
Integrated Leaching and Separation of Metals Using Mixtures of Organic Acids and Ionic Liquids




Abstract

1. Introduction
2. Results and Discussion
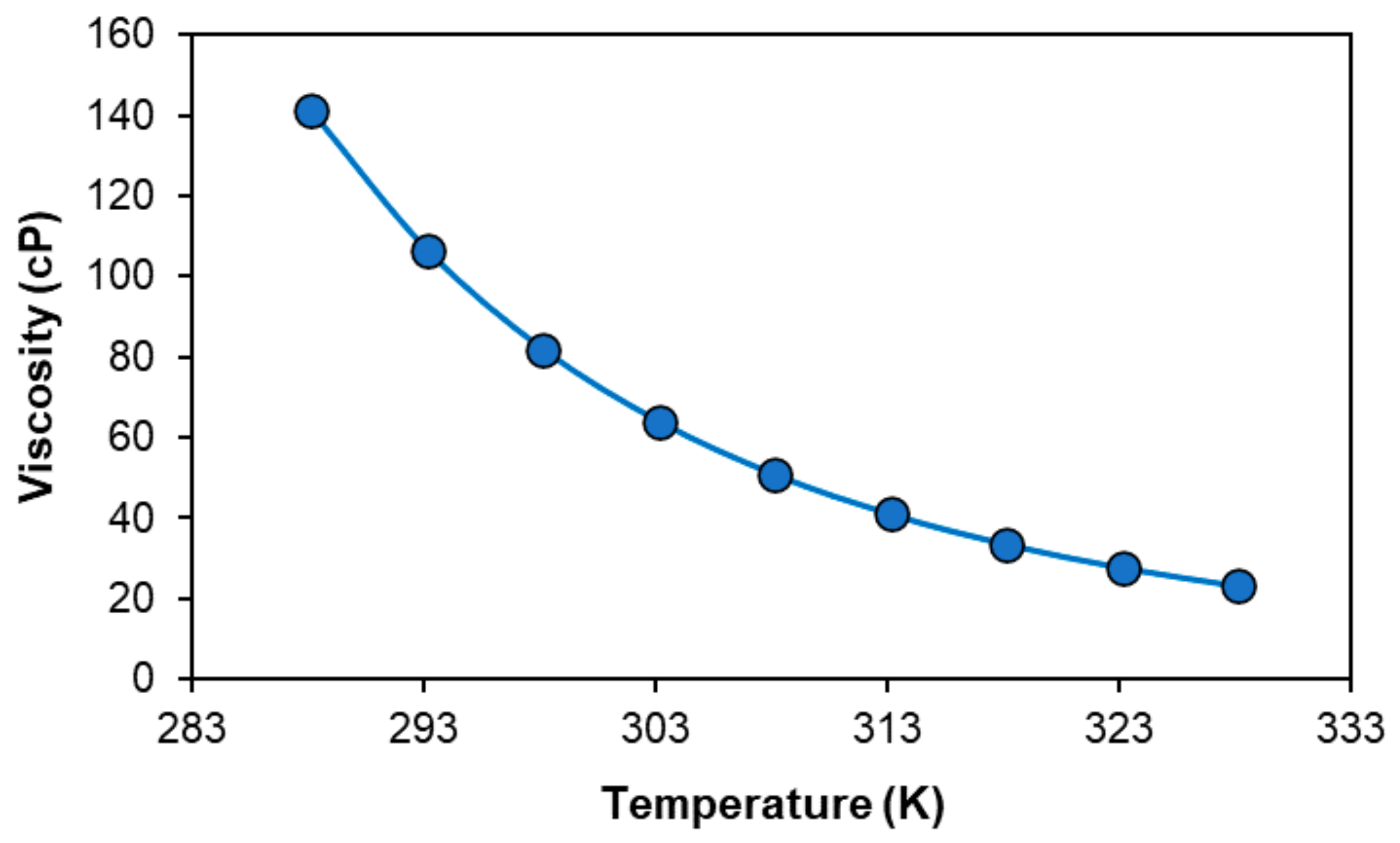
2.1. Characterisation of the [P44414]Cl + CH3COOH + H2O System
2.2. Mechanism of Phase Separation in the [P44414]Cl + CH3COOH + H2O System
2.3. Metal Oxide Leaching in the [P44414]Cl + CH3COOH System
2.4. Application of the [P44414]Cl + CH3COOH System to Waste NiMH Battery
3. Methodology
3.1. Reagent and Instrumentation
3.2. Determination and Characterisation of [P44414]Cl + Organic Acid + H2O Systems
3.3. Dissolution of Metal Oxides and Separation
3.4. Molecular Dynamic Simulations of the [P44414]Cl + CH3COOH+ H2O System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Binnemans, K.; Jones, P.T. Solvometallurgy: An Emerging Branch of Extractive Metallurgy. J. Sustain. Metall. 2017, 3, 570–600. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, R.-W.; Jang, S.E.J.S.S.; Wong, C.-P.; Hong, J.-I. “Organic Aqua Regia”-Powerful Liquids for Dissolving Noble Metals. Angew. Chem. Int. Ed. 2010, 49, 7929–7932. [Google Scholar] [CrossRef] [PubMed]
- Räisänen, M.; Heliövaara, E.; Al-Qaisi, F.; Muuronen, M.; Eronen, A.; Liljeqvist, H.; Nieger, M.; Kemell, M.; Moslova, K.; Hämäläinen, J.; et al. Pyridinethiol-Assisted Dissolution of Elemental Gold in Organic Solutions. Angew. Chem. Int. Ed. 2018, 57, 17104–17109. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Frisch, G.; Gurman, S.J.; Hillman, A.R.; Hartley, J.; Holyoak, F.; Ryder, K.S. Ionometallurgy: Designer redox properties for metal processing. Chem. Comm. 2011, 47, 10031–10033. [Google Scholar] [CrossRef]
- Hartley, J.M.; Ip, C.-M.; Forrest, G.C.H.; Singh, K.; Gurman, S.J.; Ryder, K.S.; Abbott, A.P.; Frisch, G. EXAFS Study into the Speciation of Metal Salts Dissolved in Ionic Liquids and Deep Eutectic Solvents. Inorg. Chem. 2014, 53, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K. Ionic Liquid Analogues Formed from Hydrated Metal Salts. Chem. Eur. J. 2004, 10, 3769–3774. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Harris, R.C.; Holyoak, F.; Frisch, G.; Hartley, J.; Jenkin, G.R.T. Electrocatalytic recovery of elements from complex mixtures using deep eutectic solvents. Green Chem. 2015, 17, 2172–2179. [Google Scholar] [CrossRef]
- Jenkin, G.R.; Al-Bassam, A.Z.; Harris, R.C.; Abbott, A.P.; Smith, D.J.; Holwell, D.A.; Chapman, R.J.; Stanley, C.J. The application of deep eutectic solvent ionic liquids for environmentally-friendly dissolution and recovery of precious metals. Miner. Eng. 2016, 87, 18–24. [Google Scholar] [CrossRef]
