
Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments




Abstract
1. Introduction
2. Basic Principles of the Scattering Techniques
3. Small Angle Scattering of Neutrons (SANS) and X-rays (SAXS)
3.1. SAXS and SANS Experimental Approaches and Technical Features
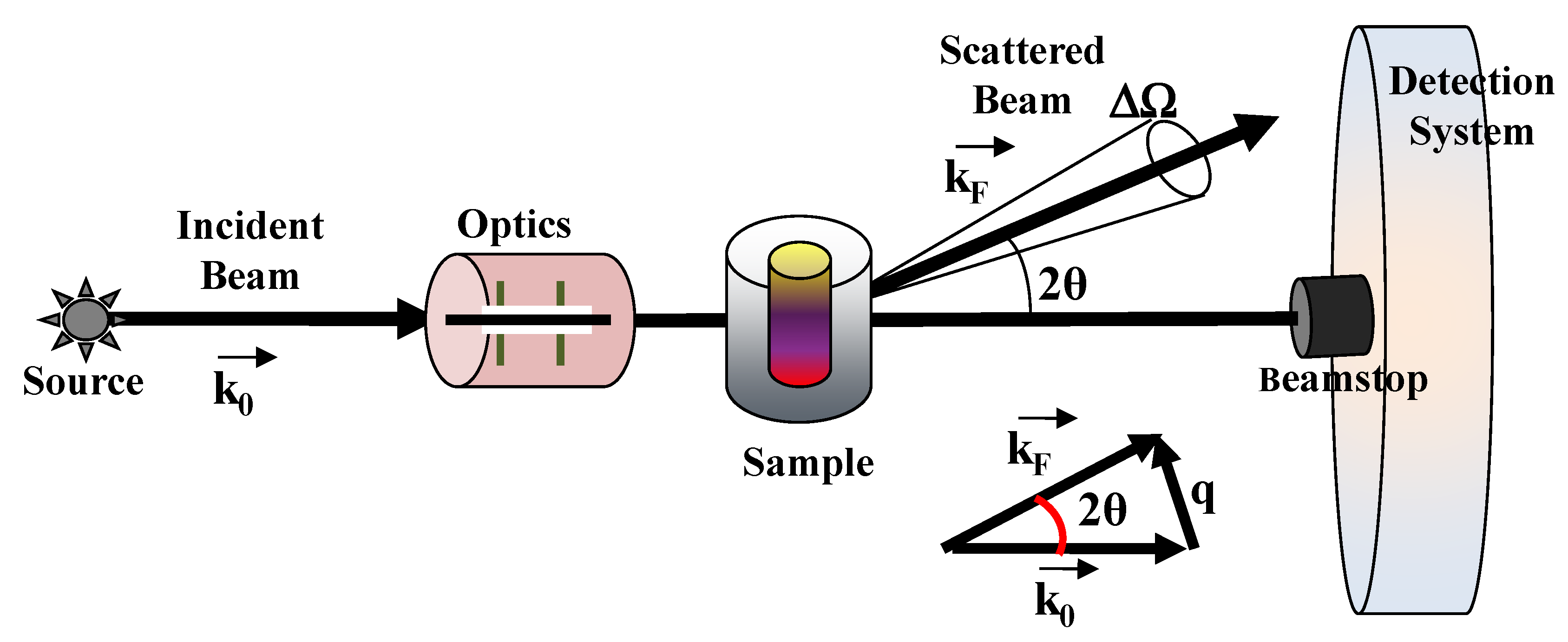
3.1.1. Experimental Setup and Preliminary Data Treatment
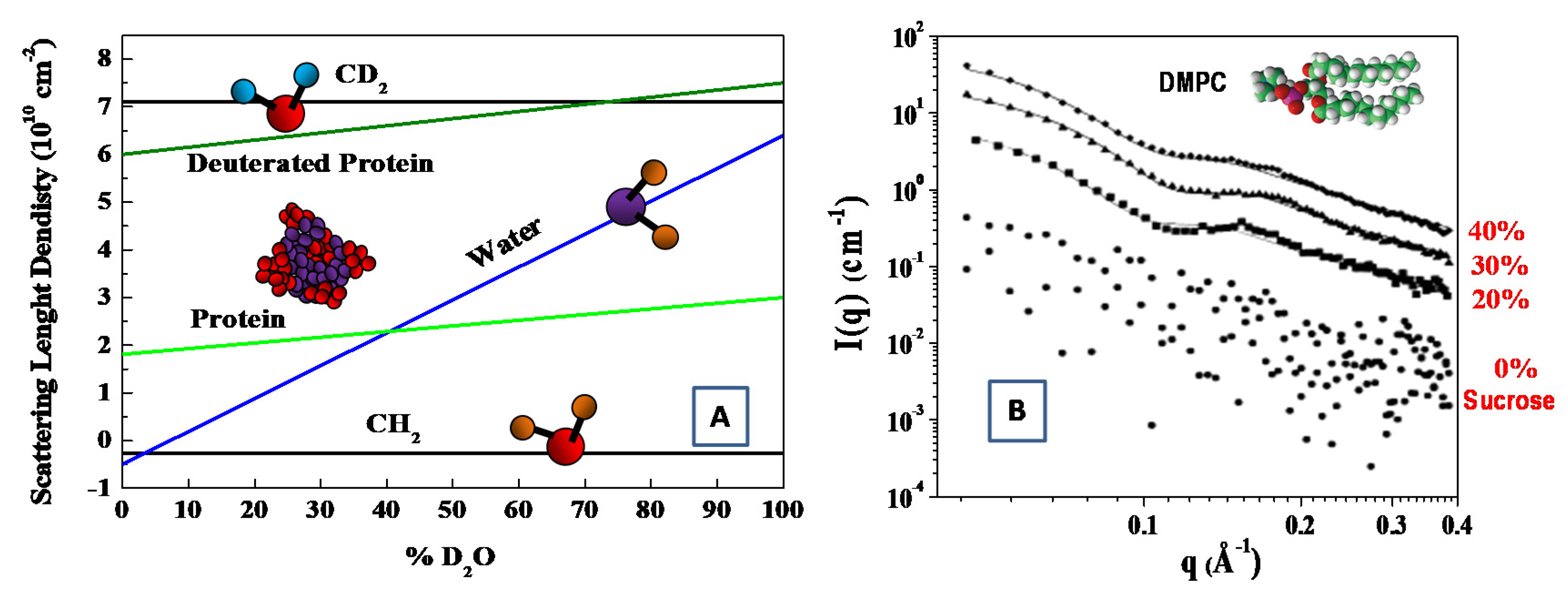
3.1.2. Atomic Scattering Length and Scattering Length Density
3.1.3. The Contrast Variation Method
3.1.4. Complementary Aspects of SAXS and SANS Techniques
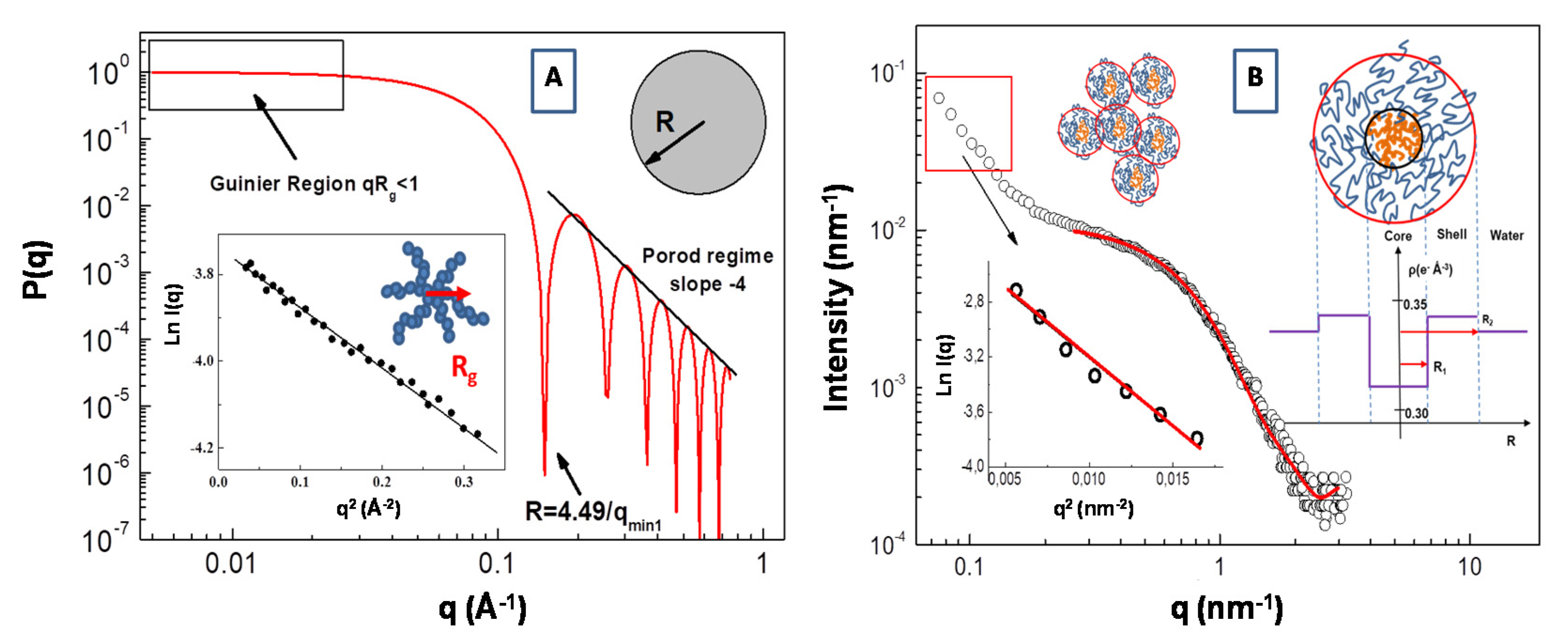
3.2. Form Factor P(Q) Analysis
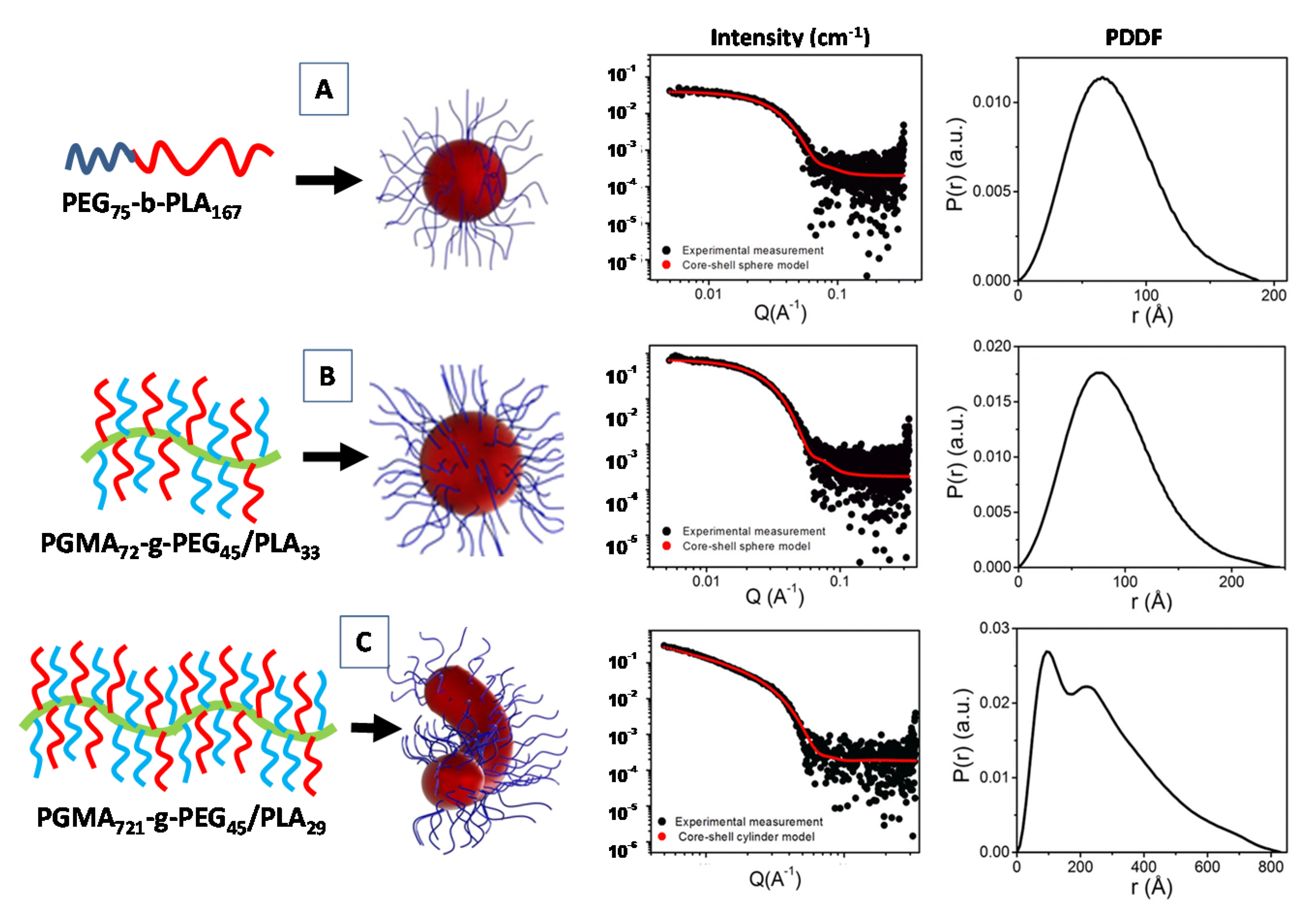
3.3. Indirect Fourier Transform Method: Pair Distance Distribution Function (PDDF)
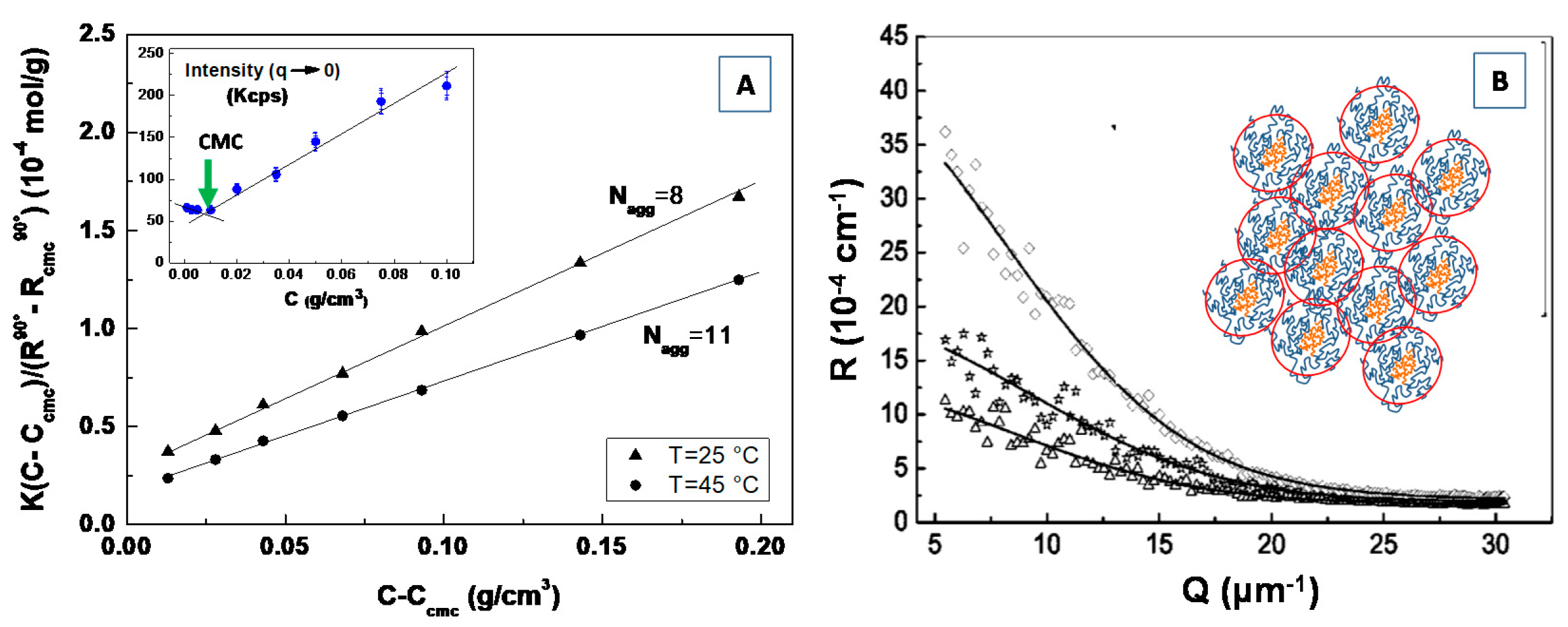
3.4. Study of the Self-Assembly Processes, Phase Transitions Mechanisms and Aggregation Kinetics
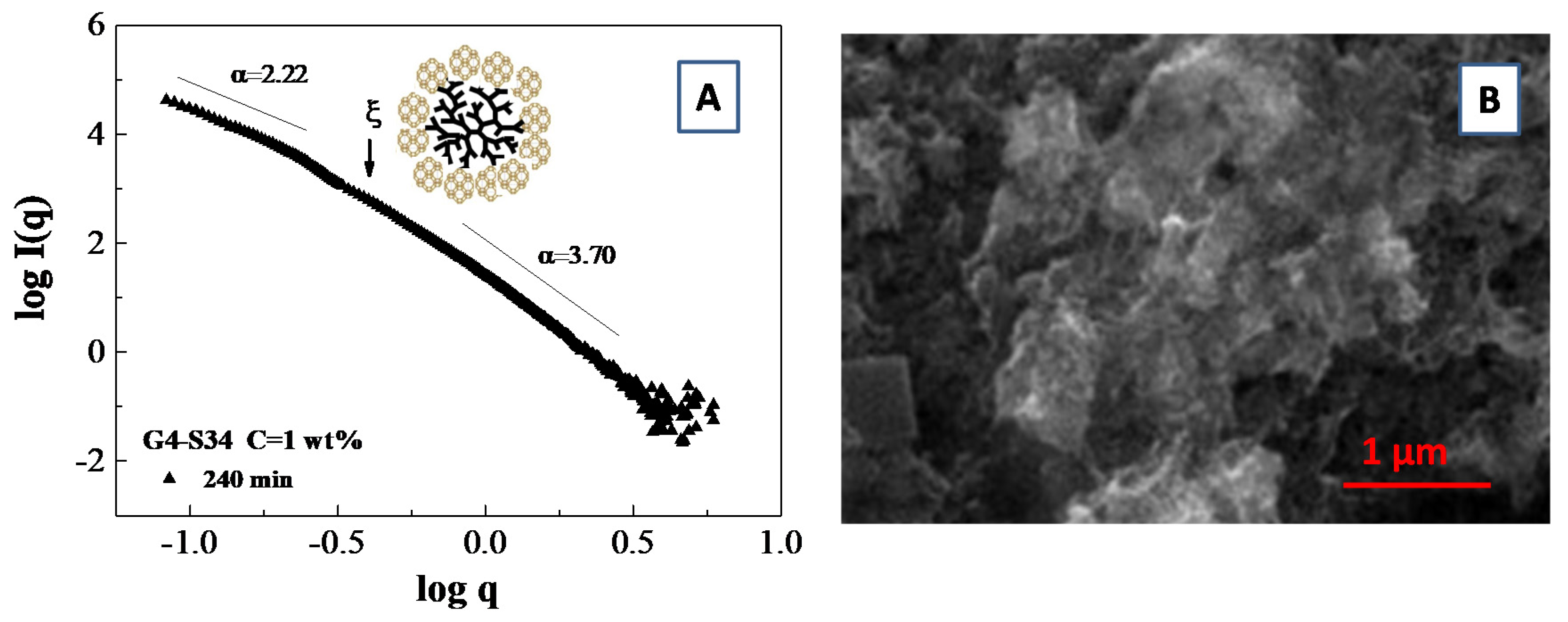
3.5. Scattering from Fractal Aggregate: Porod Limit
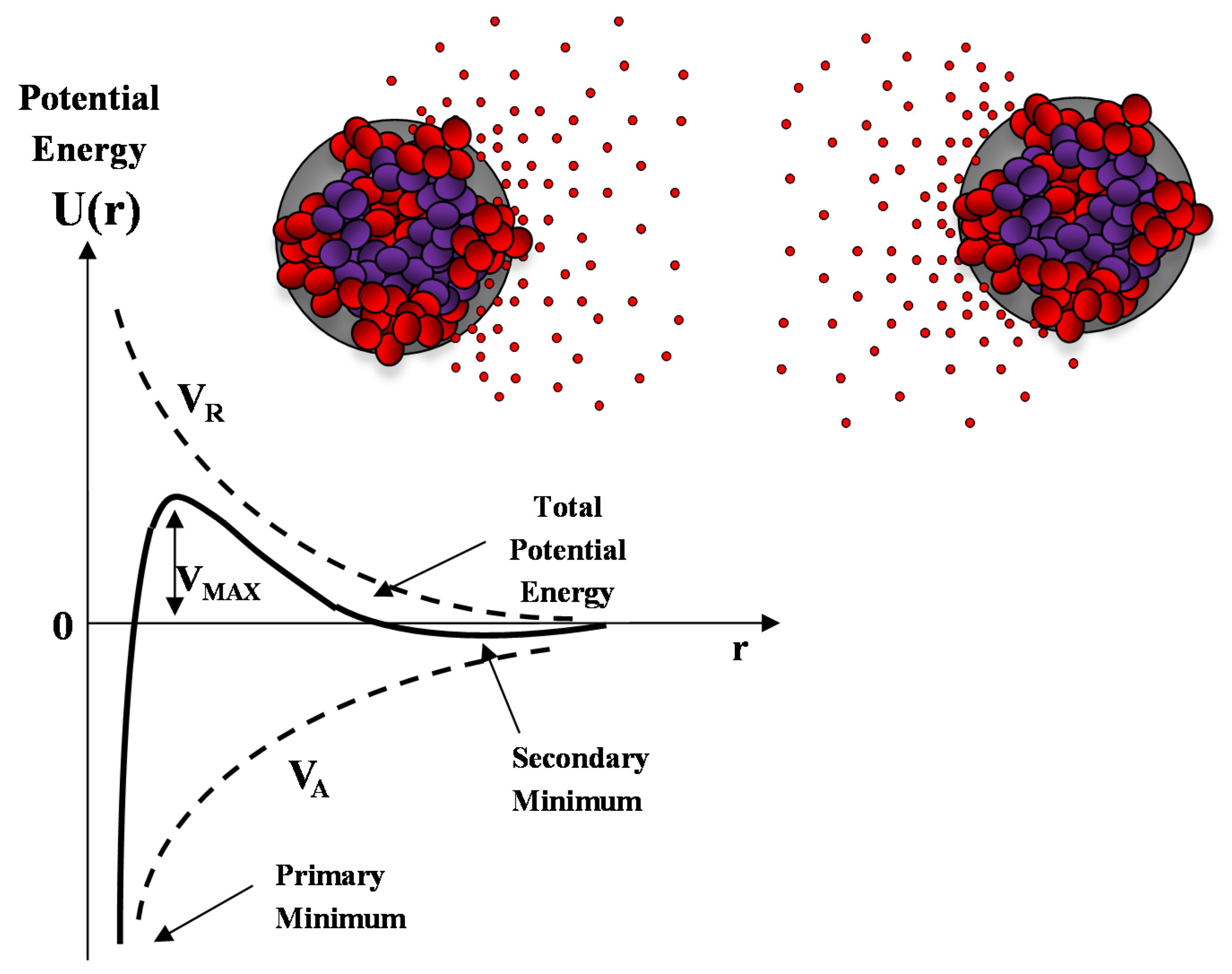
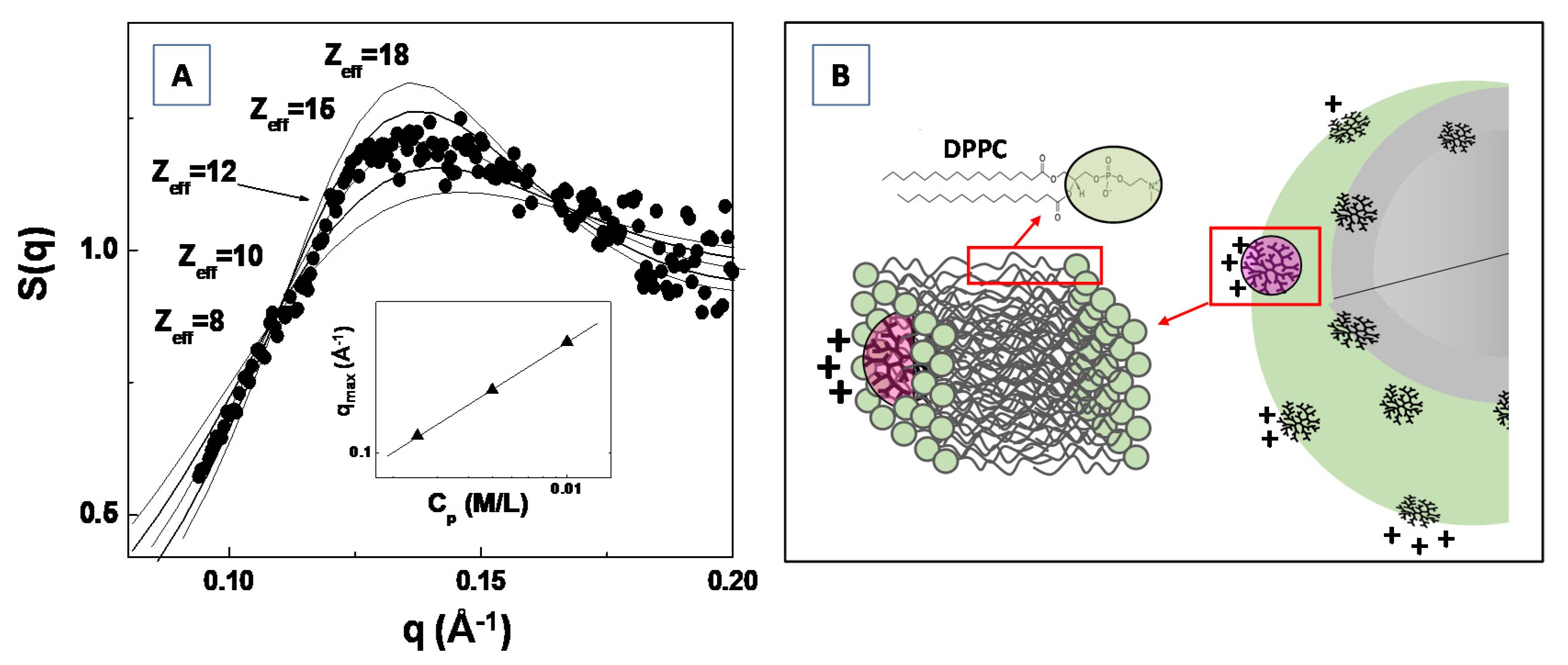
3.6. Analysis of the Structure Factor S(q): Study of the Interpaticles Interaction
3.7. SAS Investigation of Structure-Function Relationship in Biomolecules and Biomaterials
3.7.1. Drug–Biomembranes Interaction
3.7.2. Conformational Changes in Flexible Biomolecules: Ensemble Fitting of the SAS Data
3.7.3. SAS Structural Investigation of Bio-Polymers and Hydrogels
4. Light Scattering Techniques
4.1. Static Light Scattering (SLS) and Small Angle Light Scattering (SALS)
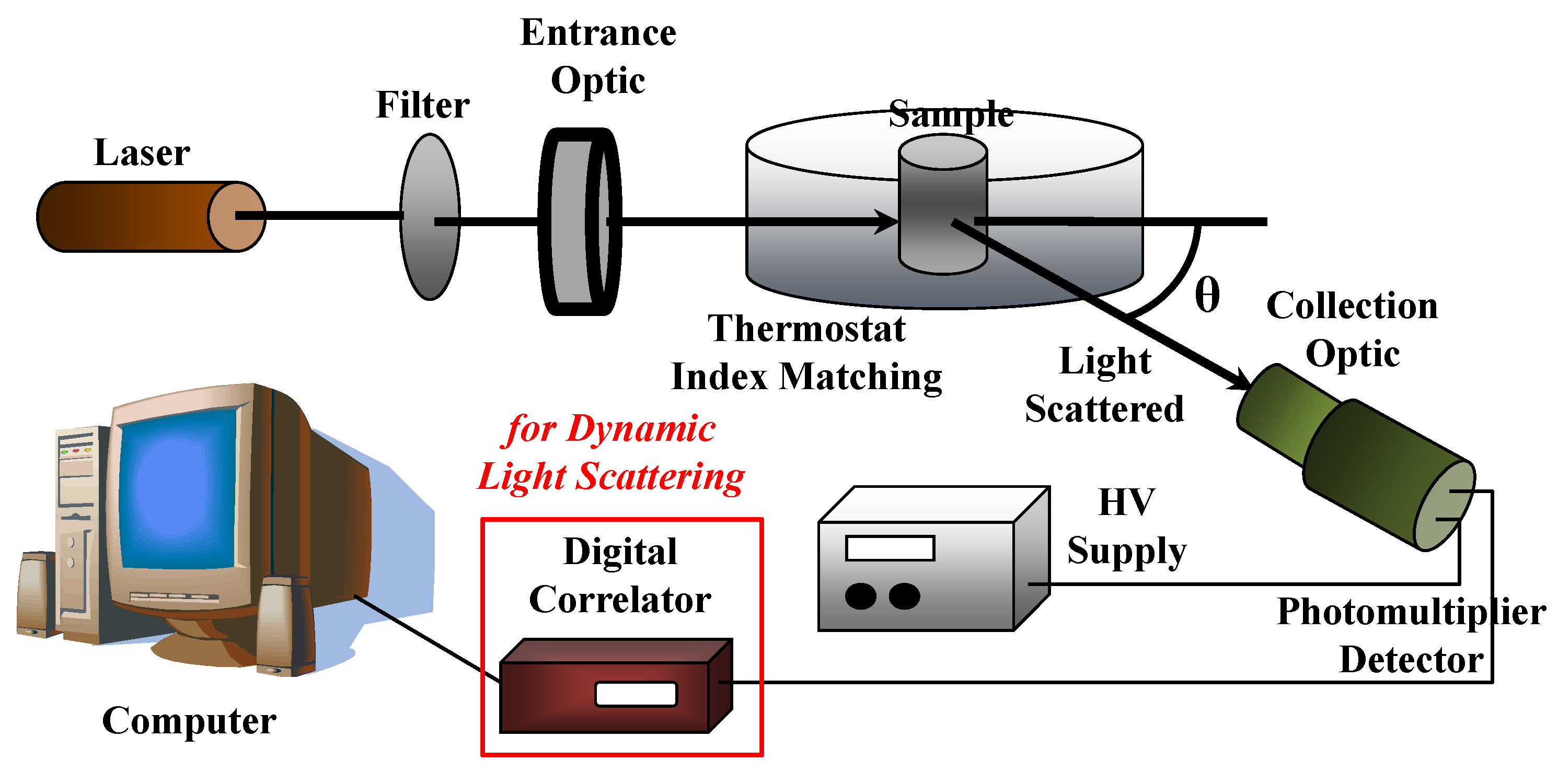
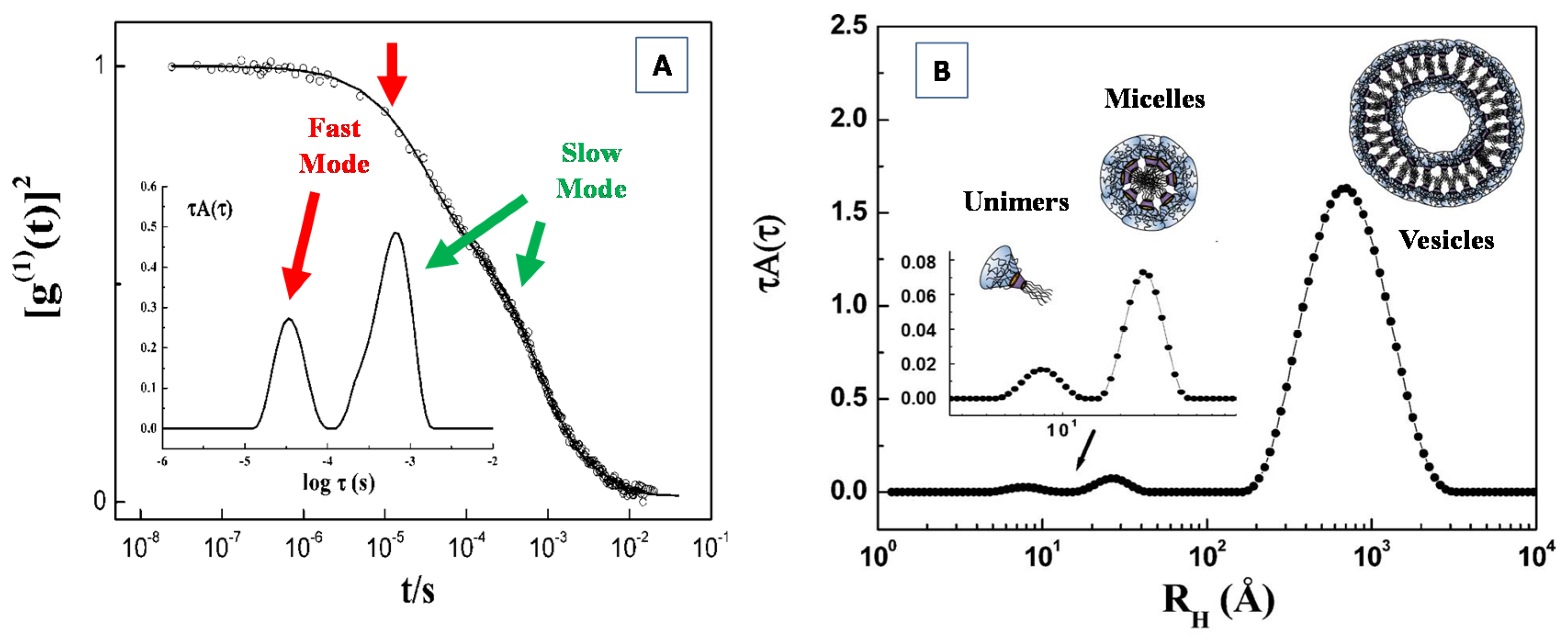
4.2. Dynamic Light Scattering (DLS)
5. Characterization with Complementary Techniques
6. Concluding Remarks and Future Developments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gale, P.; Steed, J. Supramolecular Chemistry: From Molecules to Nanomaterials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Grzelczak, M.; Liz-Marzán, L.M.; Klajn, R. Stimuli-responsive self-assembly of nanoparticles. Chem. Soc. Rev. 2019, 48, 1342–1361. [Google Scholar] [CrossRef] [PubMed]
