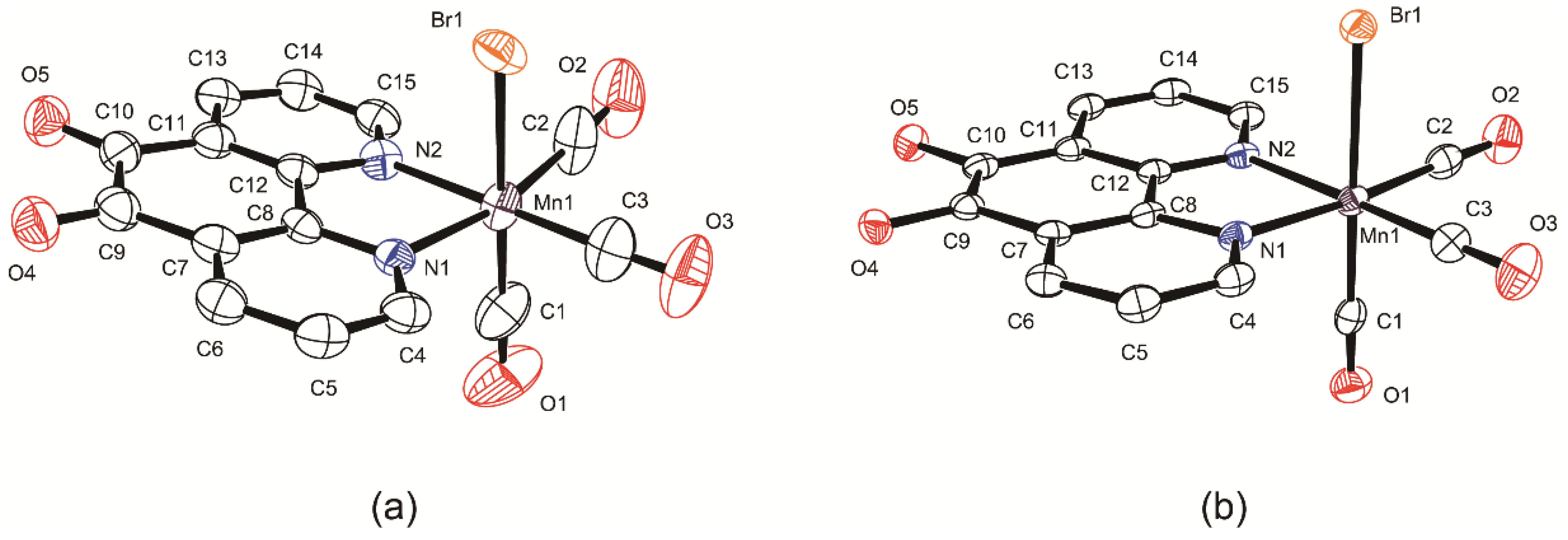
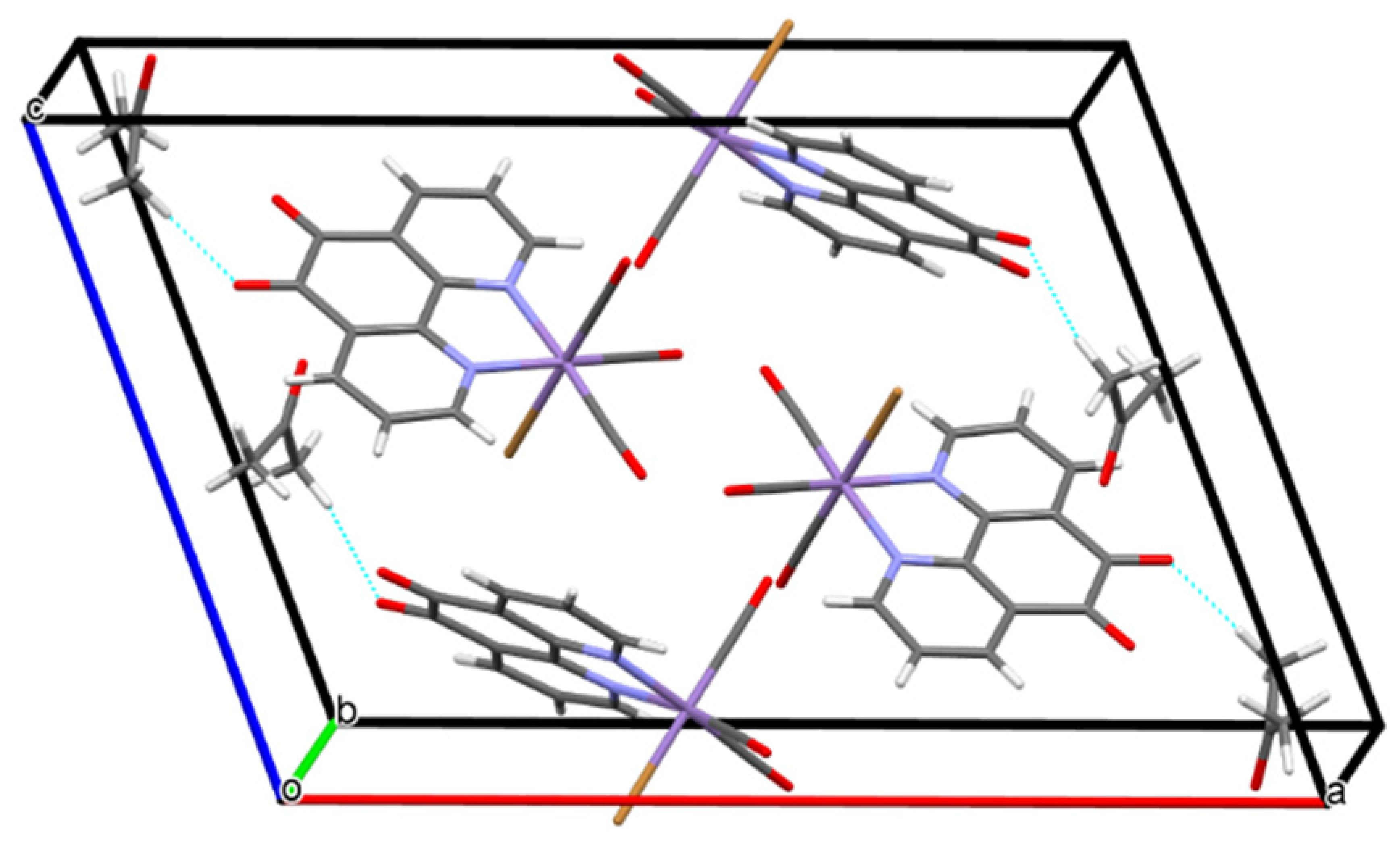
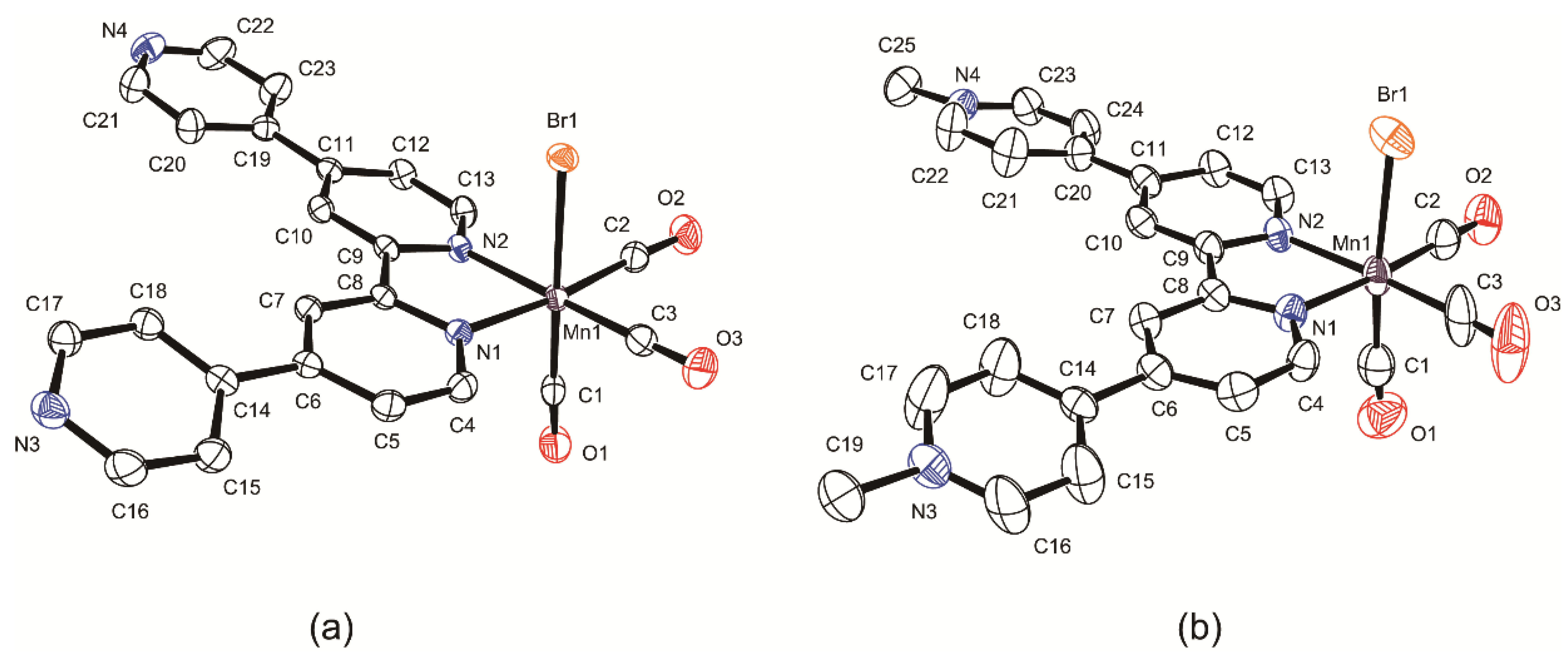
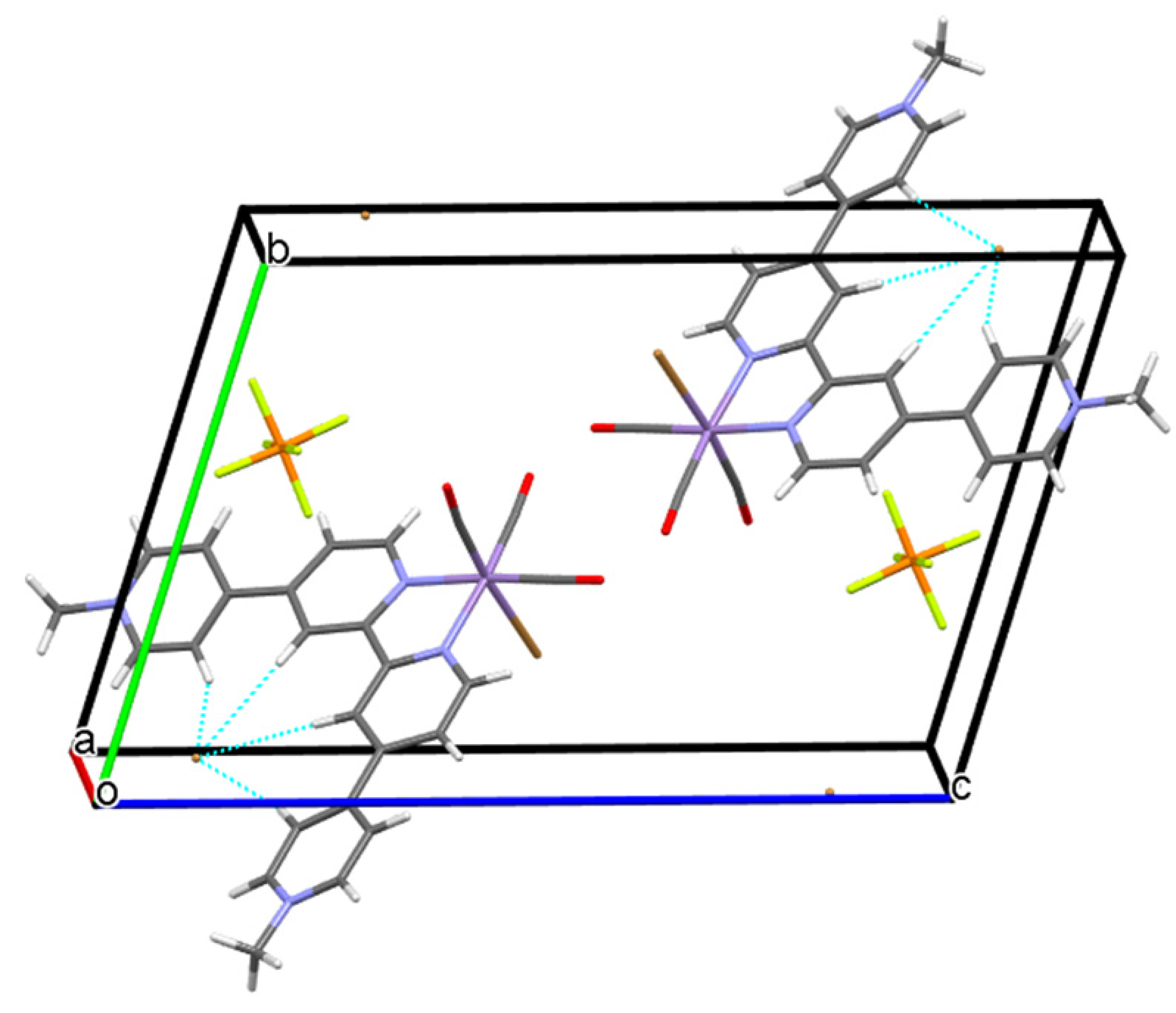
2.2. Structural Characterization of the Tricarbonylmanganese Complexes
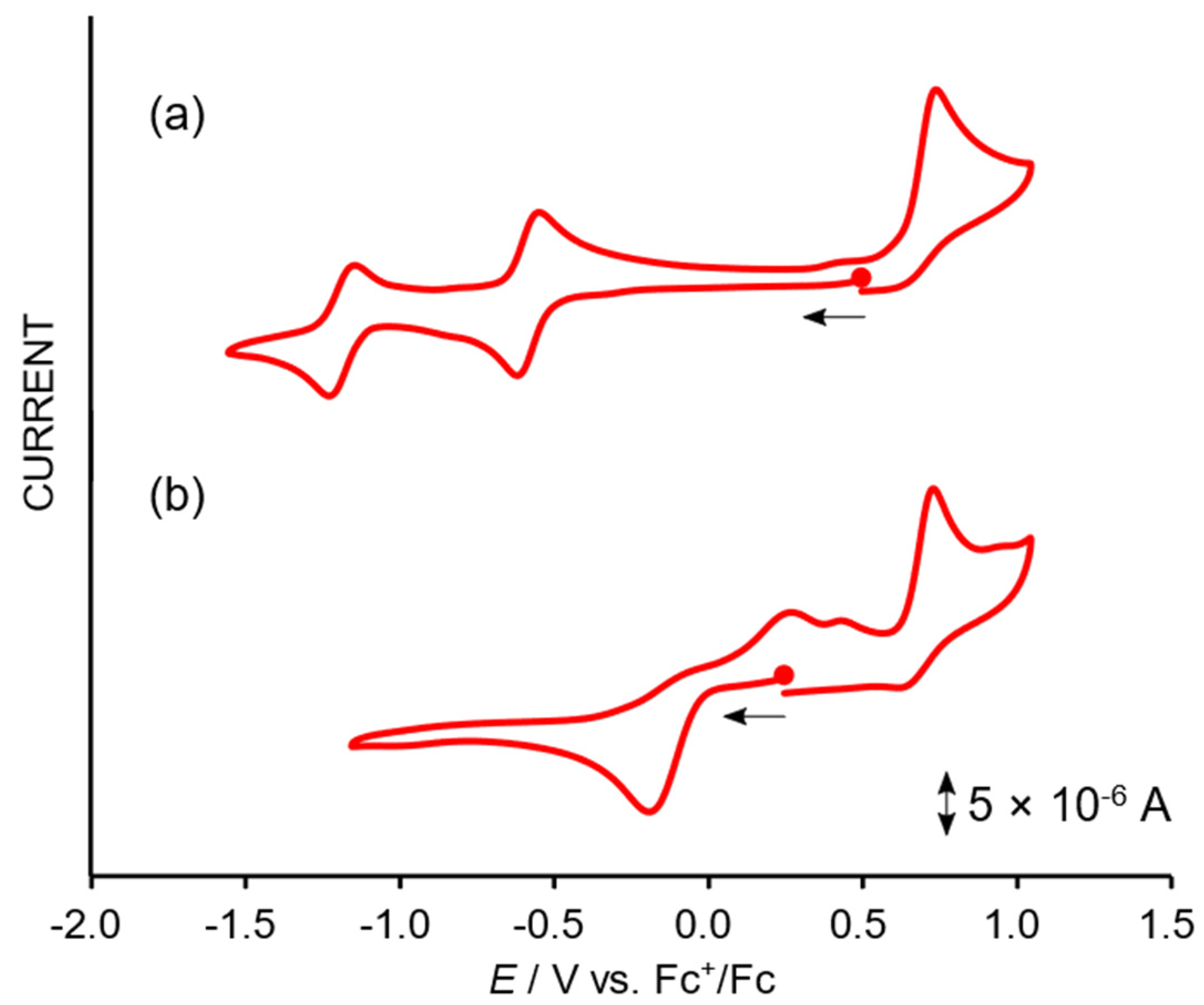
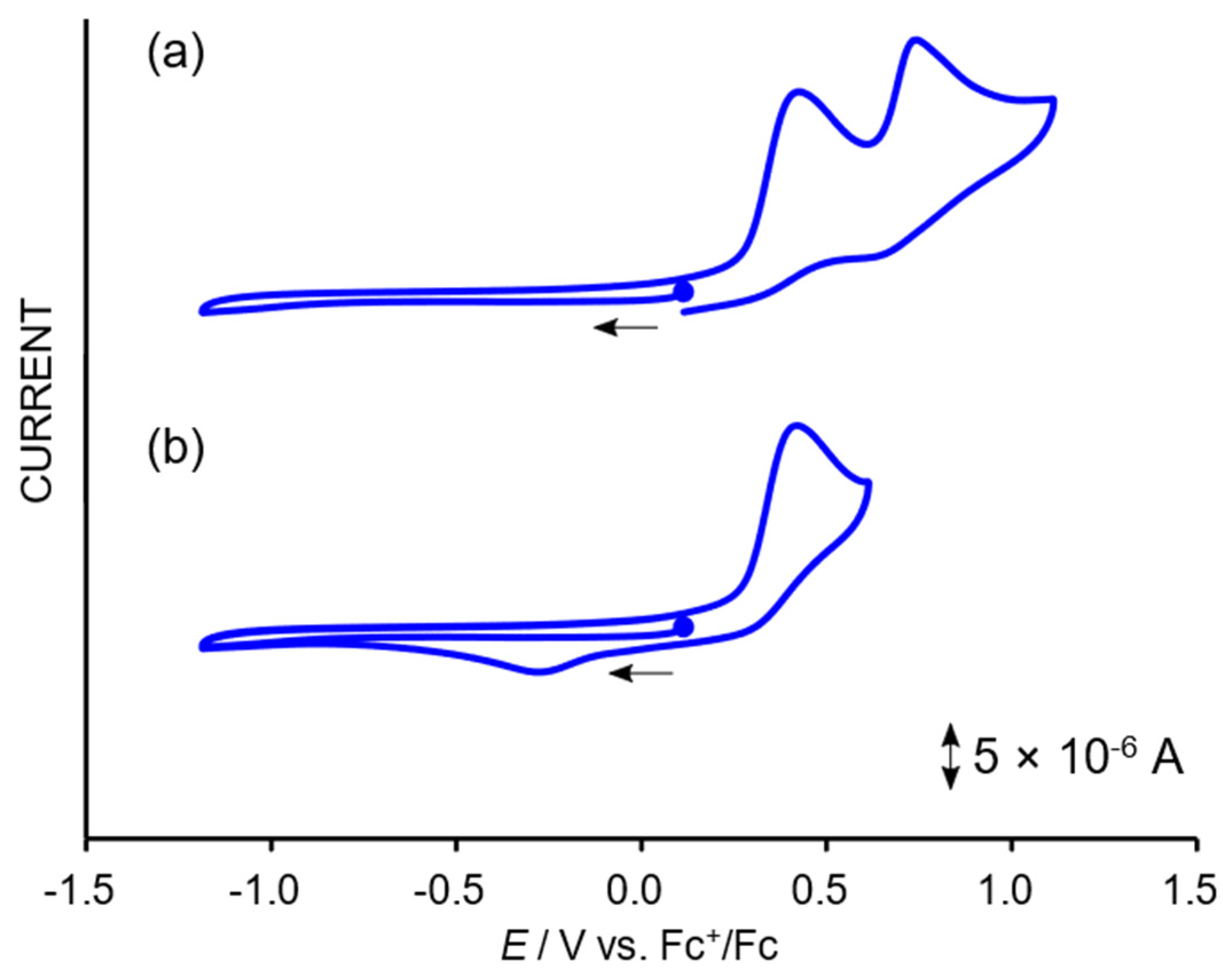
2.3. Redox Properties of the Tricarbonylmanganese Complexes
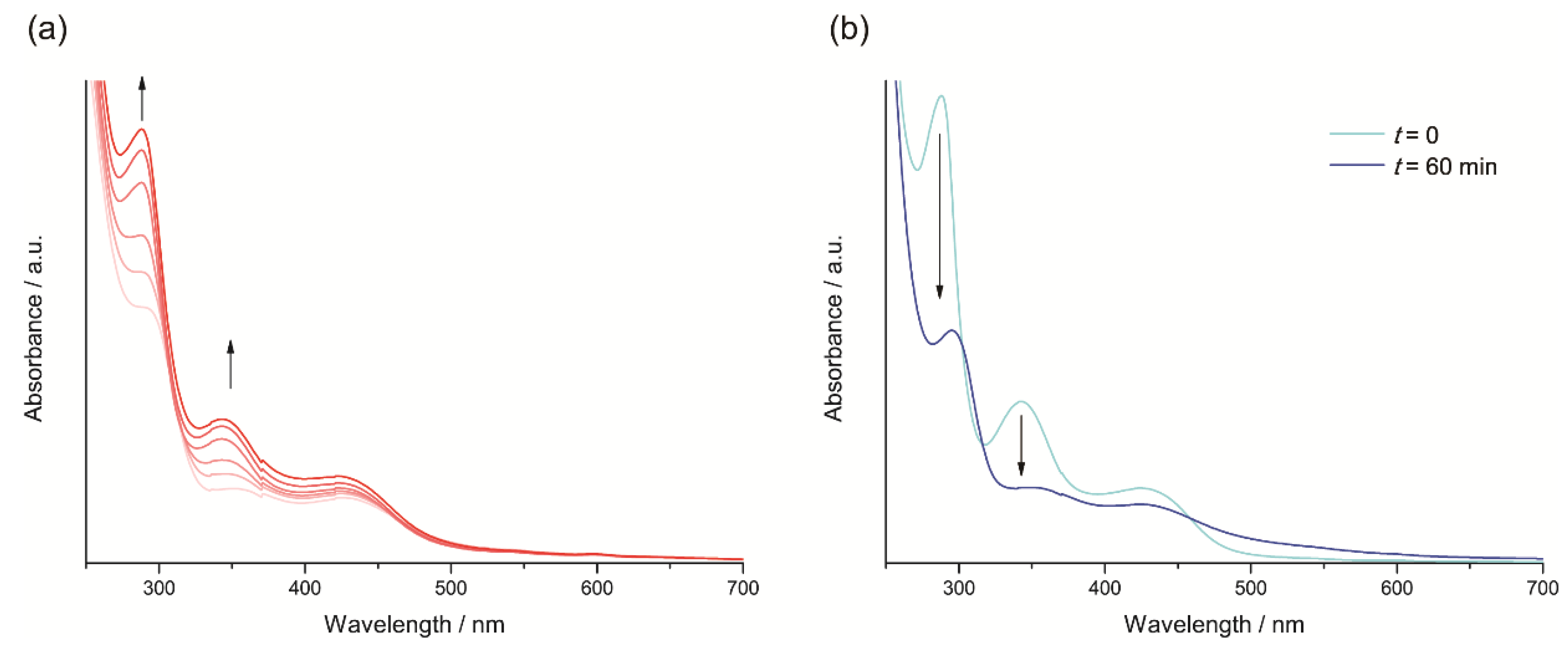
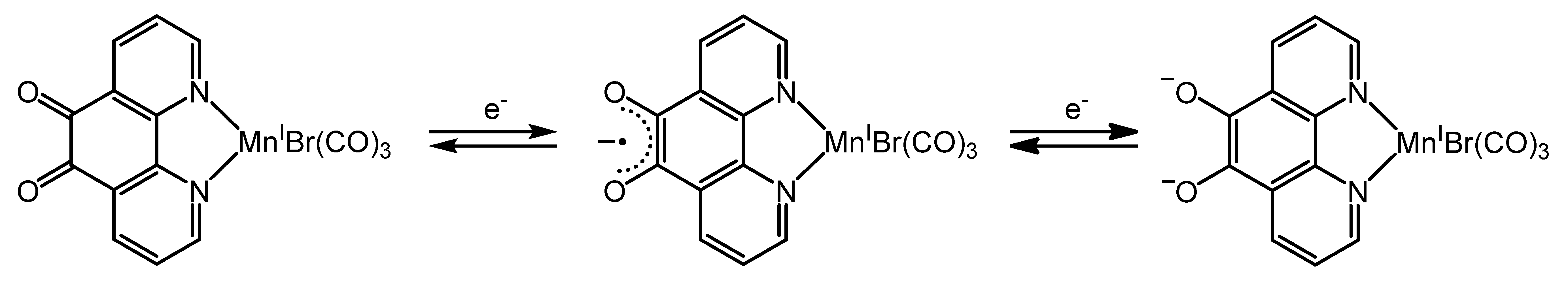
2.3.1. Phenanthroline-Based Complexes: Interconversion between Quinone and Catechol Units
2.3.2. Bipyridine-Based Complexes
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of the Complexes
3.2.1. Synthesis of fac-[MnBr(CO)3(dpc)]
3.2.2. Synthesis of fac-[MnBr(CO)3(qpy)] and fac-[MnBr(CO)3(dmqpy)](PF6)2
3.3. X-ray Crystallographic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef]
- Takeda, H.; Koizumi, H.; Okamoto, K.; Ishitani, O. Photocatalytic CO2 reduction using a Mn complex as a catalyst. Chem. Commun. 2014, 50, 1491–1493. [Google Scholar] [CrossRef]
- Torralba-Peñalver, E.; Luo, Y.; Compain, J.-D.; Chardon-Noblat, S.; Fabre, B. Selective catalytic electroreduction of CO2 at silicon nanowires (SiNWs) photocathodes using non-noble metal-based manganese carbonyl bipyridyl molecular catalysts in solution and grafted onto SiNWs. ACS Catal. 