- Abbott, A.P.; Al-Bassam, A.Z.M.; Goddard, A.; Harris, R.C.; Jenkin, G.R.T.; Nisbet, F.J.; Wieland, M. Dissolution of pyrite and other Fe–S–As minerals using deep eutectic solvents. Green Chem. 2017, 19, 2225–2233. [Google Scholar] [CrossRef]
- Anggara, S.; Bevan, F.; Harris, R.C.; Hartley, J.M.; Frisch, G.; Jenkin, G.R.T.; Abbott, A.P. Direct extraction of copper from copper sulfide minerals using deep eutectic solvents. Green Chem. 2019, 21, 6502–6512. [Google Scholar] [CrossRef]
- Zürner, P.; Frisch, G. Leaching and Selective Extraction of Indium and Tin from Zinc Flue Dust Using an Oxalic Acid-Based Deep Eutectic Solvent. ACS Sustain. Chem. Eng. 2019, 7, 5300–5308. [Google Scholar] [CrossRef]
- Abbott, A.P.; Frisch, G.; Hartley, J.; Ryder, K.S. Processing of metals and metal oxides using ionic liquids. Green Chem. 2011, 13, 471–481. [Google Scholar] [CrossRef]
- Rodriguez, N.R.; Machiels, L.; Binnemans, K. p-Toluenesulfonic Acid-Based Deep-Eutectic Solvents for Solubilizing Metal Oxides. ACS Sustain. Chem. Eng. 2019, 7, 3940–3948. [Google Scholar] [CrossRef]
- Damilano, G.; Laitinen, A.; Willberg-Keyriläinen, P.; Lavonen, T.; Häkkinen, R.; Dehaen, W.; Binnemans, K.; Kuutti, L. Effects of thiol substitution in deep-eutectic solvents (DESs) as solvents for metal oxides. RSC Adv. 2020, 10, 23484–23490. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; McKenzie, K.J.; Obi, S.U. Solubility of Metal Oxides in Deep Eutectic Solvents Based on Choline Chloride. J. Chem. Eng. Data 2006, 51, 1280–1282. [Google Scholar] [CrossRef]
- Jiang, J.; Bai, X.; Zhao, X.; Chen, W.; Yu, T.; Li, Y.; Mu, T. Poly-quasi-eutectic solvents (PQESs): Versatile solvents for dissolving metal oxides. Green Chem. 2019, 21, 5571–5578. [Google Scholar] [CrossRef]
- Smith, D.F.; A Gucinski, J. Synthetic silver oxide and mercury-free zinc electrodes for silver–zinc reserve batteries. J. Power Sources 1999, 80, 66–71. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Q.; Zhang, S.; Lu, X.; Zhang, X. Electrodeposition in Ionic Liquids. ChemPhysChem 2015, 17, 335–351. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Van Osch, D.J.; Zubeir, L.F.; Bruinhorst, A.A.V.D.; Da Rocha, M.M.A.; Kroon, M.C. Hydrophobic deep eutectic solvents as water-immiscible extractants. Green Chem. 2015, 17, 4518–4521. [Google Scholar] [CrossRef]