- Chircov, C.; Spoiala, A.; Păun, C.; Crăciun, L.; Ficai, D.; Ficai, A.; Andronescu, E.; Turculeƫ, Ș.C. Mesoporous Silica Platforms with Potential Applications in Release and Adsorption of Active Agents. Molecules 2020, 25, 3814. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hudalla, G.A. Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials. Molecules 2019, 24, 1450. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Magazù, S.; Wanderlingh, U.; Barreca, D.; Pasqua, L.; Kiselev, M.A. Soft nanoparticles charge expression within lipid membranes: The case of amino terminated dendrimers in bilayers vesicles. Colloids Surf. B Biointerfaces 2018, 170, 609–616. [Google Scholar] [CrossRef]
- Tiwari, A.; Tiwari, A. Nanomaterials in Drug Delivery, Imaging, and Tissue Engineerin; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Lombardo, D.; Kiselev, M.A.; Magazu, S.; Calandra, P. Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Adv. Condens. Matter Phys. 2015, 2015, 151683. [Google Scholar] [CrossRef]
- Ilie, A.; Ghiţulică, C.; Andronescu, E.; Cucuruz, A.; Ficai, A. New composite materials based on alginate and hydroxyapatite as potential carriers for ascorbic acid. Int. J. Pharm. 2016, 510, 501–507. [Google Scholar] [CrossRef]
- Wang, L.; Gong, C.; Yuan, X.; Wei, G. Controlling the Self-Assembly of Biomolecules into Functional Nanomaterials through Internal Interactions and External Stimulations: A Review. Nanomaterials 2019, 9, 285. [Google Scholar] [CrossRef]
- Salehi, B.; Calina, D.; Docea, A.O.; Koirala, N.; Aryal, S.; Lombardo, D.; Pasqua, L.; Taheri, Y.; Castillo, C.M.S.; Martorell, M.; et al. Curcumin’s Nanomedicine Formulations for Therapeutic Application in Neurological Diseases. J. Clin. Med. 2020, 9, 430. [Google Scholar] [CrossRef]
- Kaga, S.; Truong, N.P.; Esser, L.; Senyschyn, D.; Sanyal, A.; Sanyal, R.; Quinn, J.F.; Davis, T.P.; Kaminskas, L.M.; Whittaker, M.R. Influence of Size and Shape on the Biodistribution of Nanoparticles Prepared by Polymerization-Induced Self-Assembly. Biomacromolecules 2017, 18, 3963–3970. [Google Scholar] [CrossRef]
- Lanza, R.; Langer, R.; Vacanti, J.; Atala, A. Principles of Tissue Engineering; Academic Press: Waltham, MA, USA, 2020. [Google Scholar]
