
In Silico Discovery of Antimicrobial Peptides as an Alternative to Control SARS-CoV-2



Abstract
1. Introduction
2. Results and Discussion
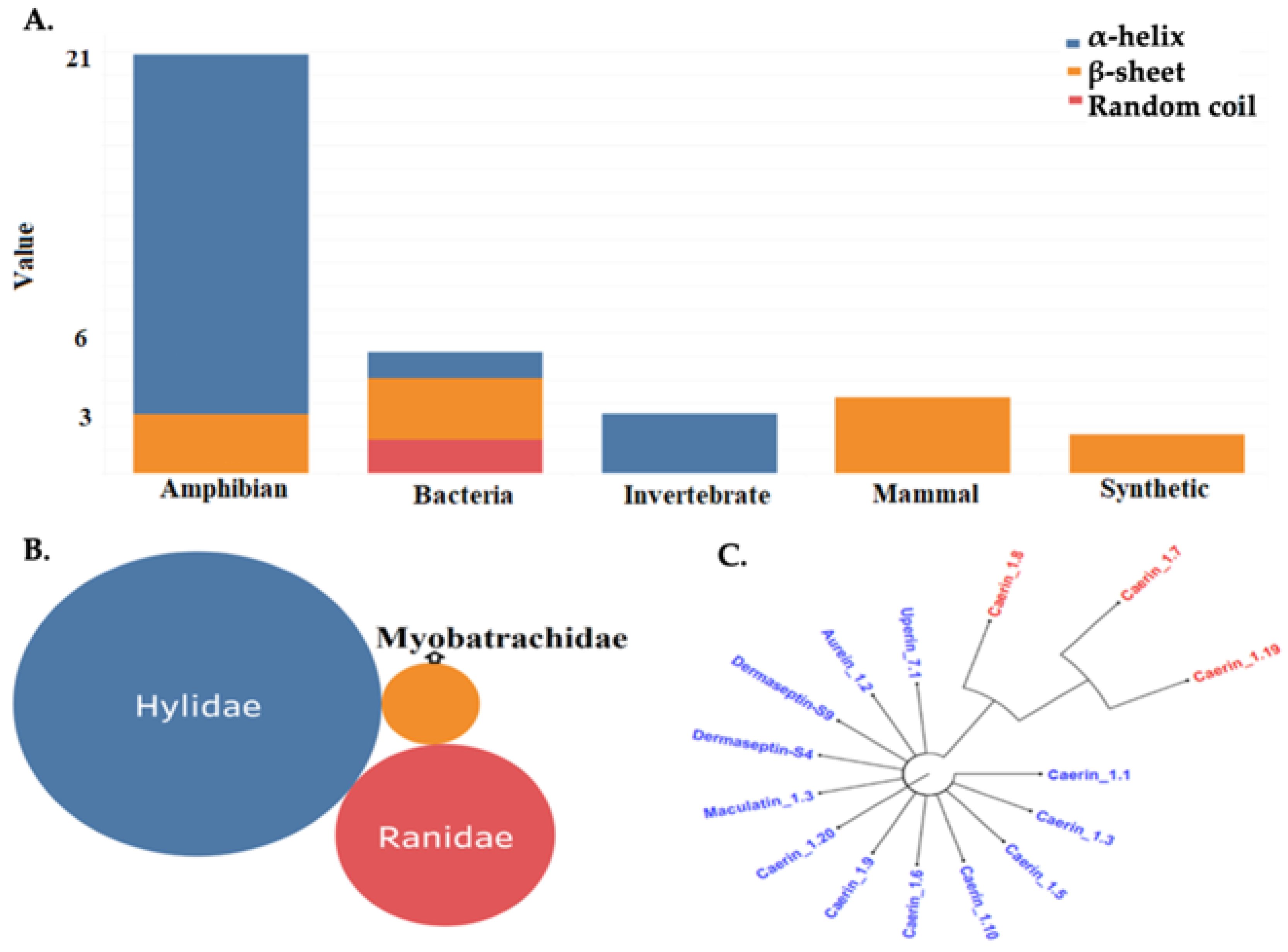
2.1. AMPs Clusters, Structural Prediction and Validation
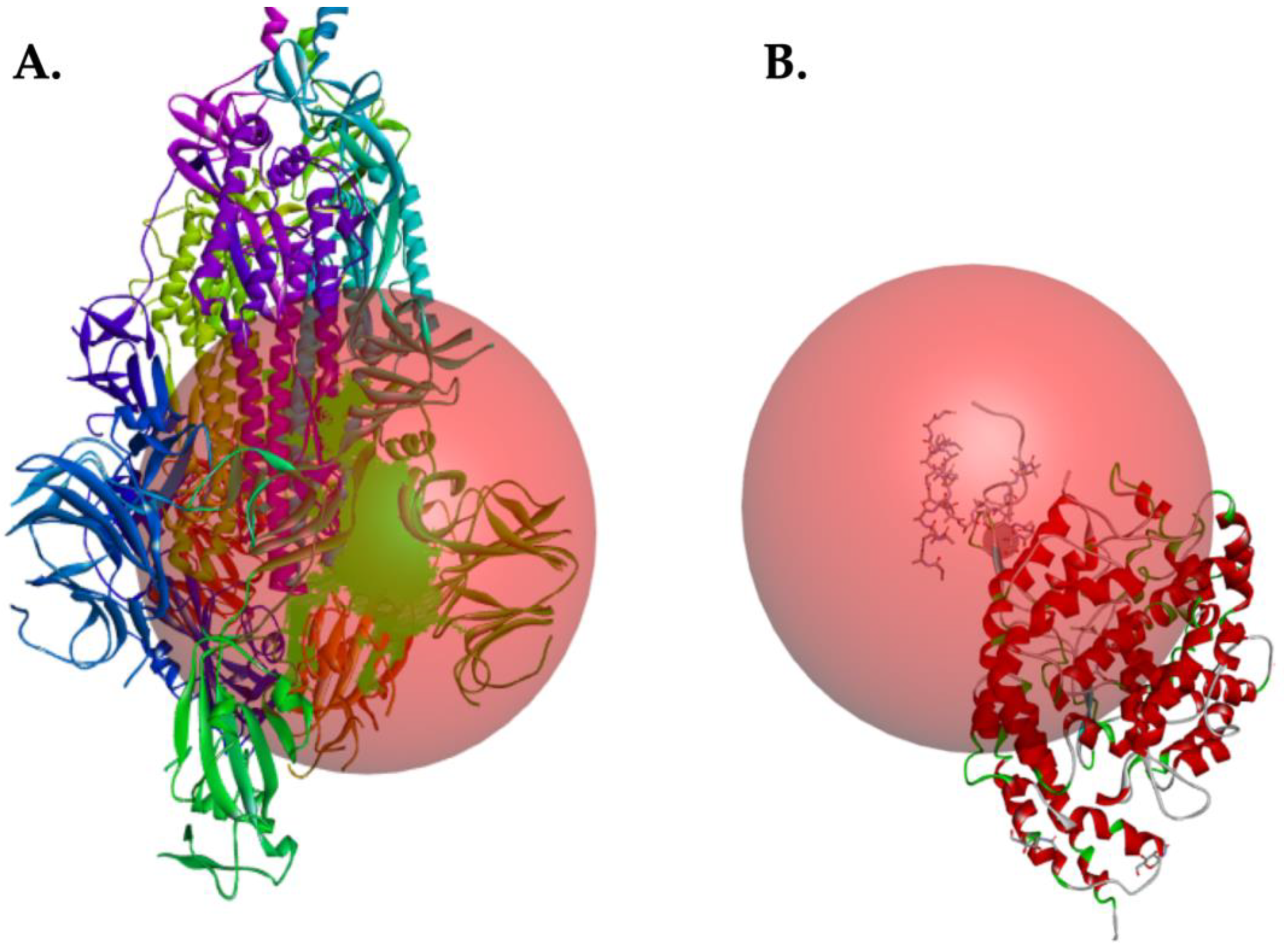
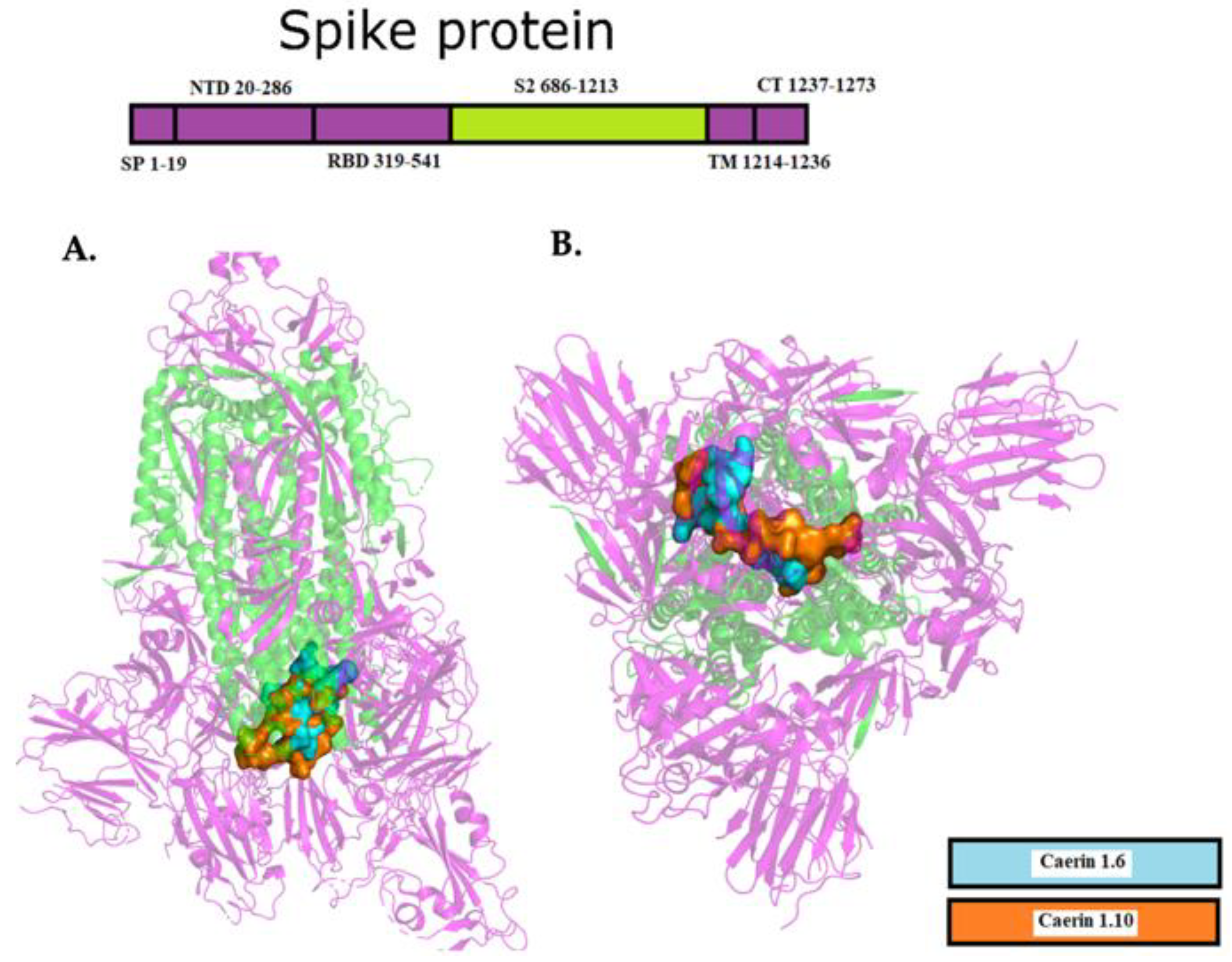
2.2. Coordinates for Gridbox of Target Proteins