2015, 5, 6138–6147. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Electrocatalytic dihydrogen production by an earth-abundant manganese bipyridine catalyst. Inorg. Chem. 2015, 54, 6674–6676. [Google Scholar] [CrossRef]
- Fei, H.; Sampson, M.D.; Lee, Y.; Kubiak, C.P.; Cohen, S.M. Photocatalytic CO2 reduction to formate using a Mn(I) molecular catalyst in a robust metal-organic framework. Inorg. Chem. 2015, 54, 6821–6828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-X.; Hu, C.-Y.; Wang, W.; Wang, H.; Bian, Z.-Y. Visible light driven reduction of CO2 catalyzed by an abundant manganese catalyst with zinc porphyrin photosensitizer. Appl. Catal. A Gen. 2016, 522, 145–151. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Manganese electrocatalysts with bulky bipyridine ligands: Utilizing Lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Stanbury, M.; Compain, J.-D.; Trejo, M.; Smith, P.; Gouré, E.; Chardon-Noblat, S. Mn-carbonyl molecular catalysts containing a redox-active phenanthroline-5,6-dione for selective electro- and photoreduction of CO2 to CO or HCOOH. Electrochim. Acta 2017, 240, 288–299. [Google Scholar] [CrossRef]
- Morgan, R.J.; Baker, A.D. 2,2′:4,4′’:4′,4′’’-Quaterpyridyl: A building block for the preparation of novel redox reagents. 1. Preparation and quaternization. J. Org. Chem. 1990, 55, 1986–1993. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Eckert, T.S.; Bruice, T.C. Chemical properties of phenanthrolinequinones and the mechanism of amine oxidation by o-quinones of medium redox potentials. J. Am. Chem. Soc. 1983, 105, 4431–4441. [Google Scholar] [CrossRef]
- Wu, J.-Z.; Li, H.; Zhang, J.-G.; Xu, J.-H. Synthesis and DNA binding behavior of a dipyridocatecholate bridged dicopper(II) complex. Inorg. Chem. Commun. 2002, 5, 71–75. [Google Scholar] [CrossRef]
- Liu, Y.; Hammitt, R.; Lutterman, D.A.; Thummel, R.P.; Turro, C. Marked differences in light-switch behavior of Ru(II) complexes possessing a tridentate DNA intercalating ligand. Inorg. Chem. 2007, 46, 6011–6021. [Google Scholar] [CrossRef]
- Paw, W.; Eisenberg, R. Synthesis, characterization, and spectroscopy of dipyridocatecholate complexes of platinum. Inorg. Chem. 1997, 36, 2287–2293. [Google Scholar] [CrossRef]
- Ambroise, A.; Maiya, B.G. Ruthenium(II) complexes of 6,7-dicyanodipyridoquinoxaline: Synthesis, luminescence studies, and DNA interaction. Inorg. Chem. 2000, 39, 4264–4272. [Google Scholar] [CrossRef]