- Mantha, S.; Pillai, S.; Khayambashi, P.; Upadhyay, A.; Zhang, Y.; Tao, O.; Pham, H.M.; Tran, S.D. Smart Hydrogels in Tissue Engineering and Regenerative Medicine. Materials 2019, 12, 3323. [Google Scholar] [CrossRef]
- Radulescu, M.; Ficai, D.; Oprea, O.; Ficai, A.; Andronescu, E.; Holban, A.M. Antimicrobial Chitosan based formulations with impact on different biomedical applications. Curr. Pharm. Biotechnol. 2015, 16, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Chimisso, V.; Aleman Garcia, M.A.; Yorulmaz Avsar, S.; Dinu, I.A.; Palivan, C.G. Design of Bio-Conjugated Hydrogels for Regenerative Medicine Applications: From Polymer Scaffold to Biomolecule Choice. Molecules 2020, 25, 4090. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, Z.P. Nanomaterials for Medical Applications; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Doshi, N.; Mitragotri, S. Designer Biomaterials for Nanomedicine. Adv. Funct. Mater. 2009, 19, 3843–3854. [Google Scholar] [CrossRef]
- Andronescu, E.; Ficai, M.; Voicu, G.; Ficai, D.; Maganu, M.; Ficai, A. Synthesis and characterization of collagen/hydroxyapatite: Magnetite composite material for bone cancer treatment. J. Mater. Sci. Mater. Med. 2010, 21, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Pasqua, L.; De Napoli, I.E.; De Santo, M.; Greco, M.; Catizzone, E.; Lombardo, D.; Montera, G.; Comandè, A.; Nigro, A.; Morelli, C.; et al. Mesoporous silica-based hybrid materials for bone-specific drug delivery. Nanoscale Adv. 2019, 1, 3269–3278. [Google Scholar] [CrossRef]
- Gong, C.; Sun, S.; Zhang, Y.; Sun, L.; Su, Z.; Wu, A.; Wei, G. Hierarchical nanomaterials via biomolecular self-assembly and bioinspiration for energy and environmental applications. Nanoscale 2019, 11, 4147–4182. [Google Scholar] [CrossRef]
- Malmsten, M. Surfactants and Polymers in Drug Delivery; Marcel Dekker Inc.: New York, NY, USA, 2006; pp. 116–212. [Google Scholar]
- Lombardo, D.; Calandra, P.; Pasqua, L.; Magazù, S. Self-assembly of Organic Nanomaterials and Biomaterials: The Bottom-Up Approach for Functional Nanostructures Formation and Advanced Applications. Materials 2020, 13, 1048. [Google Scholar] [CrossRef]