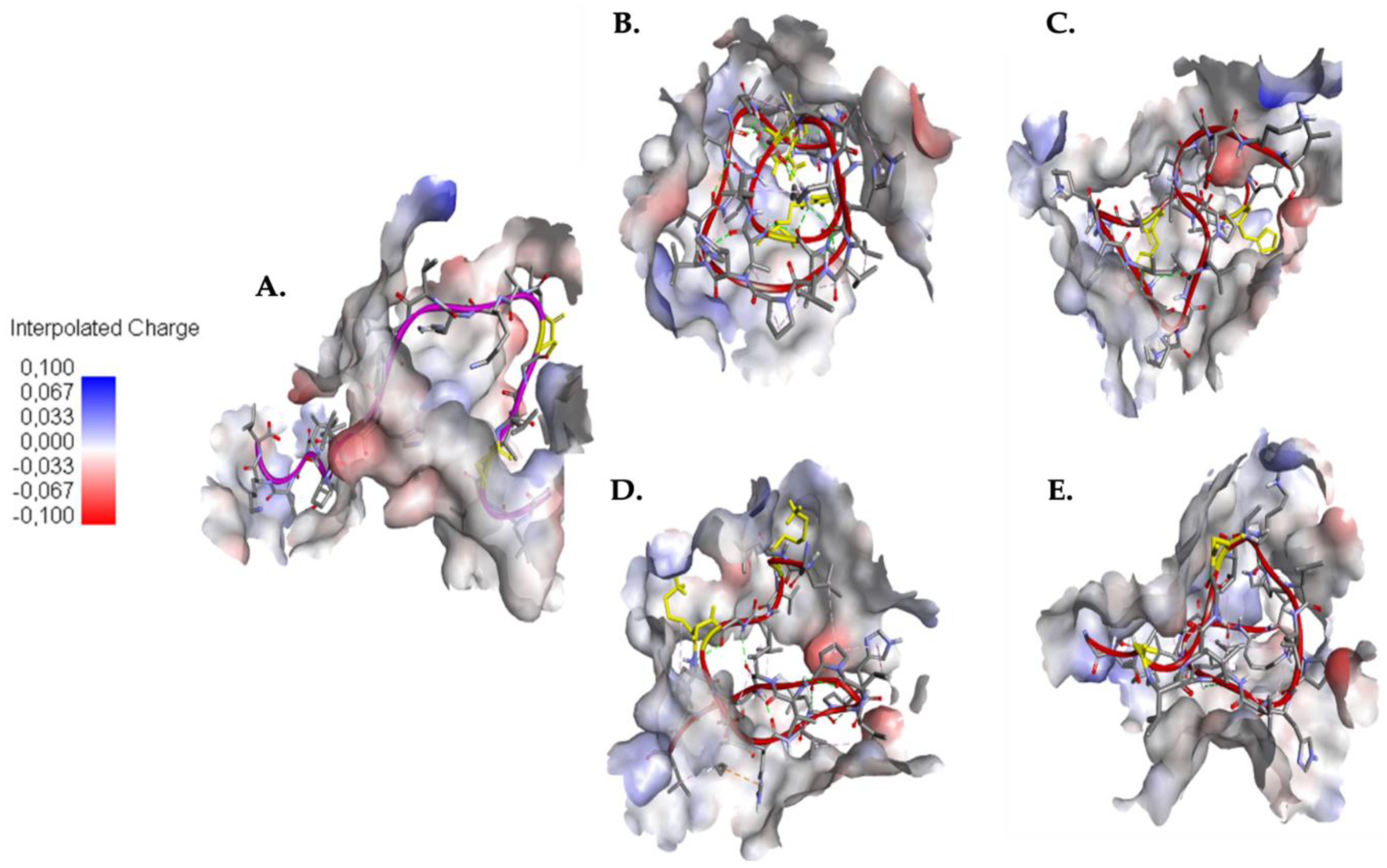
2.3. Interaction between EK1 and SARS-CoV-HR2P and Target Viral Protein
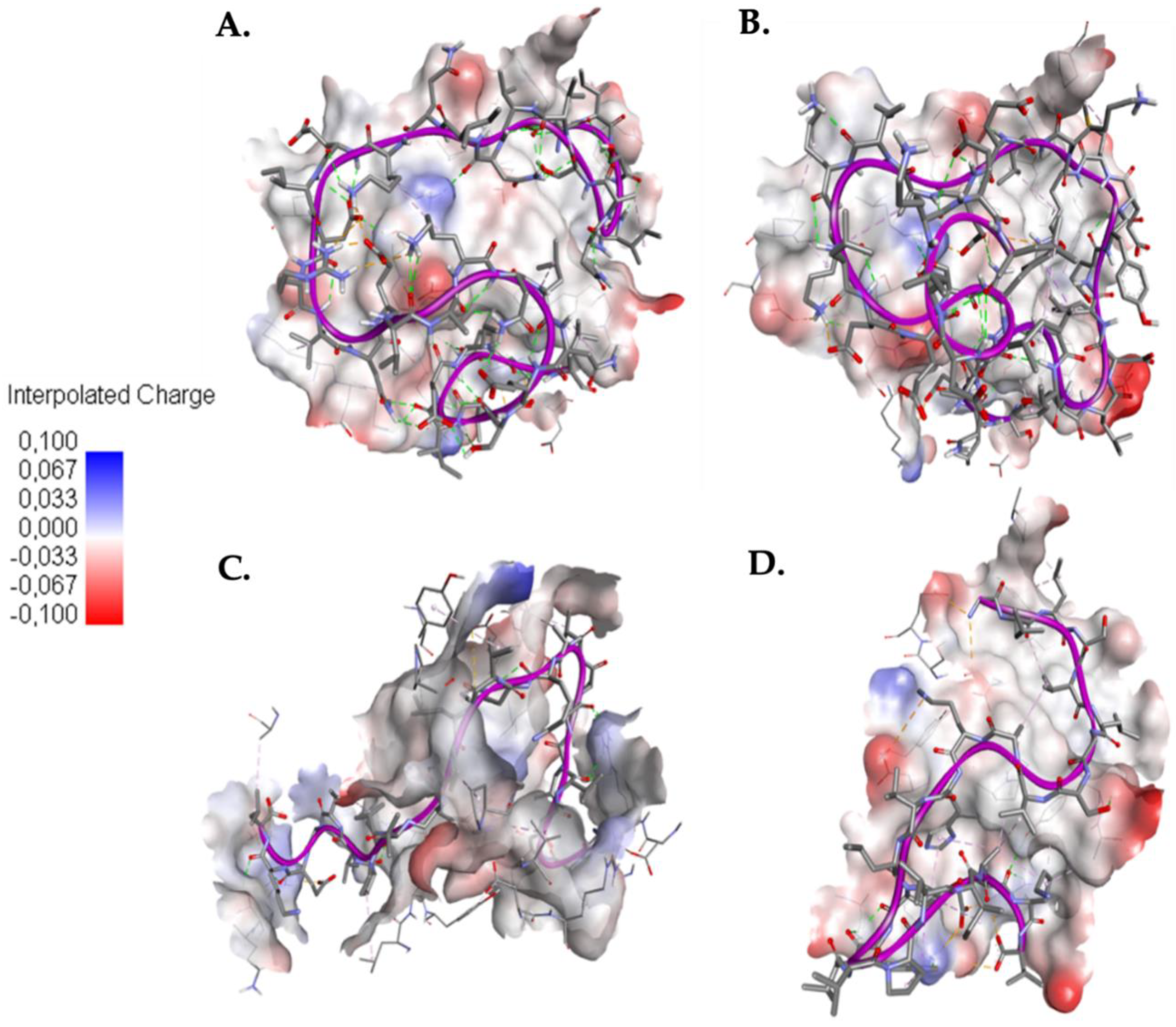
2.4. Understanding the Role of SER4 and SER8 in the Caerin 1.10
3. Materials and Methods
3.1. Public Datasets
3.2. Database Screening and Selection of Antimicrobial Peptides
3.3. In Silico Structural Modeling of AMPs and Validation
3.4. AMPs-Target Proteins Docking
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chan, J.F.; Li, K.S.; To, K.K.; Cheng, V.C.; Chen, H.; Yuen, K.-Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Hui, D.S.; I Azhar, E.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 15 October 2020).