- Phelps, T.E.; Bhawawet, N.; Jurisson, S.S.; Baker, G.A. Efficient and Selective Extraction of 99mTcO4– from Aqueous Media Using Hydrophobic Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2018, 6, 13656–13661. [Google Scholar] [CrossRef]
- Tereshatov, E.E.; Boltoeva, M.Y.; Folden, C.M. First evidence of metal transfer into hydrophobic deep eutectic and low-transition-temperature mixtures: Indium extraction from hydrochloric and oxalic acids. Green Chem. 2016, 18, 4616–4622. [Google Scholar] [CrossRef]
- Rodriguez, N.R.; Machiels, L.; Onghena, B.; Spooren, J.; Binnemans, K. Selective recovery of zinc from goethite residue in the zinc industry using deep-eutectic solvents. RSC Adv. 2020, 10, 7328–7335. [Google Scholar] [CrossRef]
- Söldner, A.; Zach, J.; König, B. Deep eutectic solvents as extraction media for metal salts and oxides exemplarily shown for phosphates from incinerated sewage sludge ash. Green Chem. 2019, 21, 321–328. [Google Scholar] [CrossRef]
- Riaño, S.; Petranikova, M.; Onghena, B.; Hoogerstraete, T.V.; Banerjee, D.; Foreman, M.R.S.-J.; Ekberg, C.; Binnemans, K. Separation of rare earths and other valuable metals from deep-eutectic solvents: A new alternative for the recycling of used NdFeB magnets. RSC Adv. 2017, 7, 32100–32113. [Google Scholar] [CrossRef]
- Landa-Castro, M.; Aldana-González, J.; De Oca-Yemha, M.M.; Romero-Romo, M.; Arce-Estrada, E.; Palomar-Pardavé, M. Ni–Co alloy electrodeposition from the cathode powder of Ni-MH spent batteries leached with a deep eutectic solvent (reline). J. Alloys Compd. 2020, 830, 154650. [Google Scholar] [CrossRef]
- Tran, M.K.; Rodrigues, M.-T.F.F.; Kato, K.; Babu, G.; Ajayan, P.M. Deep eutectic solvents for cathode recycling of Li-ion batteries. Nat. Energy 2019, 4, 339–345. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Lu, Z.; Xu, Z. A novel method for screening deep eutectic solvent to recycle the cathode of Li-ion batteries. Green Chem. 2020, 22, 4473–4482. [Google Scholar] [CrossRef]
- Peeters, N.; Binnemans, K.; Riaño, S. Solvometallurgical recovery of cobalt from lithium-ion battery cathode materials using deep-eutectic solvents. Green Chem. 2020, 22, 4210–4221. [Google Scholar] [CrossRef]
- Passos, H.; Tavares, D.J.P.; Ferreira, A.M.; Freire, M.G.; Coutinho, J.A.P. Are Aqueous Biphasic Systems Composed of Deep Eutectic Solvents Ternary or Quaternary Systems? ACS Sustain. Chem. Eng. 2016, 4, 2881–2886. [Google Scholar] [CrossRef]