- Jaekel, A.; Stegemann, P.; Saccà, B. Manipulating Enzymes Properties with DNA Nanostructures. Molecules 2019, 24, 3694. [Google Scholar] [CrossRef]
- Russina, O.; Gontrani, L.; Fazio, B.; Lombardo, D.; Triolo, A.; Caminiti, R. Selected chemical–physical properties and structural heterogeneities in 1-ethyl-3-methylimidazolium alkyl-sulfate room temperature ionic liquids. Chem. Phys. Lett. 2010, 493, 259–262. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry; WILEY-VCH Verlag GmbH: Weinheim, Germany, 1995. [Google Scholar]
- Lee, Y.S. Self-Assembly and Nanotechnology, a Force Balance Approach; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Calandra, P.; Caschera, D.; Liveri, V.T.; Lombardo, D. How self-assembly of amphiphilic molecules can generate complexity in the nanoscale. Colloids Surf. A Physicochem. Eng. Asp. 2015, 484, 164–183. [Google Scholar] [CrossRef]
- Ariga, K.; Ito, M.; Mori, T.; Watanabe, S.; Takeya, J. Atom/molecular nanoarchitectonics for devices and related applications. Nano Today 2019, 28, 100762. [Google Scholar] [CrossRef]
- Pica, A.; Guran, C.; Andronescu, E.; Oprea, O.; Ficai, D.; Ficai, A. Antimicrobial performances of some film forming materials based on silver nanoparticles. J. Optoelectron. Adv. Mater. 2012, 14, 863–868. [Google Scholar]
- Salam, A.; Makhlouf, H.; Barhoum, A. DNA Nanostructures: Chemistry, Self-Assembly, and Applications. In Emerging Applications of Nanoparticles and Architecture Nanostructures; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Plenum Press: New York, NY, USA; Springer: London, UK, 1987. [Google Scholar]
- Glatter, O.; Kratky, O. Small-Angle X-ray Scattering; Academic Press: London, UK, 1982. [Google Scholar]
- Squires, G.L. Introduction to the Theory of Thermal Neutron Scattering; Dover Publications: New York, NY, USA, 1997. [Google Scholar]
- Fitter, J.; Gutberlet, T.; Katsaras, J. Neutron Scattering in Biology Techniques and Applications; Springer Biological: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- X-ray Data Booklet, A.C.; Thompson Lawrence Berkeley National Laboratory: Berkeley, NY, USA, 2009.