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Andersen, P.I.; Ianevski, A.; Lysvand, H.; Vitkauskiene, A.; Oksenych, V.; Bjørås, M.; Telling, K.; Lutsar, I.; Dumpis, U.; Irie, Y.; et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 2020, 93, 268–276. [Google Scholar] [CrossRef]
- Ton, A.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 1–8. [Google Scholar] [CrossRef]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Yedery, R.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef]
- Fry, D.E. Antimicrobial Peptides. Surg. Infect. 2018, 19, 804–811. [Google Scholar] [CrossRef]
- Guaní-Guerra, E.; Santos-Mendoza, T.; Lugo-Reyes, S.O.; Tamayo, L.F.T. Antimicrobial peptides: General overview and clinical implications in human health and disease. Clin. Immunol. 2010, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Futur. Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, S.; Balkhy, H.; Gabere, M.N. Current treatment options and the role of peptides as potential therapeutic components for Middle East Respiratory Syndrome (MERS): A review. J. Infect. Public Health 2018, 11, 9–17. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Mustafa, S.; Balkhy, H.; Gabere, M.N. Peptide-Protein Interaction Studies of Antimicrobial Peptides Targeting Middle East Respiratory Syndrome Coronavirus Spike Protein: An In Silico Approach. Adv. Bioinform. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.-M.; Chan, C.C.-S.; Leung, H.-C.; Fai, N.; Lin, Y.-P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Wohlford-Lenane, C.L.; Meyerholz, D.K.; Perlman, S.; Zhou, H.; Tran, D.; Selsted, M.E.; McCray, P.B. Rhesus Theta-Defensin Prevents Death in a Mouse Model of Severe Acute Respiratory Syndrome Coronavirus Pulmonary Disease. J. Virol. 2009, 83, 11385–11390. [Google Scholar] [CrossRef]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.-H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.-W.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef]
- Ho, T.-Y.; Wu, S.-L.; Chen, J.-C.; Wei, Y.-C.; Cheng, S.-E.; Chang, Y.-H.; Liu, H.-J.; Hsiang, C.-Y. Design and biological activities of novel inhibitory peptides for SARS-CoV spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2006, 69, 70–76. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef]
- Medhi, B.; Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; et al. Drug for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56–65. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Cava, C.; Bertoli, G.; Castiglioni, I. In Silico Discovery of Candidate Drugs against Covid-19. Viruses 2020, 12, 404. [Google Scholar] [CrossRef]
- Marimuthu, S.K.; Nagarajan, K.; Perumal, S.K.; Palanisamy, S.; Subbiah, L. Insilico Alpha-Helical Structural Recognition of Temporin Antimicrobial Peptides and Its Interactions with Middle East Respiratory Syndrome-Coronavirus. Int. J. Pept. Res. Ther. 2020, 26, 1473–1483. [Google Scholar] [CrossRef]
- Du, L.; Yang, Y.; Zhouc, Y.; Lud, L.; Li, F.; Jiang, S. MERS-CoV Spike Protein: A Key Target for Antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. [Google Scholar] [CrossRef]
- Vanhoye, D.; Bruston, F.; Nicolas, P.; Amiche, M. Antimicrobial Peptides from Hylid and Ranin Frogs Originated from a 150-Million-Year-Old Ancestral Precursor with a Conserved Signal Peptide but a Hypermutable Antimicrobial Domain. Eur. J. Biochem. 2003, 270, 2068–2081. [Google Scholar] [CrossRef]
- Xu, X.; Lai, R. The Chemistry and Biological Activities of Peptides from Amphibian Skin Secretions. Chem. Rev. 2015, 115, 1760–1846. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M. Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell. Mol. Life Sci. 2011, 68, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.; Bowie, J.H.; Carver, J.A. The Solution Structure and Activity of Caerin 1.1, an Antimicrobial Peptide from the Australian Green Tree Frog, Litoria Splendida. Eur. J. Biochem. 1997, 247, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Steinborner, S.T.; Waugh, R.J.; Bowie, J.H.; Wallace, J.C.; Tyler, M.J.; Ramsay, S.L. New caerin antibacterial peptides from the skin glands of the Australian tree frogLitoria xanthomera. J. Pept. Sci. 1997, 3, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Steinborner, S.T.; Currie, G.J.; Bowie, J.H.; Wallace, J.C.; Tyler, M.J. New antibiotic caerin 1 peptides from the skin secretion of the Australian tree frog Litoria chloris. Comparison of the activities of the caerin 1 peptides from the genus Litoria. J. Pept. Res. 1998, 51, 121–126. [Google Scholar] [CrossRef]