- Spathariotis, S.; Peeters, N.; Ryder, K.S.; Abbott, A.P.; Binnemans, K.; Riaño, S. Separation of iron(iii), zinc(ii) and lead(ii) from a choline chloride–ethylene glycol deep eutectic solvent by solvent extraction. RSC Adv. 2020, 10, 33161–33170. [Google Scholar] [CrossRef]
- Cherigui, E.A.M.; Sentosun, K.; Mamme, M.H.; Lukaczynska, M.; Terryn, H.A.; Bals, S.; Ustarroz, J. On the Control and Effect of Water Content during the Electrodeposition of Ni Nanostructures from Deep Eutectic Solvents. J. Phys. Chem. C 2018, 122, 23129–23142. [Google Scholar] [CrossRef]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. The Effect of Water upon Deep Eutectic Solvent Nanostructure: An Unusual Transition from Ionic Mixture to Aqueous Solution. Angew. Chem. Int. Ed. 2017, 56, 9782–9785. [Google Scholar] [CrossRef]
- Florindo, C.; Branco, L.C.; Marrucho, I.M. Quest for Green-Solvent Design: From Hydrophilic to Hydrophobic (Deep) Eutectic Solvents. ChemSusChem 2019, 12, 1549–1559. [Google Scholar] [CrossRef]
- Saita, S.; Kohno, Y.; Ohno, H. Detection of small differences in the hydrophilicity of ions using the LCST-type phase transition of an ionic liquid–water mixture. Chem. Commun. 2012, 49, 93–95. [Google Scholar] [CrossRef]
- Dong, D.; Hooper, J.B.; Bedrov, D. Structural and Dynamical Properties of Tetraalkylammonium Bromide Aqueous Solutions: A Molecular Dynamics Simulation Study Using a Polarizable Force Field. J. Phys. Chem. B 2017, 121, 4853–4863. [Google Scholar] [CrossRef]
- Kumar, S.; Aswal, V.K.; Naqvi, A.Z.; Goyal, P.S.; Din, K.U. Cloud Point Phenomenon in Ionic Micellar Solutions: A SANS Study. Langmuir 2001, 17, 2549–2551. [Google Scholar] [CrossRef]
- Schaeffer, N.; Pérez-Sánchez, G.; Passos, H.; Gomes, J.R.; Papaiconomou, N.; Coutinho, J.A.P. Mechanisms of phase separation in temperature-responsive acidic aqueous biphasic systems. Phys. Chem. Chem. Phys. 2019, 21, 7462–7473. [Google Scholar] [CrossRef]
- Longeras, O.; Gautier, A.; Ballerat-Busserolles, K.; Andanson, J.-M. Deep Eutectic Solvent with Thermo-Switchable Hydrophobicity. ACS Sustain. Chem. Eng. 2020, 8, 12516–12520. [Google Scholar] [CrossRef]
- European Commission; The Commission to the European Parliament, the Council; The European Economic and Social Committee; The Committee of the Regions. Critical Raw Materials Resilience: Charting a Path towards Greater Security and Sustainability; European Commission: Brussels, Belgium, 2020; Available online: https://ec.europa.eu/info/index_en (accessed on 3 September 2020).