- Stuhrmanna, H.B. Contrast variation in X-ray and neutron scattering. J. Appl. Cryst. 2007, 40, s23–s27. [Google Scholar] [CrossRef]
- Luo, G.; Zhang, Q.; Del Castillo, A.R.; Urban, V.; O’Neill, H. Characterization of sol gel-encapsulated proteins using small-angle neutron scattering. ACS Appl. Mater. Interfaces 2009, 1, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Jacrot, B. Study of biological structures by neutron-scattering from solution. Rep. Prog. Phys. 1976, 39, 911–953. [Google Scholar] [CrossRef]
- Sztucki, M.; Di Cola, E.; Narayanan, T. Anomalous small-angle X-ray scattering from charged soft matter. Eur. Phys. J. Spec. Top. 2012, 208, 319–331. [Google Scholar] [CrossRef]
- Gebel, G. Structure of Membranes for Fuel Cells: SANS and SAXS Analyses of Sulfonated PEEK Membranes and Solutions. Macromolecules 2013, 46, 6057–6066. [Google Scholar] [CrossRef]
- Varga, Z.; Bóta, A.; Goerigk, G. Localization of Dibromophenol in DPPC/Water Liposomes Studied by Anomalous Small-Angle X-ray Scattering. J. Phys. Chem. B 2006, 110, 11029–11032. [Google Scholar] [CrossRef][Green Version]
- Ballauff, M.; Jusufi, A. Anomalous small-angle X-ray scattering: Analyzing correlations and fluctuations in polyelectrolytes. Colloid Polym. Sci. 2006, 284, 1303–1311. [Google Scholar] [CrossRef]
- Nakasako, M.; Wada, M.; Tokutomi, S.; Yamamoto, K.; Sakai, J.; Kataoka, M.; Tokunaga, F.; Furuya, M. Quaternary structure of a PEA phytochrome I dimer studied with small-angle X-ray scattering and rotatory shadowing electron microscopy. Photochem. Photobiol. 1990, 52, 3–12. [Google Scholar] [CrossRef]
- Dingenouts, N.; Ballauff, M. Small-angle x-ray analysis of latex particles using contrast variation. Acta Polym. 1993, 44, 178–183. [Google Scholar] [CrossRef]
- Kiselev, M.A.; Lesieur, P.; Kisselev, A.M.; Lombardo, D.; Killany, M.; Lesieur, S.; Ollivon, M. A sucrose solutions application to the study of model biological membranes. Nucl. Instrum. Methods Phys. Res. A 2001, 470, 409–416. [Google Scholar] [CrossRef]
- Kiselev, M.; Lesieur, P.; Kisselev, A.M.; Lombardo, D.; Killany, M.; Lesieur, S. Sucrose solutions as prospective medium to study the vesicle structure: SAXS and SANS study. J. Alloys Compd. 2001, 328, 71–76. [Google Scholar] [CrossRef]
- Naruse, K.; Eguchi, K.; Akiba, I.; Sakurai, K. Hiroyasu, Flexibility and Cross-Sectional Structure of an Anionic Dual-Surfactant Wormlike Micelle Explored with Small-Angle X-ray Scattering Coupled with Contrast Variation Technique. J. Phys. Chem. B 2009, 113, 10222–10229. [Google Scholar] [CrossRef]
- Hickl, P.; Ballauff, M.; Jada, A. Small-Angle X-ray Contrast-Variation Study of Micelles Formed by Poly (styrene)−Poly (ethylene oxide) Block Copolymers in Aqueous Solution. Macromolecules 1996, 29, 4006–4014. [Google Scholar] [CrossRef]
- Bolze, J.; Hörner, K.D.; Ballauff, M. Adsorption of the Nonionic Surfactant Triton X-405 on Polystyrene Latex Particles As Monitored by Small-Angle X-ray Scattering. Langmuir 1996, 12, 2906–2912. [Google Scholar] [CrossRef]
- Fernandez, R.M.; Riske, K.A.; Amaral, L.Q.; Itri, R.; Lamy, M.T. Influence of salt on the structure of DMPG studied by SAXS and optical microscopy. Biochim. Biophys. Acta 2008, 1778, 907–916. [Google Scholar] [CrossRef]
- Schmidt, P.W. Modern Aspects of Small-Angle Scattering; Brumberger, H., Ed.; Kluwer Academic: Dordrecht, The Netherlands, 1995; p. 1. [Google Scholar]
- Di Cola, E.; Grillo, I.; Ristori, S. Small Angle X-ray and Neutron Scattering: Powerful Tools for Studying the Structure of Drug-Loaded Liposomes. Pharmaceutics 2016, 8, 10. [Google Scholar] [CrossRef]
- Zemb, T.; Lindner, P. Neutron, X-rays and Light Scattering Methods Applied to Soft Condensed Matter; North-Holland Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Kiselev, M.A.; Lombardo, D. Structural characterization in mixed lipid membrane systems by neutron and X-ray scattering. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3700–3717. [Google Scholar] [CrossRef]
- Alexandridis, P.; Lindman, B. Amphiphilic Block Copolymers: Self-Assembly and Applications (Studies in Surface Science and Catalysis); Elsevier Science, B.V.: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Mallamace, F.; Beneduci, R.; Gambadauro, P.; Lombardo, D.; Chen, S.H. Glass and percolation transitions in dense attractive micellar system. Phys. A Stat. Mech. Appl. 2001, 302, 202–219. [Google Scholar] [CrossRef]
- Chen, S.H.; Mallamace, F.; Faraone, A.; Gambadauro, P.; Lombardo, D.; Chen, W.R. Observation of a re-entrant kinetic glass transition in a micellar system with temperature-dependent attractive interaction. Eur. Phys. J. E Soft Matter. 2002, 9, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Eisenberg, A. Self-Assembly of Block Copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Nelemans, L.C.; Gurevich, L. Drug Delivery with Polymeric Nanocarriers-Cellular Uptake Mechanisms. Materials 2020, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.; Castelletto, V. Small-Angle Scattering of Block Copolymers; Borsali, R., Pecora, R., Eds.; Soft Matter Characterization; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Lombardo, D.; Munaò, M.; Calandra, P.; Pasqua, L.; Caccamo, M.T. Evidence of pre-micellar aggregates in water solution of amphiphilic PDMS-PEO block copolymer. Phys. Chem. Chem. Phys. 2019, 21, 11983–11991. [Google Scholar] [CrossRef]
- Narayanan, T.; Sztucki, M.; Van Vaerenbergh, P.; Léonardon, J.; Gorini, J.; Claustre, L.; Sever, F.; Morse, J.; Boesecke, P. A multipurpose instrument for time-resolved ultra-small-angle and coherent X-ray scattering. J. Appl. Crystallogr. 2018, 51, 1511–1524. [Google Scholar] [CrossRef]
- Amenitsch, H.; Bernstorff, S.; Kriechbaum, M.; Lombardo, D.; Mio, H.; Rappolt, M.; Laggner, P. Performance and first results of the ELETTRA high-flux beamline for small-angle X-ray scattering. J. Appl. Cryst. 1997, 30, 872–876. [Google Scholar] [CrossRef]
- Lobry, L.; Micali, N.; Mallamace, F.; Liao, C.; Chen, S.H. Interaction and percolation in the L64 triblock copolymer micellar system. Phys. Rev. E 1999, 60, 7076. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Gerstenberg, M.C. Scattering Form Factor of Block Copolymer Micelles. Macromolecules 1996, 29, 1363–1365. [Google Scholar] [CrossRef]
- Kotlarchyk, M.; Chen, S.-H. Analysis of small angle neutron scattering spectra from polydisperse interacting colloids. J. Chem. Phys. 1983, 79, 2461. [Google Scholar] [CrossRef]
- Kiselev, M.A.; Janich, M.; Hildebrand, A.; Strunz, P.; Neubert, R.H.H.; Lombardo, D. Structural transition in aqueous lipid/bile salt [DPPC/NaDC] supramolecular aggregates: SANS and DLS study. Chem. Phys. 2013, 424, 93–99. [Google Scholar] [CrossRef]
- Svergun, D.I.; Semenyuk, A.V.; Feigin, L.A. Small-angle-scattering-data treatment by the regularization method. Acta Cryst. 1988, A44, 244–250. [Google Scholar] [CrossRef]
- Svergun, D.I.; Koch, M.H.J. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 2003, 66, 1735–1782. [Google Scholar] [CrossRef]
- Weyerich, B.; Brunner-Popela, J.; Glatter, O. Small-angle scattering of interacting particles. II. Generalized indirect Fourier transformation under consideration of the effective structure factor for polydisperse systems. J. Appl. Crystallogr. 1999, 32, 197–209. [Google Scholar] [CrossRef]
- Szymusiak, M.; Kalkowski, J.; Luo, H. Core-shell Structure and Aggregation Number of Micelles Composed of Amphiphilic Block Copolymers and Amphiphilic Heterografted Polymer Brushes Determined by Small-Angle X-ray Scattering. ACS Macro Lett. 2017, 19, 1005–1012. [Google Scholar] [CrossRef]