- Fujihara, T.; Okamura, R.; Wada, T.; Tanaka, K. Coordination ability of 1,10-phenanthroline-5,6-dione: Syntheses and redox behavior of a Ru(II) complex with an o-quinoid moiety and of bridged Ru(II)–M(II) complexes (M = Pd, Pt). Dalton Trans. 2003, 3221–3226. [Google Scholar] [CrossRef]
- We could not observe signals derived from the OH groups of the catechol units even in DMSO-d6.
- Shi, A.; Pokhrel, M.R.; Bossmann, S.H. Synthesis of highly charged ruthenium(II)-quaterpyridinium complexes: A bottom-up approach to monodisperse nanostructures. Synthesis 2007, 4, 505–514. [Google Scholar]
- Coe, B.J.; Harper, E.C.; Helliwell, M.; Ta, Y.T. Syntheses and properties of complexes with bis(2,2′-bipyridyl)ruthenium(II) moieties coordinated to 4,4′:2′,2′’:4′’,4′’’-quaterpyridinium ligands. Polyhedron 2011, 30, 1830–1841. [Google Scholar] [CrossRef]
- Kurtz, D.A.; Dhakal, B.; Hulme, R.J.; Nichol, G.S.; Felton, G.A.N. Correlations between photophysical and electrochemical properties for a series of new Mn carbonyl complexes containing substituted phenanthroline ligands. Inorg. Chim. Acta 2015, 427, 22–26. [Google Scholar] [CrossRef]