- Fu, X.; Beatty, D.N.; Gaustad, G.; Ceder, G.; Roth, R.; Kirchain, R.E.; Bustamante, M.; Babbitt, C.W.; Olivetti, E. Perspectives on Cobalt Supply through 2030 in the Face of Changing Demand. Environ. Sci. Technol. 2020, 54, 2985–2993. [Google Scholar] [CrossRef]
- Pateli, I.M.; Thompson, D.; Alabdullah, S.S.M.; Abbott, A.P.; Jenkin, G.R.T.; Hartley, J.M. The effect of pH and hydrogen bond donor on the dissolution of metal oxides in deep eutectic solvents. Green Chem. 2020, 22, 5476–5486. [Google Scholar] [CrossRef]
- Golmohammadzadeh, R.; Faraji, F.; Rashchi, F. Recovery of lithium and cobalt from spent lithium ion batteries (LIBs) using organic acids as leaching reagents: A review. Resour. Conserv. Recycl. 2018, 136, 418–435. [Google Scholar] [CrossRef]
- Ruggeri, S.; Poletti, F.; Zanardi, C.; Pigani, L.; Zanfrognini, B.; Corsi, E.; Dossi, N.; Salomäki, M.; Kivelä, H.; Lukkari, J.; et al. Chemical and electrochemical properties of a hydrophobic deep eutectic solvent. Electrochim. Acta 2019, 295, 124–129. [Google Scholar] [CrossRef]
- Florindo, C.; Branco, L.; Marrucho, I. Development of hydrophobic deep eutectic solvents for extraction of pesticides from aqueous environments. Fluid Ph. Equilib. 2017, 448, 135–142. [Google Scholar] [CrossRef]
- Gras, M.; Papaiconomou, N.; Schaeffer, N.; Chainet, E.; Tedjar, F.; Coutinho, J.A.P.; Billard, I. Ionic-Liquid-Based Acidic Aqueous Biphasic Systems for Simultaneous Leaching and Extraction of Metallic Ions. Angew. Chem. Int. Ed. 2018, 57, 1563–1566. [Google Scholar] [CrossRef]
- Tomé, L.I.N.; Pereira, J.F.B.; Rogers, R.D.; Freire, M.G.; Gomes, J.R.; Coutinho, J.A.P. “Washing-out” ionic liquids from polyethylene glycol to form aqueous biphasic systems. Phys. Chem. Chem. Phys. 2014, 16, 2271. [Google Scholar] [CrossRef]
- Neves, C.M.S.S.; Shahriari, S.; Lemus, J.; Pereira, J.F.B.; Freire, M.G.; Coutinho, J.A.P. Aqueous biphasic systems composed of ionic liquids and polypropylene glycol: Insights into their liquid–liquid demixing mechanisms. Phys. Chem. Chem. Phys. 2016, 18, 20571–20582. [Google Scholar] [CrossRef]
- Qiao, Y.; Ma, W.; Theyssen, N.; Chen, C.; Hou, Z. Temperature-Responsive Ionic Liquids: Fundamental Behaviors and Catalytic Applications. Chem. Rev. 2017, 117, 6881–6928. [Google Scholar] [CrossRef]
- Sangster, J. Octanol-Water Partition Coefficients of Simple Organic Compounds. J. Phys. Chem. Ref. Data 1989, 18, 1111–1229. [Google Scholar] [CrossRef]
- Cláudio, A.F.M.; Neves, M.C.; Shimizu, K.; Lopes, J.N.C.; Freire, M.G.; Coutinho, J.A.P. The magic of aqueous solutions of ionic liquids: Ionic liquids as a powerful class of catanionic hydrotropes. Green Chem. 2015, 17, 3948–3963. [Google Scholar] [CrossRef]
- Posada, E.; López-Salas, N.; Riobóo, R.J.J.; Ferrer, M.L.; Gutiérrez, M.C.; Del Monte, F. Reline aqueous solutions behaving as liquid mixtures of H-bonded co-solvents: Microphase segregation and formation of co-continuous structures as indicated by Brillouin and 1H NMR spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 17103–17110. [Google Scholar] [CrossRef] [PubMed]