- Glatter, O. The Interpretation of Real-Space Information from Small-Angle Scattering Experiments. J. Appl. Crystallogr. 1979, 12, 166–175. [Google Scholar] [CrossRef]
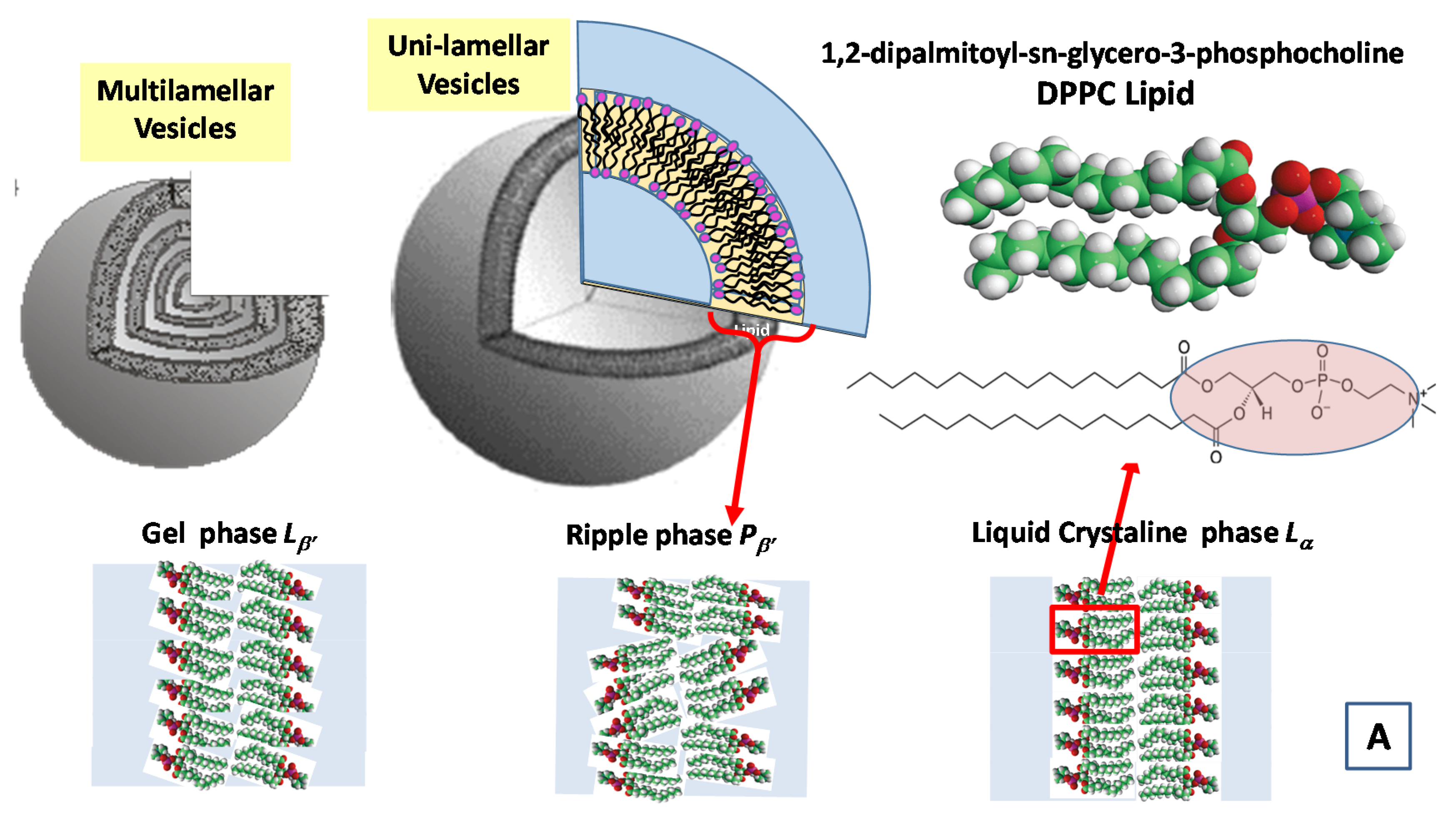
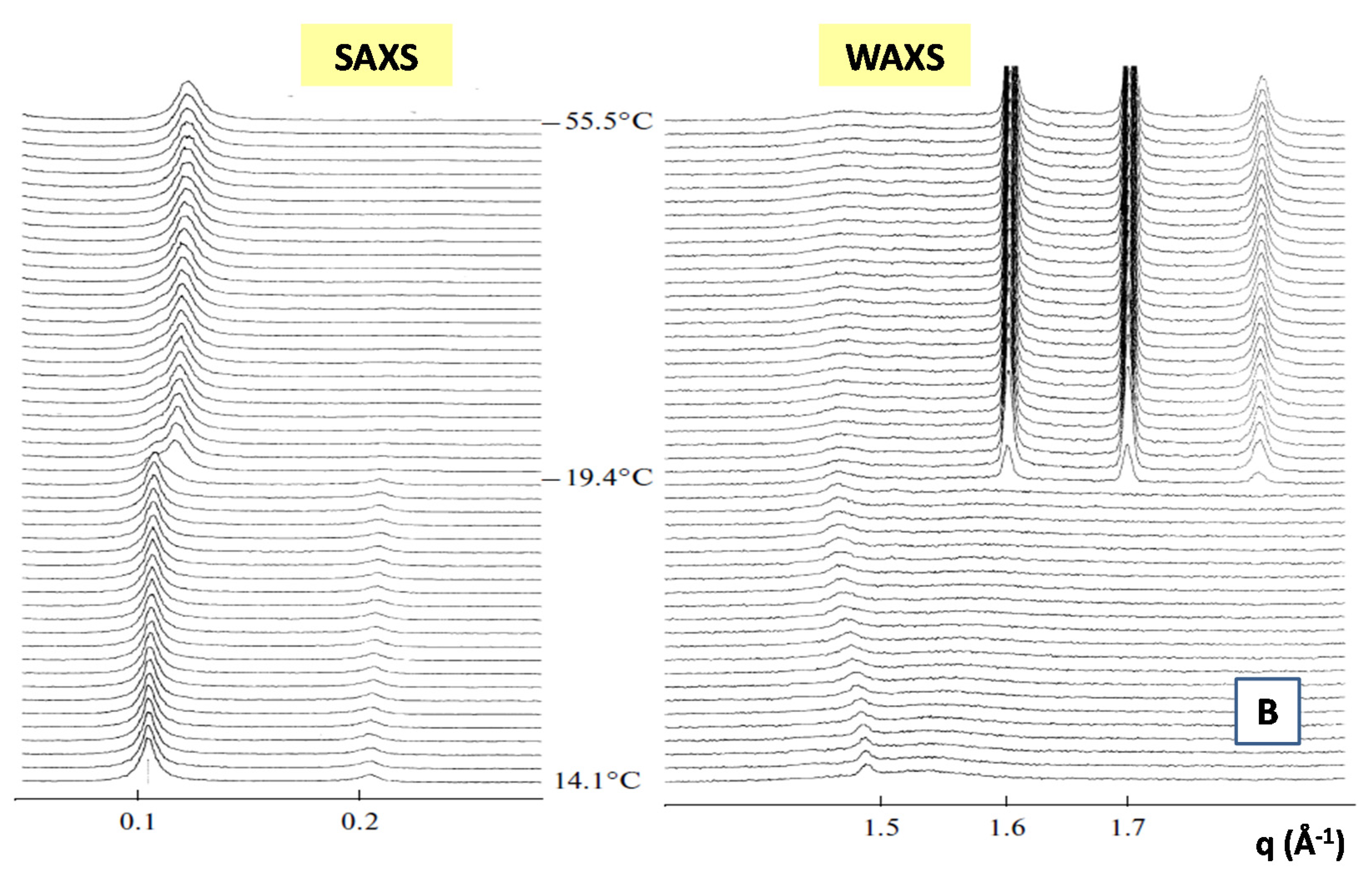
- Heftberger, P.; Kollmitzer, B.; Heberle, F.A.; Pan, J.; Rappolt, M.; Amenitsch, H.; Kučerka, N.; Katsaras, J.; Pabst, G. Global small-angle X-ray scattering data analysis for multilamellar vesicles: The evolution of the scattering density profile model. J. Appl. Crystallogr. 2013, 47, 173–180. [Google Scholar] [CrossRef]
- Koch, M.H.; Vachette, P.; Svergun, D.I. Small-angle scattering: A view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 2003, 36, 147–227. [Google Scholar] [CrossRef]
- Calandra, P.; Ruggirello, A.; Mele, A.; Liveri, V.T. Self-assembly in surfactant-based liquid mixtures: Bis(2-ethylhexyl)phosphoric acid/bis(2-ethylhexyl)amine systems. J. Colloid Interface Sci. 2010, 348, 183–188. [Google Scholar] [CrossRef]
- Micali, N.; Scolaro, L.M.; Romeo, A.; Lombardo, D.; Lesieur, P.; Mallamace, F. Structural properties of methanol-polyamidoamine dendrimer solutions. Phys. Rev. E 1998, 58, 6229–6235. [Google Scholar] [CrossRef]
- Lesieur, P.; Kiselev, M.A.; Barsukov, L.I.; Lombardo, D. Temperature-induced micelle to vesicle transition: Kinetic effects in the DMPC/NaC system. J. Appl. Cryst. 2000, 33, 623–627. [Google Scholar] [CrossRef]
- Winter, R.; Koehling, R. Static and time-resolved synchrotron small-angle x-ray scattering studies of lyotropic lipid mesophases, model biomembranes and proteins in solution. J. Condens. Matter Phys. 2004, 16, S327–S352. [Google Scholar] [CrossRef]
- Chaudhuri, B.; Muñoz, I.G.; Qian, S.; Urban, V.S. Biological Small Angle Scattering: Techniques, Strategies and Tips; Springer: Singapore, 2017. [Google Scholar]
- Calandra, P.; Liveri, V.T.; Riello, P.; Freris, I.; Mandanici, A. Self-assembly in surfactant-based liquid mixtures: Octanoic acid/Bis(2-ethylhexyl)amine systems. J. Colloid Interface Sci. 2012, 367, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D. Liquid-like ordering of negatively charged poly (amidoamine) (PAMAM) dendrimers in solution. Langmuir 2009, 25, 3271–3275. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, M.; Lesieur, P.; Kisselev, A.M.; Lombardo, D.; Aksenov, V. Model of separated form factors for unilamellar vesicles. Appl. Phys. A 2002, 74, s1654–s1656. [Google Scholar] [CrossRef]
- Holmberg, K.; Jonsson, B.; Kronberg, B.; Lindman, B. Surfactants and Polymers in Aqueous Solution, 2nd ed.; Wiley and Sons Ltd.: Chichester, UK, 2002. [Google Scholar]
- Laughlin, R.G. The Aqueous Phase Behavior of Surfactants; Academic Press: London, UK, 1994. [Google Scholar]
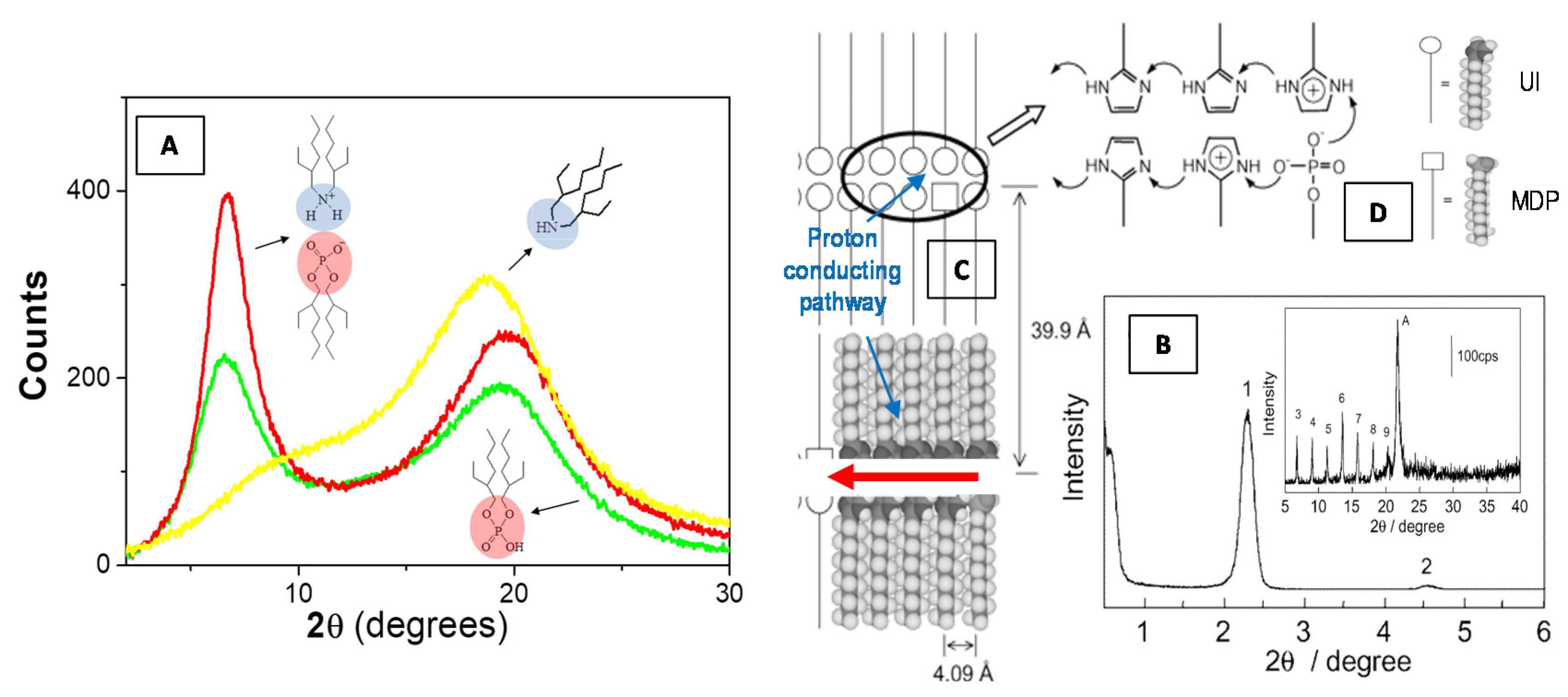
- Yamada, M.; Honma, I.J. Anhydrous protonic conductivity of a self-assembled acid−base composite material. J. Phys. Chem. B 2004, 108, 5522–5526. [Google Scholar] [CrossRef]
- Kim, J.D.; Honma, I. Anhydrous solid state proton conductor based on enzimidazole/monododecyl phosphate molecular hybrids. Solid State Ion. 2005, 176, 979–984. [Google Scholar] [CrossRef]
- Calandra, P.; Turco Liveri, V.; Ruggirello, A.M.; Licciardi, M.; Lombardo, D.; Mandanici, A. Anti-Arrhenian behaviour of conductivity in octanoic acid–bis (2-ethylhexyl) amine systems: A physico-chemical study. J. Mater. Chem. C 2015, 3, 3198–3210. [Google Scholar] [CrossRef]
- Pochylski, M.; Lombardo, D.; Calandra, P. Optical Birefringence Growth Driven by Magnetic Field in Liquids: The Case of Dibutyl Phosphate/Propylamine System. Appl. Sci. 2019, 10, 164. [Google Scholar] [CrossRef]
- Rikken, R.; Kerkenaar, H.; Nolte, R.; Maan, J.; Van Hest, J.; Christianen, P.; Wilson, D. Probing morphological changes in polymersomes with magnetic birefringence. Chem. Commun. 2014, 50, 5394–5396. [Google Scholar] [CrossRef]
- Calandra, P.; Caputo, P.; De Santo, M.P.; Todaro, L.; Turco Liveri, V.; Rossi, C.O. Effect of additives on the structural organization of asphaltene aggregates in bitumen. Constr. Build. Mater. 2019, 199, 288–297. [Google Scholar] [CrossRef]
- Turco Liveri, V.; Lombardo, D.; Pochylski, M.; Calandra, P. Molecular association of small amphiphiles: Origin of ionic liquid properties in dibutyl phosphate/propylamine binary mixtures. J. Mol. Liq. 2018, 263, 274–281. [Google Scholar] [CrossRef]
- Calandra, P. On the physico-chemical basis of self-nanosegregation giving magnetically-induced birefringence in dibutyl phosphate/bis(2-ethylhexyl) amine systems. J. Mol. Liq. 2020, 310, 113186. [Google Scholar] [CrossRef]
- Cho, H.S.; Dashdorj, N.; Schotte, F.; Graber, T.; Henning, R.; Anfinrud, P. Protein structural dynamics in solution unveiled via 100-ps time-resolved x-ray scattering. Proc. Natl. Acad. Sci. USA 2010, 107, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Pollack, L. Time resolved SAXS and RNA folding. Biopolymers 2011, 95, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Bruetzel, L.K.; Walker, P.U.; Gerling, T.; Dietz, H.; Lipfert, J. Time-Resolved Small-Angle X-ray Scattering Reveals Millisecond Transitions of a DNA Origami Switch. Nano Lett. 2018, 18, 2672–2676. [Google Scholar] [CrossRef]
- Narayanan, T.; Wacklin, H.; Konovalov, O.; Lund, R. Recent applications of synchrotron radiation and neutrons in the study of soft matter. Crystallogr. Rev. 2017, 23, 160–226. [Google Scholar] [CrossRef]
- Amenitsch, H.; Rappolt, M.; Kriechbaum, M.; Mio, H.; Laggner, P.; Bernstorff, S. First performance assessment of the small-angle X-ray scattering beamline at ELETTRA. J. Synchrotron Radiat. 1998, 5, 506–508. [Google Scholar] [CrossRef]
- Möller, J.; Léonardon, J.; Gorini, J.; Dattani, R.; Narayanan, T. A sub-ms pressure jump setup for time-resolved X-ray scattering. Rev. Sci. Instrum. 2016, 87, 125116. [Google Scholar] [CrossRef]
- Kiselev, M.A. Methods for lipid nanostructure investigation at neutron and synchrotron sources. Phys. Part. Nucl. 2011, 42, 302–331. [Google Scholar] [CrossRef]
- Ludescher, L.; Morak, R.; Balzer, C.; Waag, A.M.; Braxmeier, S.; Putz, F.; Busch, S.; Gor, G.Y.; Neimark, A.V.; Husing, N.; et al. In Situ Small-Angle Neutron Scattering Investigation of Adsorption-Induced Deformation in Silica with Hierarchical Porosity. Langmuir 2019, 35, 11590–11600. [Google Scholar] [CrossRef]
- Bonaccorsi, L.; Lombardo, D.; Longo, A.; Proverbio, E.; Triolo, A. Dendrimer template directed self-assembly during zeolite formation. Macromolecules 2009, 42, 1239–1243. [Google Scholar] [CrossRef]
- Gommes, C.J.; Prieto, G.; de Jongh, P.E. Small-Angle Scattering Analysis of Empty or Loaded Hierarchical Porous MaterialsJ. Phys. Chem. C 2016, 120, 1488–1506. [Google Scholar] [CrossRef]
- Bonaccorsi, L.; Calandra, P.; Amenitsch, H.; Proverbio, E.; Lombardo, D. Growth of fractal aggregates during template directed SAPO-34 zeolite formation. Micropor. Mesopor. Mater. 2013, 167, 3–9. [Google Scholar] [CrossRef]
- Huang, X.; Zheng, S.; Kim, I. Hyperbranched Polymers and Dendrimers as Templates for Organic/Inorganic Hybrid Nanomaterials. J. Nanosci. Nanotechnol. 2014, 14, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Belleville, P.; Popall, M.; Nicole, L. Applications of advanced hybrid organic-inorganic nanomaterials: From laboratory to market. Chem. Soc. Rev. 2011, 40, 696–753. [Google Scholar] [CrossRef] [PubMed]
- Michaux, F.; Baccile, N.; Imperor-Clerc, M.; Malfatti, L.; Folliet, N.; Gervais, C.; Manet, S.; Meneau, F.; Pedersen, J.S.; Babonneau, F. In situ time-resolved SAXS study of the formation of mesostructured organically modified silica through modeling of micelles evolution during surfactant-templated self-assembly. Langmuir 2012, 28, 17477–17493. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi, L.; Calandra, P.; Kiselev, M.A.; Amenitsch, H.; Proverbio, E.; Lombardo, D. Self-assembly in poly(dimethylsiloxane)-poly(ethylene oxide) block copolymer template directed synthesis of linde type A zeolite. Langmuir 2013, 29, 7079–7086. [Google Scholar] [CrossRef]