- Henke, W.C.; Otolski, C.J.; Moore, W.N.G.; Elles, C.G.; Blakemore, J.D. Ultrafast spectroscopy of [Mn(CO)3] complexes: Tuning the kinetics of light-driven CO release and solvent binding. Inorg. Chem. 2020, 59, 2178–2187. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Chabolla, S.A.; Dang, T.; Kubiak, C.P. Developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 2014, 33, 4550–4559. [Google Scholar] [CrossRef]
- Jimenez, J.; Chakraborty, I.; Mascharak, P.K. Synthesis and assessment of CO-release capacity of manganese carbonyl complexes derived from rigid α-diimine ligands of varied complexity. Eur. J. Inorg. Chem. 2015, 30, 5021–5026. [Google Scholar] [CrossRef]
- Lense, S.; Guzei, I.A.; Andersen, J.; Thao, K.C. Crystal structures of a manganese(I) and a rhenium(I) complex of a bipyridine ligand with a non-coordinating benzoic acid moiety. Acta Cryst. 2018, E74, 731–736. [Google Scholar] [CrossRef]
- Walsh, J.J.; Smith, C.L.; Neri, G.; Whitehead, G.F.S.; Robertson, C.M.; Cowan, A.J. Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 2015, 183, 147–160. [Google Scholar] [CrossRef]
- Wadayama, K.; Takase, T.; Oyama, D. fac-Bromido/chlorido(0.50/0.50)[3-carbamoyl-1-(1,10-phenanthrolin-2-ylmethyl)pyridinium-κ2N,N’]tricarbonylmanganese(I) 0.49-bromide 0.51-chloride methanol monosolvate. IUCrData 2019, 4, x181792. [Google Scholar] [CrossRef]