- Wabnitz, P.A.; Bowie, J.H.; Tyler, M.J.; Wallace, J.C.; Smith, B.P. Differences in the Skin Peptides of the Male and Female Australian Tree Frog Litoria Splendida. The Discovery of the Aquatic Male Sex Pheromone Splendipherin, Together with Phe8 Caerulein and a New Antibiotic Peptide Caerin 1.10. Eur. J. Biochem. 2000, 267, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The Antibiotic and Anticancer Active Aurein Peptides from the Australian Bell Frogs Litoria Aurea and Litoria Raniformis the Solution Structure of Aurein 1.2. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef]
- Apponyi, M.A.; Pukala, T.; Brinkworth, C.S.; Maselli, V.M.; Bowie, J.H.; Tyler, M.J.; Booker, G.W.; Wallace, J.C.; Carver, J.A.; Separovic, F.; et al. Host-defence peptides of Australian anurans: Structure, mechanism of action and evolutionary significance. Peptides 2004, 25, 1035–1054. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.-T.; Hou, M.-C.; Xiao, Z.-X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2019, 41, 138–144. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Norrie, J.D. Remdesivir for COVID-19: Challenges of underpowered studies. Lancet 2020, 395, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Erickson, T.B.; Chai, P.R.; Boyer, E.W. Chloroquine, hydroxychloroquine and COVID-19. Toxicol. Commun. 2020, 4, 40–42. [Google Scholar] [CrossRef]
- Tecle, T.; Tripathi, S.; Hartshorn, K.L. Review: Defensins and cathelicidins in lung immunity. Innate Immun. 2010, 16, 151–159. [Google Scholar] [CrossRef]
- Budge, P.J.; Lebowitz, J.; Graham, B.S. Antiviral Activity of RhoA-Derived Peptides against Respiratory Syncytial Virus Is Dependent on Formation of Peptide Dimers. Antimicrob. Agents Chemother. 2003, 47, 3470–3477. [Google Scholar] [CrossRef]
- Kozhikhova, K.V.; Shilovskiy, I.P.; Shatilov, A.A.; Timofeeva, A.V.; Turetskiy, E.A.; Vishniakova, L.I.; Nikolskii, A.A.; Barvinskaya, E.D.; Karthikeyan, S.; Smirnov, V.V.; et al. Linear and dendrimeric antiviral peptides: Design, chemical synthesis and activity against human respiratory syncytial virus. J. Mater. Chem. B 2020, 8, 2607–2617. [Google Scholar] [CrossRef]
- Boas, L.C.P.V.; Campos, M.L.; Berlanda, R.L.A.; Neves, N.D.C.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, D. In-Silico Molecular Binding Prediction for Human Drug Targets Using Deep Neural Multi-Task Learning. Genes 2019, 10, 906. [Google Scholar] [CrossRef]
- Li, Z.; Han, P.; You, Z.-H.; Li, X.; Zhang, Y.; Yu, H.; Nie, R.; Chen, X. In silico prediction of drug-target interaction networks based on drug chemical structure and protein sequences. Sci. Rep. 2017, 7, 11174. [Google Scholar] [CrossRef]
- Sonawane, P.; Patel, K.; Vishwakarma, R.K.; Singh, S.; Khan, B.M. In Silico mutagenesis and docking studies of active site residues suggest altered substrate specificity and possible physiological role of Cinnamoyl CoA Reductase 1 (Ll-CCRH1). Bioinformation 2013, 9, 224–232. [Google Scholar] [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. A systematic study of the effects of structural and physical properties on biological activity. JBIC J. Biol. Inorg. Chem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H. A Clustering Method Based on K-Means Algorithm. Phys. Procedia 2012, 25, 1104–1109. [Google Scholar] [CrossRef]
- Syakur, M.A.; Khotimah, B.K.; Rochman, E.M.S.; Satoto, B.D. Integration K-Means Clustering Method and Elbow Method for Identification of The Best Customer Profile Cluster. IOP Conf. Ser. Mater. Sci. Eng. 2018, 336, 12017. [Google Scholar] [CrossRef]