- Teixeira, J. Small-Angle Scattering by Fractal Systems. J. Appl. Cryst. 1988, 21, 781–785. [Google Scholar] [CrossRef]
- Hunter, R.J. Foundations of Colloid Science; Oxford University Press: New York, NY, USA, 1986; Volumes I–II. [Google Scholar]
- Hansen, J.P.; Mc Donald, I.A. Theory of Simple Liquids; Academic Press: New York, NY, USA, 1986. [Google Scholar]
- Caccamo, C. Integral Equation Theory Description of Phase Equilibria in Classical Fluids. Phys. Rep. 1996, 274, 1–105. [Google Scholar] [CrossRef]
- Lombardo, D. Modeling Dendrimers Charge Interaction in Solution: Relevance in Biosystems. Biochem. Res. Int. 2014, 837651, 22014. [Google Scholar] [CrossRef]
- Sato, K.; Anzai, J. Dendrimers in layer-by-layer assemblies: Synthesis and applications. Molecules 2013, 18, 8440–8460. [Google Scholar] [CrossRef] [PubMed]
- Mhlwatika, Z.; Aderibigbe, B.A. Application of Dendrimers for the Treatment of Infectious Diseases. Molecules 2018, 23, 2205. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Bellocco, E.; Laganà, G.; Barreca, D.; Magazù, S.; Wanderlingh, U.; Kiselev, M.A. Effect of anionic and cationic polyamidoamine (PAMAM) dendrimers on a model lipid membrane. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Ekimoto, T.; Kokabu, Y.; Oroguchi, T.; Ikeguchi, M. Combination of coarse-grained molecular dynamics simulations and small-angle X-ray scattering experiments. Biophys. Physicobiol. 2019, 29, 377–390. [Google Scholar] [CrossRef]
- Björling, A.; Niebling, S.; Marcellini, M.; Van Der Spoel, D.; Westenhoff, S. Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations. J. Chem. Theory Comput. 2015, 11, 780–787. [Google Scholar] [CrossRef]
- Sachs, J.; Petrache, H.; Woolf, T. Interpretation of small angle X-ray measurements guided by molecular dynamics simulations of lipid bilayers. Chem. Phys. Lipids 2003, 26, 211–223. [Google Scholar] [CrossRef]
- Pan, J.; Cheng, X.; Monticelli, L.; Heberle, F.A.; Kučerka, N.; Tieleman, D.P.; Katsara, S.J. The molecular structure of a phosphatidylserine bilayer determined by scattering and molecular dynamics simulations. Soft Matter 2014, 10, 3716–3725. [Google Scholar] [CrossRef]
- Arbe, A.; Alvareza, F.; Colmenero, J. Neutron scattering and molecular dynamics simulations: Synergetic tools to unravel structure and dynamics in polymers. Soft Matter 2012, 8, 8257–8270. [Google Scholar] [CrossRef]
- Alford, A.; Kozlovskaya, V.; Kharlampieva, E. Small Angle Scattering for Pharmaceutical Applications: From Drugs to Drug Delivery Systems. Adv. Exp. Med. Biol. 2017, 1009, 239–262. [Google Scholar] [CrossRef]
- Narayanan, T. Synchrotron small-angle X-ray scattering. In Soft Matter: Characterization; Springer: Heidelberg, Germany, 2008; Volume II. [Google Scholar]
- Kiselev, M.; Zemlyanaya, E.; Ryabova, N.Y.; Hauss, T.; Almásy, L.; Funari, S.; Zbytovská, J.; Lombardo, D. Influence of ceramide on the internal structure and hydration of the phospholipid bilayer studied by neutron and X-ray scattering. Appl. Phys. A 2014, 116, 319–325. [Google Scholar] [CrossRef]
- Schilt, Y.; Berman, T.; Wei, X.; Barenholz, Y.; Raviv, U. Using solution X-ray scattering to determine the high-resolution structure and morphology of PEGylated liposomal doxorubicin nanodrugs. Biochim. Biophys. Acta 2016, 1860, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Yaghmur, A.; Rappolt, M.; Jonassen, A.L.; Schmitt, M.; Larsen, S. In situ monitoring of the formation of lipidic non-lamellar liquid crystalline depot formulations in synovial fluid. J. Colloid Interface Sci. 2020, 582 Pt B, 773–781. [Google Scholar] [CrossRef]
- Giulimondi, F.; Digiacomo, L.; Pozzi, D.; Palchetti, S.; Vulpis, E.; Capriotti, A.L.; Chiozzi, R.Z.; Laganà, A.; Amenitsch, H.; Masuelli, L.; et al. Interplay of protein corona and immune cells controls blood residency of liposomes. Nat. Commun. 2019, 10, 3686. [Google Scholar] [CrossRef]
- Kennedy, M.T.; Pozharski, E.V.; Rakhmanova, V.A.; MacDonald, R.C. Factors governing the assembly of cationic phospholipid-DNA complexes. Biophys. J. 2000, 78, 1620–1633. [Google Scholar] [CrossRef]
- Simberg, D.; Danino, D.; Talmon, Y.; Minsky, A.; Ferrari, M.E.; Wheeler, C.J.; Barenholz, Y. Phase behavior, DNA ordering, and size instability of cationic lipoplexes Relevance to optimal transfection activity. J. Biol. Chem. 2001, 276, 47453–47459. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Caccamo, M.T.; Magazù, S.; Kiselev, M.A. Colloidal stability of liposomes. AIMS Mater. Sci. 2019, 6, 200. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Mishra, A.; Wang, J.; DeGrado, W.F.; Wong, G.C. Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission. J. Am. Chem. Soc. 2013, 135, 13710–13719. [Google Scholar] [CrossRef]
- Schmidt, N.; Wong, G. Antimicrobial peptides and induced membrane curvature: Geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mater. Sci. 2013, 17, 151–163. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by A-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Lombardo, D.; Caccamo, M.T.; Magazù, S.; Kiselev, M.A.; Calandra, P. Enhancement of colloidal stability of drug nanocarriers in complex biological environment. AAPP Phys. Math. Nat. Sci. 2019, 97, A25. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Laganà, A. Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Caccamo, M.T.; Magazù, S.; Pasqua, L.; Kiselev, M.A. Interdisciplinary approaches to the study of biological membranes. AIMS Biophys. 2020, 7, 267–290. [Google Scholar] [CrossRef]
- Franke, D.; Jeffries, C.M.; Svergun, D.I. Machine Learning Methods for X-Ray Scattering Data Analysis from Biomacromolecular Solutions. Biophys. J. 2018, 114, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Frewein, M.P.; Rumetshofer, M.; Pabst, G. Global small-angle scattering data analysis of inverted hexagonal phases. J. Appl. Crystallogr. 2019, 52, 403–414. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2 Pt 2, 207–217. [Google Scholar] [CrossRef]
- Bernadó, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Ii, F.L.P.; Tsutakawa, S.E.; Jr, F.E.J.; Classen, S.; Frankel, K.A.; Hopkins, R.C.; et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 2009, 6, 606–612. [Google Scholar] [CrossRef]
- von Gundlach, A.R.; Garamus, V.M.; Gorniak, T.; Davies, H.A.; Reischl, M.; Mikut, R.; Hilpert, K.; Rosenhahn, A. Small angle X-ray scattering as a high-throughput method to classify antimicrobial modes of action. Biochim. Biophys. Acta 2016, 1858, 918–925. [Google Scholar] [CrossRef]
- Hyland, L.L.; Taraban, M.; Yu, Y.B. Using Small-Angle Scattering Techniques to Understand Mechanical Properties of Biopolymer-Based Biomaterials. Soft Matter 2013, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Hule, R.A.; Nagarkar, R.P.; Hammouda, B.; Schneider, J.P.; Pochan, D.J. Dependence of Self-Assembled Peptide Hydrogel Network Structure on Local Fibril Nanostructure. Macromolecules 2009, 42, 7137–7145. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Shurtenberger, P. Scattering Functions of Semiflexible Polymers with and without Excluded Volume Effects. Macromolecules 1996, 29, 7602–7612. [Google Scholar] [CrossRef]
- MacKintosh, F.C.; Käs, J.; Janmey, P.A. Elasticity of semiflexible biopolymer networks. Phys. Rev. Lett. 1995, 75, 4425–4428. [Google Scholar] [CrossRef] [PubMed]