- Kanno, T.; Takase, T.; Oyama, D. Synthesis and crystal structures of manganese(I) carbonyl complexes bearing ester-substituted α-diimine ligands. Acta Cryst. 2020, E76, 1433–1436. [Google Scholar]
- Calderazzo, F.; Marchetti, F.; Pampaloni, G.; Passarelli, V. Co-ordination properties of 1,10-phenanthroline-5,6-dione towards group 4 and 5 metals in low and high oxidation states. J. Chem. Soc. Dalton Trans. 1999, 4389–4396. [Google Scholar] [CrossRef]
- Lin, X.-Y.; Tang, S.-J.; Wu, W.-S. 5,6-Dihydroxy-1,10-phenanthrolin-1-ium chloride dihydrate. Acta Cryst. 2009, E65, o2367. [Google Scholar] [CrossRef] [PubMed]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- When Mn-dpq was measured with a Pt working electrode, only an oxidation wave was observed at 0.43 V. Thus, the electrode reactions taking place under these conditions may depend on the material employed (i.e., carbon or platinum); Lei, Y.; Anson, F.C. Hydration of the carbonyl groups in 1,10-phenanthroline-5,6-dione induced by binding protons or metal cations to the pyridine nitrogen sites. J. Am. Chem. Soc. 1995, 117, 9849–9854.