- Guermeur, Y.; Geourjon, C.; Gallinari, P.; Deléage, G. Improved Performance in Protein Secondary Structure Prediction by Inhomogeneous Score Combination. Bioinformatics 1999, 15, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network Protein Sequence Analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sipp, M.J. ProSA-web: Interactive web service for the recognition of errors in three dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins Struct. Funct. Genet. 2002, 50, 437–450. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2020; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Length | Net Charge | % Hydrophobicity | Total Peptides | Peptides with Antiviral Activity | |||
|---|---|---|---|---|---|---|---|---|
| Low | High | Low | High | Low | High | |||
| 1 | 17 | 38 | −2 | 13 | 35 | 56 | 293 | 48 |
| 2 | 5 | 38 | −7 | 13 | 0 | 40 | 88 | 6 |
| 3 | 33 | 64 | −12 | 20 | 20 | 58 | 178 | 12 |
| 4 | 6 | 28 | −3 | 6 | 45 | 100 | 218 | 36 |
| 5 | 69 | 147 | −11 | 33 | 19 | 49 | 23 | 5 |
| Peptide | Specie | Structure | Length | Net Charge | % Hydrophobicity | Cluster |
|---|---|---|---|---|---|---|
| Aurein 1.2 | Litoria raniformis | α-helix | 13 | 1 | 53 | 4 |
| Caerin 1.1 | Litoria splendida | α-helix | 25 | 1 | 56 | 4 |
| Caerin 1.3 | Litoria caerula | α-helix | 25 | 0 | 56 | 4 |
| Caerin 1.5 | Litoria caerula | α-helix | 25 | 1 | 56 | 4 |
| Caerin 1.6 | Litoria xanthomera | α-helix | 24 | 2 | 58 | 4 |
| Caerin 1.7 | Litoria xanthomera | α-helix | 24 | 3 | 54 | 4 |
| Caerin 1.8 | Litoria chloris | α-helix | 24 | 3 | 54 | 4 |
| Caerin 1.9 | Litoria chloris | α-helix | 24 | 2 | 54 | 4 |
| Caerin 1.10 | Litoria splendida | α-helix | 25 | 2 | 56 | 4 |
| Dermaseptin-S4 | Phyllomedusa sauvagii | α-helix | 28 | 4 | 71 | 4 |
| Uperin 7.1 | Litoria ewingi | β-sheet | 13 | 1 | 61 | 4 |
| Caerin 1.20 | Litoria caerulea | α-helix | 25 | 1 | 56 | 4 |
| Caerin 1.19 | Litoria gracilenta | α-helix | 25 | 3 | 56 | 4 |
| Dermaseptin-S9 | Phyllomedusa sauvagei | α-helix | 24 | 4 | 54 | 4 |
| Maculatin 1.3 | Litoria eucnemis | α-helix | 21 | 1 | 57 | 4 |
| Peptide | Sequence | Itasser | Modeller | Z-Score | |
|---|---|---|---|---|---|
| Number of Residues in Favored Region (%) | DOPE Score | Number of Residues in Favored Region (%) | |||
| Aurein 1.2 | GLFDIIKKIAESF | 100 | NA | NA | −0.91 |
| Caerin 1.1 | GLLSVLGSVAKHVLPHVVPVIAEHL | 78,3 | −1399 | 95 | 1.09 |
| Caerin 1.10 | GLLSVLGSVAKHVLPHVVPVIAEKL | 78 | −1405 | 96 | 0.97 |
| Caerin 1.19 | GLFKVLGSVAKHLLPHVAPIIAEKL | 91 | −1517 | 100 | 0.05 |
| Caerin 1.20 | GLFGILGSVAKHVLPHVIPVVAEHL | 56,5 | −1324 | 96 | 1.59 |
| Caerin 1.3 | GLLSVLGSVAQHVLPHVVPVIAEHL | 100 | NA | NA | 1.27 |
| Caerin 1.5 | GLLSVLGSVVKHVIPHVVPVIAEHL | 100 | NA | NA | 1.37 |
| Caerin 1.6 | GLFSVLGAVAKHVLPHVVPVIAEK | 91 | −1422 | 96 | −0.04 |
| Caerin 1.7 | GLFKVLGSVAKHLLPHVAPVIAEK | 77 | −1405 | 100 | −0.18 |
| Caerin 1.8 | GLFKVLGSVAKHLLPHVVPVIAEK | 96 | −1313 | 96 | −1.02 |
| Caerin 1.9 | GLFGVLGSIAKHVLPHVVPVIAEK | 68 | −1577 | 100 | 1.34 |
| Dermaseptin-S4 | ALWMTLLKKVLKAAAKAALNAVLVGANA | 65 | −1892 | 96 | −1.73 |
| Dermaseptin-S9 | GLRSKIWLWVLLMIWQESNKFKKM | 86 | −1586 | 86 | 0.57 |
| Maculatin 1.3 | GLLGLLGSVVSHVVPAIVGHF | 89 | −1046 | 100 | 1.18 |
| Uperin 7.1 | GWFDVVKHIASAV | 100 | NA | NA | 1.02 |
| Sgp | ACE 2 | ||
|---|---|---|---|
| Peptide | Binding Energies (ΔG) k.cal/mol | Peptide | Binding Energies (ΔG) k.cal/mol |
| Caerin 1.10 | −7.7 | Uperin 7.1 | −7.1 |
| Caerin 1.6 | −7.5 | Maculatin 1.3 | −6.4 |
| Caerin 1.9 | −7.4 | Aurein 1.2 | −5.9 |
| Uperin 7.1 | −7.4 | Caerin 1.20 | −5.8 |
| Caerin 1.20 | −6.9 | Caerin 1.3 | −5.7 |
| Maculatin 1.3 | −6.9 | Caerin 1.1 | −5.6 |
| Caerin 1.1 | −6.7 | Caerin 1.5 | −5.5 |
| Caerin 1.3 | −6.5 | Dermaseptin−S4 | −5.5 |
| Dermaseptin-S4 | −6.4 | Caerin 1.6 | −5.4 |
| Dermaseptin-S9 | −6.4 | Caerin 1.9 | −5.4 |
| Caerin 1.19 | −6.2 | Caerin 1.10 | −5.2 |
| Caerin 1.5 | −6.1 | Caerin 1.19 | −4.8 |
| Aurein 1.2 | −5.8 | Dermaseptin-S9 | −4.2 |
| Caerin 1.8 | −6.0 | Caerin 1.7 | −6.2 |
| Caerin 1.7 | −6.3 | Caerin 1.8 | −6.2 |
| Peptides | Binding Energy (kcal/mol) | Hydrogen Bond * | Electrostatic Bond * | Hydrophobic Bond * |