- Kuddushi, M.; Nangala, G.S.; Rajput, S.; Ijardar, S.P.; Malek, N.I. Understanding the peculiar effect of water on the physicochemical properties of choline chloride based deep eutectic solvents theoretically and experimentally. J. Mol. Liq. 2019, 278, 607–615. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS - A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Weber, H.; Thomas, M.; Hollóczki, O.; Kirchner, B. Domain Analysis in Nanostructured Liquids: A Post-Molecular Dynamics Study at the Example of Ionic Liquids. ChemPhysChem 2015, 16, 3271–3277. [Google Scholar] [CrossRef]
- Konings, R.J.M.; Beneš, O. The Thermodynamic Properties of the f-Elements and Their Compounds. I. The Lanthanide and Actinide Metals. J. Phys. Chem. Ref. Data 2010, 39, 043102. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, J.; Lan, X.; Zhao, X.; Mou, H.; Mu, T. A strategy for the dissolution and separation of rare earth oxides by novel Brønsted acidic deep eutectic solvents. Green Chem. 2019, 21, 4748–4756. [Google Scholar] [CrossRef]
- Liu, W.; Etschmann, B.; Brugger, J.; Spiccia, L.; Foran, G.; McInnes, B. UV–Vis spectrophotometric and XAFS studies of ferric chloride complexes in hyper-saline LiCl solutions at 25–90 °C. Chem. Geol. 2006, 231, 326–349. [Google Scholar] [CrossRef]
- Zhang, N.; Brugger, J.; Etschmann, B.; Ngothai, Y.; Zeng, D. Thermodynamic Modeling of Poorly Complexing Metals in Concentrated Electrolyte Solutions: An X-Ray Absorption and UV-Vis Spectroscopic Study of Ni(II) in the NiCl2-MgCl2-H2O System. PLoS ONE 2015, 10, e0119805. [Google Scholar] [CrossRef]
- Nockemann, P.; Thijs, B.; Parac-Vogt, T.N.; Van Hecke, K.; Van Meervelt, L.; Tinant, B.; Hartenbach, I.; Schleid, T.; Ngan, V.T.; Nguyen, M.T.; et al. Carboxyl-Functionalized Task-Specific Ionic Liquids for Solubilizing Metal Oxides. Inorg. Chem. 2008, 47, 9987–9999. [Google Scholar] [CrossRef]
- Fagnant, J.D.P.; Goff, G.S.; Scott, B.L.; Runde, W.; Brennecke, J.F. Switchable Phase Behavior of [HBet][Tf2N]–H2O upon Neodymium Loading: Implications for Lanthanide Separations. Inorg. Chem. 2013, 52, 549–551. [Google Scholar] [CrossRef]
- Zanonato, P.L.; Di Bernardo, P.; Bismondo, A.; Rao, L.; Choppin, G.R. Thermodynamic Studies of the Complexation between Neodymium and Acetate at Elevated Temperatures. J. Solut. Chem. 2001, 30, 1–18. [Google Scholar] [CrossRef]
- Gammons, C.; Wood, S.; Williams-Jones, A. The aqueous geochemistry of the rare earth elements and yttrium: VI. Stability of neodymium chloride complexes from 25 to 300°C. Geochim. Cosmochim. Acta 1996, 60, 4615–4630. [Google Scholar] [CrossRef]
- Fernandes, A.; Afonso, J.C.; Dutra, A.J.B. Separation of nickel(II), cobalt(II) and lanthanides from spent Ni-MH batteries by hydrochloric acid leaching, solvent extraction and precipitation. Hydrometallurgy 2013, 133, 37–43. [Google Scholar] [CrossRef]
- Schaeffer, N.; Gras, M.; Passos, H.; Mogilireddy, V.; Mendonça, C.M.N.; Pereira, E.; Chainet, E.; Billard, I.; Coutinho, J.A.P.; Papaiconomou, N. (N.) Synergistic Aqueous Biphasic Systems: A New Paradigm for the “One-Pot” Hydrometallurgical Recovery of Critical Metals. ACS Sustain. Chem. Eng. 2018, 7, 1769–1777. [Google Scholar] [CrossRef]