- We could not carry out CV measurements for dpc due to its low solubility in almost all solvents.
- Stallings, M.D.; Morrison, M.M.; Sawyer, D.T. Redox chemistry of metal-catechol complexes in aprotic media. 1. Electrochemistry of substituted catechols and their oxidation products. Inorg. Chem. 1981, 20, 2655–2660. [Google Scholar] [CrossRef]
- Pordel, S.; White, J.K. Impact of Mn(I) photoCORM ligand set on photochemical intermediate formation during visible light-activated CO release. Inorg. Chim. Acta 2020, 500, 119206. [Google Scholar] [CrossRef]
- A newly emerged oxidation wave (0.35 V) accompanying the reductions was assignable to the oxidation of Br– since another CV measurement using n-Bu4NBr showed an oxidation wave at 0.34 V under identical conditions. Therefore, the electrochemical results strongly suggest dissociation of the Br ligand and the following Mn-Mn dimerization.
- Yamada, M.; Tanaka, Y.; Yoshimoto, Y.; Kuroda, S.; Shimao, I. Synthesis and properties of diamino-substituted dipyrido [3,2-a:2′,3′-c]phenazine. Bull. Chem. Soc. Jpn. 1992, 65, 1006–1011. [Google Scholar] [CrossRef]
- Chakraborty, I.; Carrington, S.J.; Mascharak, P.K. Photodelivery of CO by designed photoCORMs: Correlation between absorption in the visible region and metal–CO bond labilization in carbonyl complexes. ChemMedChem 2014, 9, 1266–1274. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- We could not obtain any satisfactory results in the elemental analysis of Mn-dpc, due to its incombustibility.
- This is a service of the International Union of Crystallography (IUCr).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
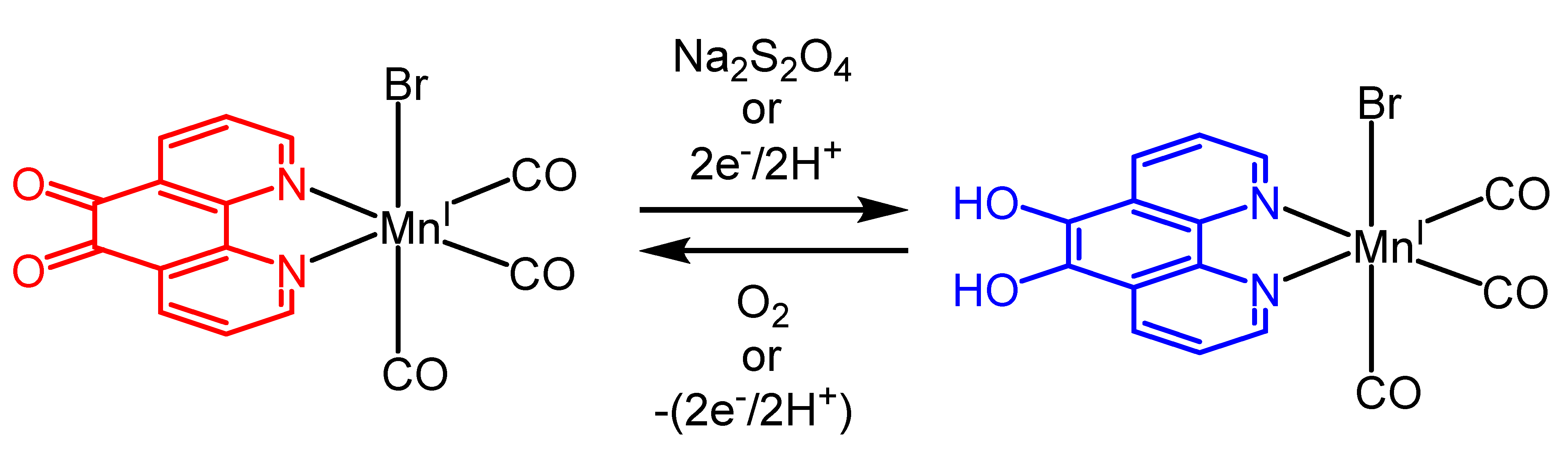{kind=link}
| Technique | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy |
|---|---|---|---|---|
| IR (νCO/cm−1) | 2033 | 2032 | 2027 | 2029 |
| 1944 1928 1698 | 1961 1938 | 1936 1914 | 1944 1927 | |
| UV-vis (λmax/nm) | 427 (2.4) | 425 (1.9) | 427 (1.4) | 468 (4.1) |
| (ε/×103 M−1cm−1) | 352 (2.9) 294 (9.0) | 343 (4.1) 288 (12) | 297 (11) | 319 (17) |
| Parameter | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy | ||||
|---|---|---|---|---|---|---|---|---|
| Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | |
| Mn1-C1 | 1.799(5) | 1.801 | 1.812(2) | 1.797 | 1.813(2) | 1.797 | 1.806(8) | 1.801 |
| Mn1-C2 | 1.817(7) | 1.815 | 1.804(2) | 1.813 | 1.8123(19) | 1.814 | 1.829(8) | 1.817 |
| Mn1-C3 | 1.810(7) | 1.815 | 1.809(2) | 1.813 | 1.8054(17) | 1.815 | 1.834(12) | 1.817 |
| Mn1-N1 | 2.054(4) | 2.077 | 2.0452(17) | 2.080 | 2.0344(15) | 2.066 | 2.051(6) | 2.063 |
| Mn1-N2 | 2.049(4) | 2.077 | 2.0614(19) | 2.080 | 2.0369(12) | 2.065 | 2.045(7) | 2.063 |
| Mn1-Br1 | 2.5385(7) | 2.599 | 2.5628(10) | 2.610 | 2.5250(4) | 2.610 | 2.5617(12) | 2.600 |
| C1-O1 | 1.133(7) | 1.156 | 1.121(3) | 1.157 | 1.120(3) | 1.156 | 1.124(11) | 1.155 |
| C2-O2 | 1.145(9) | 1.154 | 1.141(3) | 1.155 | 1.142(2) | 1.155 | 1.140(11) | 1.154 |
| C3-O3 | 1.151(8) | 1.154 | 1.143(3) | 1.155 | 1.145(2) | 1.155 | 1.091(16) | 1.154 |