- Taraban, M.B.; Feng, Y.; Hammouda, B.; Hyland, L.L.; Yu, Y.B. Chirality-Mediated Mechanical and Structural Properties of Oligopeptide Hydrogels. Chem. Mater. 2012, 24, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Hyland, L.L.; Taraban, M.B.; Hammouda, B.; Bruce Yu, Y. Mutually reinforced multicomponent polysaccharide networks. Biopolymers 2011, 95, 840–851. [Google Scholar] [CrossRef]
- Hyland, L.L.; Taraban, M.B.; Feng, Y.; Hammouda, B.; Yu, Y.B. Viscoelastic properties and nanoscale structures of composite oligopeptide-polysaccharide hydrogels. Biopolymers 2012, 97, 177–188. [Google Scholar] [CrossRef]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering; Wiley-Interscience: New York, NY USA, 1976. [Google Scholar]
- Brown, W. Light Scattering: Principles and Developmen; Clarendon: Oxford, UK, 1996. [Google Scholar]
- Chu, B. Light Scattering. Basic Principle and Practice; Academic Press: San Diego, CA, USA, 1991. [Google Scholar]
- Lombardo, D.; Micali, N.; Villari, V.; Kiselev, M.A. Large structures in diblock copolymer micellar solution. Phys. Rev. E 2004, 70, 021402. [Google Scholar] [CrossRef]
- Alexander, M.; Ross, F. Hallett Small-angle light scattering: Instrumental design and application to particle sizing. Appl. Opt. 1999, 38, 4158–4163. [Google Scholar] [CrossRef]
- Chen, S.H.; Lombardo, D.; Mallamace, F.; Micali, N.; Trusso, S.; Vasi, C. Small-angle light scattering in microemulsions (spinodal decomposition). In Trends in Colloid and Interface Science VII; Laggner, P., Glatter, O., Eds.; Progress in Colloid & Polymer Science; Springer: Berlin, Germany, 1993; Volume 93, pp. 311–316. [Google Scholar] [CrossRef]
- Castelletto, V.; Hamley, I.W. Capillary flow behavior of worm-like micelles studied by small-angle X-ray scattering and small angle light scattering. Polym. Adv. Technol. 2006, 17, 137–144. [Google Scholar] [CrossRef]
- Butler, M.F. Mechanism and kinetics of phase separation in a gelatin/maltodextrin mixture studied by small-angle light scattering. Biomacromolecules 2002, 3, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Izumitani, T.; Hasegawa, H.; Hashimoto, T.; Han, C.C. Small-angle neutron scattering and light scattering study on the miscibility of poly(styrene-ran-butadiene)/polybutadiene blends. Macromolecules 1991, 24, 4844–4851. [Google Scholar] [CrossRef]
- Sacks, M.S.; Smith, D.B.; Hiester, E.D. A small angle light scattering device for planar connective tissue microstructural analysis. Ann. Biomed. Eng. 1997, 25, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, M.C.; Zareian, R.; DiMarzio, C.A.; Wan, C.-T.; Ruberti, J.W. Small-angle light scattering to detect strain-directed collagen degradation in native tissue. Interface Focus. 2011, 1, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Dashtimoghadam, E.; Mirzadeh, H.; Taromia, F.A.; Nyström, B. Thermoresponsive biopolymer hydrogels with tunable gel characteristics. RSC Adv. 2014, 4, 39386–39393. [Google Scholar] [CrossRef]
- Lombardo, D.; Longo, A.; Darcy, R.; Mazzaglia, A. Structural Properties of Nonionic Cyclodextrin Colloids in Water. Langmuir 2004, 20, 1057–1064. [Google Scholar] [CrossRef]
- Stepanek, P. Data analysis in dynamic light scattering. In Dynamic Light Scattering: The Method and Some Applications; Brown, W., Ed.; Clarendon Press: Oxford, UK, 1993; pp. 175–241. [Google Scholar]
- Jakes, J. Regularized Positive Exponential Sum (REPES) Program—A Way of Inverting Laplace Transform Data Obtained by Dynamic Light Scattering. Collect. Czechoslov. Chem. Commun. 1995, 60, 1781–1797. [Google Scholar] [CrossRef]
- Provencher, S.W. CONTIN: A general purpose constrained regularization program for inverting noisy linear algebraic and integral equations. Comput Phys Commun 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Ruhe, A. Fitting empirical data by positive sums of exponentials. SIAM J. Sci. Stat. Comput. 1980, 1, 481–498. [Google Scholar] [CrossRef]
- Grant, T.D.; Luft, J.R.; Wolfley, J.R.; Tsuruta, H.; Martel, A.; Montelione, G.T.; Snell, E.H. Small angle X-ray scattering as a complementary tool for high-throughput structural studies. Biopolymers 2011, 95, 517–530. [Google Scholar] [CrossRef]
- Narayanan, T.; Konovalov, O. Synchrotron Scattering Methods for Nanomaterials and Soft Matter Research. Materials 2020, 13, 752. [Google Scholar] [CrossRef] [PubMed]
- Nieh, M.P.; Heberle, F.A.; Katsaras, J. Characterization of Biological Membranes Structure and Dynamics; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2019. [Google Scholar]
- Pignatello, R.; Musumeci, T.; Basile, L. Biomembrane models and drug-biomembrane interaction studies: Involvement in drug design and development. J. Pharm. Bioallied. Sci. 2011, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Mazzaglia, A.; Angelini, N.; Darcy, R.; Donohue, R.; Lombardo, D.; Micali, N.; Sciortino, M.T.; Villari, V.; Scolaro, L.M. Novel heterotopic colloids of anionic porphyrins entangled in cationic amphiphilic cyclodextrins: Spectroscopic investigation and intracellular delivery. Chem. Eur. J. 2003, 9, 5762–5769. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Maiuolo, L.; Lombardo, D.; Szerb, E.I.; Calandra, P. Luminescent Supramolecular Nano-or Microstructures Formed in Aqueous Media by Amphiphile-Noble Metal Complexes. J. Nanomater. 2020, 5395048. [Google Scholar] [CrossRef]
- Pignatello, R. Drug-Biomembrane Interaction Studies, the Application of Calorimetric Techniques; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Abraham, T.; Lewis, R.N.A.H.; Hodges, R.S.; McElhaney, R.N. Isothermal titration calorimetry studies of the binding of a rationally designed analogue of the antimicrobial peptide gramicidin s to phospholipid bilayer membranes. Biochemistry 2005, 44, 2103–2112. [Google Scholar] [CrossRef]
- Bozzola, J.J.; Russell, L.D. Electron Microscopy: Principles and Techniques for Biologists; Jones and Bartlett: Burlington, MA, USA, 1998. [Google Scholar]
- Hayat, M. Principles and Techniques of Electron Microscopy: Biological Applications; Cambridge University: Cambridge, UK, 2000. [Google Scholar]
- Helvig, S.; Azmi, I.D.M.; Moghimi, S.M.; Yaghmur, A. Recent advances in cryo-TEM imaging of soft lipid nanoparticles. AIMS Biophys. 2015, 2, 116–130. [Google Scholar] [CrossRef]
- Variola, F. Atomic force microscopy in biomaterials surface science. Phys. Chem. Chem. Phys. 2015, 17, 2950–2959. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Z | SANS | SAXS |
|---|---|---|---|
| Hydrogen (1H) | 1 | −0.374 | 0.28 |
| Deuterium (2D) | 1 | 0.67 | 0.28 |
| Carbon (12C) | 6 | 0.66 | 1.69 |
| Nitrogen (14N) | 7 | 0.94 | 1.97 |
| Oxygen (16O) | 8 | 0.58 | 2.25 |
| Aluminium (27N) | 13 | 0.35 | 3.65 |
| Phosphorous (31P) | 15 | 0.51 | 4.23 |
| Sulfur (32N) | 16 | 0.28 | 4.50 |
| Molecule | SANS | SAXS |
|---|---|---|
| H2O | −0.56 | 9.39 |
| D2O | 6.73 | 9.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardo, D.; Calandra, P.; Kiselev, M.A. Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments. Molecules 2020, 25, 5624. https://doi.org/10.3390/molecules25235624
Lombardo D, Calandra P, Kiselev MA. Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments. Molecules. 2020; 25(23):5624. https://doi.org/10.3390/molecules25235624
Chicago/Turabian StyleLombardo, Domenico, Pietro Calandra, and Mikhail A. Kiselev. 2020. "Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments" Molecules 25, no. 23: 5624. https://doi.org/10.3390/molecules25235624
APA StyleLombardo, D., Calandra, P., & Kiselev, M. A. (2020). Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments. Molecules, 25(23), 5624. https://doi.org/10.3390/molecules25235624