- Larsson, K.; Binnemans, K. Selective extraction of metals using ionic liquids for nickel metal hydride battery recycling. Green Chem. 2014, 16, 4595–4603. [Google Scholar] [CrossRef]
- Lommelen, R.; Hoogerstraete, T.V.; Onghena, B.; Billard, I.; Binnemans, K. Model for Metal Extraction from Chloride Media with Basic Extractants: A Coordination Chemistry Approach. Inorg. Chem. 2019, 58, 12289–12301. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Hockney, R.; Goel, S.; Eastwood, J. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- A Darden, T.; York, D.M.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Lopes, J.N.C.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids III: Imidazolium, Pyridinium, and Phosphonium Cations; Chloride, Bromide, and Dicyanamide Anions. J. Phys. Chem. B 2006, 110, 19586–19592. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture Point | Total Composition (wt.%) | Top Phase (wt.%) | Bottom Phase (wt.%) | TLL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL | Acid | H2O | IL | Acid | H2O | Vol (mL) | IL | Acid | H2O | Vol (mL) | ||
| 1 | 55.00 | 20.00 | 25.00 | 49.02 | 12.00 | 38.98 | 2.0 | 1.41 | 25.31 | 73.27 | 0.4 | 49.43 |
| 1* | 55.00 | 20.00 | 23.00 | 64.81 | 26.15 | 9.04 | 1.9 | 0.01 | 13.72 | 86.27 | 0.5 | - |
| 2 | 50.00 | 25.00 | 25.00 | 69.77 | 24.08 | 6.15 | 2.0 | 0.10 | 30.72 | 69.18 | 0.4 | 69.99 |
| System | n(H2O) | [IL] (wt.%) | [CH3COOH] (wt.%) | [H2O] (wt.%) | Experimental Regime |
|---|---|---|---|---|---|
| (1) | 0 | 71.4 | 28.6 | 0.0 | Monophasic |
| (2) | 363 | 67.8 | 27.1 | 5.1 | Monophasic |
| (3) | 725 | 64.5 | 25.8 | 9.7 | Monophasic |
| (4) | 1450 | 58.8 | 23.5 | 17.7 | Biphasic |
| (5) | 2900 | 50.0 | 20.0 | 30.0 | Biphasic |
| (6) | 5800 | 38.4 | 15.4 | 46.2 | Monophasic |
| (7) | 14,500 | 22.7 | 9.1 | 68.2 | Monophasic |
| Metal Oxide | ∆fGo (kJ·mol−1) | Solubility (mol·L−1) | Solubility (mol·L−1) |
|---|---|---|---|
| [P44414]Cl + CH3COOH | [Ch]Cl + CH3COOH a | ||
| CoO | −214.1 a | 0.349 ± 0.030 | ~0.10 |
| NiO | −211.7 a | 0.028 ± 0.001 | ~0.01 |
| Fe2O3 | −742.8 a | 0.015 ± 0.001 | <0.01 |
| Nd2O3 | −1854.2 b | 0.036 ± 0.002 | - |
Sample Availability: Samples of the compounds are available from the authors upon reasonable request if available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas, S.J.R.; Passos, H.; Schaeffer, N.; Coutinho, J.A.P. Integrated Leaching and Separation of Metals Using Mixtures of Organic Acids and Ionic Liquids. Molecules 2020, 25, 5570. https://doi.org/10.3390/molecules25235570
Vargas SJR, Passos H, Schaeffer N, Coutinho JAP. Integrated Leaching and Separation of Metals Using Mixtures of Organic Acids and Ionic Liquids. Molecules. 2020; 25(23):5570. https://doi.org/10.3390/molecules25235570
Chicago/Turabian StyleVargas, Silvia J. R., Helena Passos, Nicolas Schaeffer, and João A. P. Coutinho. 2020. "Integrated Leaching and Separation of Metals Using Mixtures of Organic Acids and Ionic Liquids" Molecules 25, no. 23: 5570. https://doi.org/10.3390/molecules25235570
APA StyleVargas, S. J. R., Passos, H., Schaeffer, N., & Coutinho, J. A. P. (2020). Integrated Leaching and Separation of Metals Using Mixtures of Organic Acids and Ionic Liquids. Molecules, 25(23), 5570. https://doi.org/10.3390/molecules25235570