| C9-O4 | 1.212(6) | 1.217 | 1.362(2) | 1.353 | ||||
| C10-O5 | 1.220(7) | 1.217 | 1.367(2) | 1.353 | ||||
| Mn1-C1-O1 | 177.5(6) | 179.9 | 178.53(19) | 179.9 | 176.81(17) | 179.9 | 173.1(9) | 180.0 |
| Mn1-C2-O2 | 177.5(6) | 178.9 | 177.09(18) | 178.9 | 176.06(15) | 178.9 | 178.6(7) | 178.8 |
| Mn1-C3-O3 | 176.5(5) | 178.8 | 177.56(18) | 178.9 | 177.8(2) | 178.9 | 176.5(9) | 178.8 |
| R1/R3 1 | 15.52(7) | 30.58 | 16.8(3) | 35.56 | ||||
| R2/R4 2 | 4.63(7) | 30.88 | 12.0(3) | 35.65 | ||||
| Reaction (V vs. Fc+/Fc) | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy |
|---|---|---|---|---|
| Reductions (ΔE) | −0.58 1 (0.07) | −0.28 3,4 | −1.48 3 | −1.14 1 (0.06) |
| −1.19 1 (0.08) | −1.56 3 | −1.36 3 | ||
| −1.78 1 (0.08) | ||||
| −2.20 1 (0.09) | ||||
| Oxidations | 0.74 2 | 0.43 2 | −1.26 2 | −1.34 2 |
| 0.74 2 | −0.42 2,4 | 0.34 2,5 | ||
| 0.35 2,4 |
| Parameter | Mn-dpq [MnBr(CO)3(dpq)](CH3)2CO | Mn-dpc [MnBr(CO)3(dpc)] | Mn-qpy [MnBr(CO)3(qpy)] | Mn-dmqpy [MnBr(CO)3(dmqpy)]BrPF6 |
|---|---|---|---|---|
| Formula | C18H12BrMnN2O6 | C15H8BrMnN2O5 | C23H14BrMnN4O3 | C25H20Br2F6MnN4O3P |
| Formula weight | 487.14 | 431.08 | 529.23 | 784.17 |
| Temperature (K) | 93 | 93 | 93 | 93 |
| Crystal system | monoclinic | triclinic | monoclinic | triclinic |
| Space group | P21/c | P-1 | P21/c | P-1 |
| a (Å) | 20.3113(11) | 7.077(3) | 11.8530(2) | 6.4688(3) |
| b (Å) | 6.9096(3) | 9.759(4) | 18.0938(3) | 12.4896(6) |
| c (Å) | 14.1250(7) | 11.600(5) | 10.53500(19) | 18.8132(10) |
| α (°) | 90 | 91.966(4) | 90 | 72.4654(14) |
| β (°) | 108.8940(14) | 107.572(3) | 111.2740(7) | 89.3644(14) |
| γ (°) | 90 | 104.311(5) | 90 | 82.1867(13) |
| V (Å3) | 1875.54(16) | 734.9(6) | 2105.43(7) | 1435.18(12) |
| Z | 4 | 2 | 4 | 2 |
| Calcd density (g/cm3) | 1.725 | 1.948 | 1.669 | 1.814 |
| μ (Mo Kα) (mm−1) | 2.879 | 3.655 | 2.564 | 3.383 |
| No. unique reflns | 18452 | 7485 | 21537 | 14809 |
| No. obsd reflns | 4280 | 3303 | 4815 | 6510 |
| Refinement method | Full-matrix least squares on F2 | |||
| Parameters | 255 | 217 | 289 | 379 |
| R (I > 2σ(I)) 1 | 0.0572 | 0.0247 | 0.0254 | 0.0776 |
| wR (all data) 2 | 0.1480 | 0.0667 | 0.0642 | 0.2171 |
| S | 1.042 | 1.065 | 1.072 | 1.025 |
Sample Availability: Samples of the compounds Mn-dpq, Mn-dpc, Mn-qpy, and Mn-dmqpy are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, T.; Takase, T.; Oyama, D. Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules 2020, 25, 5921. https://doi.org/10.3390/molecules25245921
Kanno T, Takase T, Oyama D. Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules. 2020; 25(24):5921. https://doi.org/10.3390/molecules25245921
Chicago/Turabian StyleKanno, Takatoshi, Tsugiko Takase, and Dai Oyama. 2020. "Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes" Molecules 25, no. 24: 5921. https://doi.org/10.3390/molecules25245921
APA StyleKanno, T., Takase, T., & Oyama, D. (2020). Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules, 25(24), 5921. https://doi.org/10.3390/molecules25245921