|---|---|---|---|---|
| SARS-CoV-HR2P | −5.5 |
B:SER50—ligand:ASN11; B:THR51—ligand:LYS24; B:TYR200—ligand:GLU21; B:TYR200—ligand:ASP17; C:THR739—ligand:GLY4; C:GLN755—ligand:GLU28; C:GLN755—ligand:ASP32; C:GLN755—ligand:ASP32; C:THR761—ligand:ILE5; C:THR761—ligand:ALA7; C:ASN764—ligand:ILE5; B:SER50—ligand:ASN11; A:LEU517—ligand:LYS14; A:GLU516—ligand:AR8; A:THR430—ligand:AR8; B:THR51—ligand:ASN25; B:GLN52—ligand:ASN27; A:ASP428—ligand:LEU36; B:HIS49:C—ligand:VAL10; C:GLY757—ligand:SER8; C:SER758—ligand:VAL9; C:ASN751—ligand:GLU28 | A:ARG567—ligand:GLU15; B:LYS202—ligand:ASP17; B:ASP979—ligand:LYS14; A:GLU516—ligand:LYS24 | A:PRO426—ligand:LEU36; B:CYS291—ligand:ILE2; B:LYS964—ligand:VAL10; C:LEU752—ligand:ILE31; B:LEU54—ligand:VAL22; A:TYR396—ligand:LEU19; B:TYR200—ligand:LEU19 |
| EK1 | −5.3 | A:THR430—ligand:SER1; A:LYS462—ligand:GLN4; A:TYR756—ligand:GLU35; A:ARG995—ligand:LEU33; B:THR51—ligand:GLU15; B:GLN52—ligand:TYR14; B:SER975—ligand:LYS17; C:SER750—ligand:THR8; C:TYR756—ligand:LEU36; C:THR761—ligand:ASP11; C:THR998—ligand:GLU35; A:SER514—ligand:SER1; A:THR81—ligand:ASN6; C:THR739—ligand:LEU10; B:ILE973—ligand:LYS25; B:TYR756—ligand:LYS34; C:GLU990—ligand:SER29; A:PHE970—ligand:LYS34; B:HIS49—ligand:GLU15 | A:ARG567—ligand:GLU20; A:ARG995—ligand:ASP32; B:ARG44—ligand:GLU15; B:ARG44—ligand:GLU20; B:LYS202—ligand:GLU21; A:ASP428—ligand:SER1; B:ASP994—ligand:LYS34; | A:ARG995—ligand:LYS34; C:VAL991—ligand:ILE31; C:VAL991—ligand:LEU36; C:LEU754—ligand:TYR14; |
| Caerin 1.6 | −7.5 | B:THR51—Ligand:SER4; B:ARG44—Ligand:GLY7; B:HIS49 —Ligand:LEU6; B:SER975—Ligand:ALA10; B:ARG983—Ligand:LYS11; B:ARG995—Ligand:VAL17; B:ARG995—Ligand:VAL18; B:THR998—Ligand:ALA22; C:GLN755—Ligand:PRO15; C:TYR756—Ligand:LYS24; A:ASP568—Ligand:GLY7; C:THR998—Ligand:LYS24; A:THR998—Ligand:LYS24; A:ASP428—Ligand:HIS16; B:HIS49—Ligand:VAL5; B:HIS49—Ligand:GLY7; C:GLN755—Ligand:GLY1 | B:ASP979—Ligand:LYS11; A:ASP994—Ligand:LYS24 | B:ILE973—Ligand:HIS16; C:LEU754—Ligand:PHE3; C:VAL991—Ligand:VAL17; B:VAL991—Ligand:PRO19; C:VAL991—Ligand:VAL20; B:LYS964—Ligand:LEU6; C:ARG995—Ligand:ILE21; C:ARG995—Ligand:LYS24; A:LEU518—Ligand:HIS12; B:HIS49—Ligand:ALA8 |
| Caerin 1.10 | −7.7 | A:ARG995—ligand:VAL5; A:ARG995—ligand:GLY7; B:HIS49—ligand:GLU23; B:THR51—ligand:LYS24; B:ASN969—ligand:VAL18; B:ASN969—ligand:PRO19; B:THR998—ligand:LEU2; C:GLN755—ligand:HIS16; A:THR998—ligand:LEU2; B:HIS49—ligand:ALA22; B:HIS49—ligand:GLU23; B:VAL991—ligand:LEU6; A:ASP994—ligand:GLY1; B:THR51—ligand:LYS24; B:GLN52—ligand:LYS24; | B:ARG44—ligand:LEU25; B:HIS49—ligand:GLU23; A:ASP994—ligand:GLY1; B:GLU988—ligand:HIS12; C:ASP994—ligand:HIS16; | A:PRO412—ligand:LEU14; B:PRO987—ligand:VAL9; C:PRO987—ligand:PRO15; C:ARG995—ligand:LEU2; B:PRO987—ligand:ALA10; C:LEU754—ligand:PRO19; A:TYR380—ligand:VAL13; C:TYR756—ligand:LEU3; C:VAL991—ligand:HIS16; |
| Molecule | Binding Energy (kcal/mol) | Hydrogen Bond * | Electrostatic Bond * | Hydrophobic Bond * |
|---|---|---|---|---|
| SARS-CoV-HR2P | −4.50 | Ligand:GLY4—A:TYR158; Ligand:ASP1—A:SER167; Ligand:VAL10—A:SER170; Ligand:ASN11—A:LYS174; Ligand:ILE31—A:GLN472; Ligand:GLN34—A:ARG482; Ligand:ASP32—A:GLU495; Ligand:ASP17—A:THR496; Ligand:ILE2—A:ASN159; Ligand:ASN20—A:ASP471; Ligand:GLN34—A:MET474; Ligand:ASP1—A:TRP163; Ligand:GLU21—A:ASP494 | Ligand:GLU35—A:ARG482 | Ligand:LYS14—A:LYS174; Ligand:ILE16—A:PRO178; Ligand:LEU19—A:LYS470; Ligand:LEU30—A:LYS475; Ligand:LEU26—A:PRO492; Ligand:LEU36—A:PRO492; Ligand:LEU36—A:ALA614; Ligand:ILE2—A:TRP163; Ligand:LEU36—A:TYR613 |
| EK1 | −4.10 | Ligand:GLU13—A:TYR158; Ligand:GLU20—A:SER170; Ligand:SER29—A:LYS174; Ligand:SER1—A:ARG482; Ligand:LYS25—A:THR496; Ligand:LEU2—A:TYR613; Ligand:SER1—A:ASP609; Ligand:ASN6—A:PRO492; Ligand:ASN6—A:GLU166; Ligand:GLU20—A:GLU171; Ligand:LYS25—A:GLU495; Ligand:GLU28—A:ASP494; Ligand:SER29—A:ASP494; Ligand:ASP3—A:LYS475; Ligand:SER1—A:SER611; Ligand:LYS25—A:GLU181 | Ligand:ASP3—A:LYS475; Ligand:LYS17—A:GLU166 | Ligand:PHE9—A:TYR613; Ligand:LYS18—A:PRO135; Ligand:LYS25—A:ARG177; Ligand:LYS24—A:PRO178; Ligand:LYS25—A:PRO178; Ligand:LEU12—A:VAL491; Ligand:LEU12—A:PRO492; Ligand:LYS17—A:LEU162; Ligand:ALA22—A:LYS174; Ligand:LEU19—A:TRP163 |
| Caerin 1.6 | −5.40 | Caerin 1.6:HIS12—A:ASP609; Caerin 1.6:SER4—A:TYR158; Caerin 1.6:GLU23—A:TRP163; Caerin 1.6:GLU23—A:SER170; Caerin 1.6:ILE21—A:LYS174; Caerin 1.6:VAL13—A:ARG482; Caerin 1.6:ALA22—A:TYR497; Caerin 1.6:LYS11—A:TYR613; Caerin 1.6:HIS12—A:ASP609; Caerin 1.6:VAL18—A:PRO492; Caerin 1.6:PRO19—A:PRO492; Caerin 1.6:GLU23—A:GLU166; Caerin 1.6:GLU23—A:SER167; Caerin 1.6:ALA8—A:ALA614 | Caerin 1.6:GLY1—A:ASP157 | Caerin 1.6:LEU2—A:LEU162; Caerin 1.6:ALA8—A:PRO492; Caerin 1.6:LYS11—A:PRO492; Caerin 1.6:PRO19—A:VAL491; Caerin 1.6:PRO19—A:PRO492; Caerin 1.6:PHE3—A:VAL491; Caerin 1.6:VAL5—A:TYR255 |
| Caerin 1.10 | −5.20 | Caerin 1.10:PRO15—A:GLN472; Caerin 1.10:HIS16—A:GLN472; Caerin 1.10:HIS16—A:LYS475; Caerin 1.10:GLU23—A:ARG482; Caerin 1.10:GLU23—A:TYR613; Caerin 1.10:SER8—A:TYR613; Caerin 1.10:LEU2—A:TRP163 | Caerin 1.10:LEU25—A:LYS475; Caerin 1.10:GLY1—A:GLU166; Caerin 1.10:LYS11—A:ASP494; | Caerin 1.10:LEU3—A:PRO135; Caerin 1.10:VAL5—A:VAL491; Caerin 1.10:LYS24—A:ALA614; Caerin 1.10:ALA10—A:VAL491; Caerin 1.10:ALA10—A:PRO492; Caerin 1.10:ALA22—A:PRO492; Caerin 1.10:LEU3—A:TRP163; Caerin 1.10:LYS11—A:TYR497; Caerin 1.10:LYS24—A:TYR613 |
| Peptide | Sequence | Net Charge | Length | Hydrophobicity (%) | Hydrophobic Moment (µH) |
|---|---|---|---|---|---|
| Caerin 1.10 | GLLSVLGSVAKHVLPHVVPVIAEKL | 1.2 | 25 | 53 | 0.28 |
| Caerin 1.10_Synthetic A (SR4, SR8) | GLLRVLGRVAKHVLPHVVPVIAEKL | 3.2 | 25 | 53 | 0.28 |
| Caerin 1.10_Synthetic B (SH4, SH8) | GLLHVLGHVAKHVLPHVVPVIAEKL | 1.4 | 25 | 52 | 0.27 |
| Caerin 1.10_Synthetic C (SK4, SK8) | GLLKVLGKVAKHVLPHVVPVIAEKL | 3.2 | 25 | 46 | 0.34 |
| Caerin 1.10_Synthetic D (SG4, SG8) | GLLGVLGGVAKHVLPHVVPVIAEKL | 1.2 | 25 | 53 | 0.28 |
| EK1 (Positive Control) | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL | −5.0 | 36 | 63 | 0.34 |
| SARS-CoV-HR2P (Positive Control) | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | −4.0 | 36 | 53 | 0.37 |
| Peptides | Binding Energy (kcal/mol) | Hydrogen Bonds * | Electrostatic Bond * | Hydrophobic Bond * |
|---|---|---|---|---|
| Caerin 1.10_Synthetic A | −5.0 | B:GLN52—Ligand:ARG4; B:GLN965—Ligand:LYS2; A:ARG567—Ligand:ILE21; B:HIS49—Ligand:GLU23; B:SER967—Ligand:GLU23; B:ASN969—Ligand:ARG8; C:GLY757—Ligand:LEU25; B:PRO39—Ligand:PRO19 | B:HIS49—Ligand:GLU23 | C:LEU754—Ligand:LEU2; B:LYS202—Ligand:VAL17; B:LYS41—Ligand:VAL18; B:LYS964—Ligand:LYS24; C:LEU754—Ligand:LEU25; A:LEU518—Ligand:LEU6; A:LEU518—Ligand:VAL18; A:LEU518—Ligand:VAL20; B:TYR200—Ligand:VAL17 |
| Caerin 1.10_Synthetic B | −6.8 | C:ASP994—Ligand:HIS4; B:ILE973—Ligand:LEU6; B:ILE973—Ligand:GLY7; A:HIS519—Ligand:LYS11; B:GLN52—Ligand:VAL13; A:ASP428—Ligand:ALA22; A:ASP428—Ligand:GLU23; A:ASP427—Ligand:LYS24; A:ASP428—Ligand:LYS24; B:ARG983—Ligand:LEU6; B:GLN992—Ligand:GLY1; B:ARG995—Ligand:LEU3; B:GLY971—Ligand:HIS4; C:GLN755—Ligand:HIS4; C:LEU754—Ligand:HIS16; B:SER968—Ligand:HIS16; B:ILE973—Ligand:HIS4 | B:ASP40—Ligand:HIS12 | B:ASP40—Ligand:HIS12; B:ILE973—Ligand:LEU6; B:VAL42—Ligand:ALA10; B:LYS41—Ligand:LYS11; A:LEU517—Ligand:ILE21; A:PRO426—Ligand:ALA22; A:PRO463—Ligand:LYS24; A:PRO412—Ligand:LEU2; A:LEU518 —Ligand:VAL9; A:LEU518—Ligand:ILE21; C:PRO986—Ligand:LEU25; C:LEU752—Ligand:HIS4 |
| Caerin 1.10_Synthetic C | −6.1 | B:SER967—Ligand:LYS11; C:LEU754—Ligand:HIS12; A:LEU517—Ligand:VAL5; B:ARG44—Ligand:LEU25; B:ARG44—Ligand:LYS24; B:SER975—Ligand:GLU23; B:VAL976—Ligand:GLU23; C:ASN751—Ligand:VAL17; C:GLN755—Ligand:VAL13; C:GLN755—Ligand:LEU14; A:PHE515—Ligand:VAL5; A:GLU516—Ligand:VAL5; B:HIS49—Ligand:LEU25; A:ASP427—Ligand:HIS16 | NA | B:TYR200—Ligand:LEU3; A:ASP427—Ligand:HIS16; C:GLN755—Ligand:HIS12; C:LEU752—Ligand:VAL17; B:VAL42—Ligand:LYS24; A:PRO426—Ligand:LEU2; A:PRO463—Ligand:LEU2; A:LEU518—Ligand:LYS8; A:PRO463—Ligand:HIS16; A:PHE464—Ligand:LYS4 |
| Caerin 1.10_Synthetic D | −6.7 | B:CYS291—Ligand:GLY1; B:GLN52—Ligand:GLY4; B:GLN52—Ligand:LEU6; B:GLN52—Ligand:VAL9; B:GLY971—Ligand:HIS16; A:ARG567—Ligand:LYS24; B:ARG44—Ligand:ALA22; B:GLN52—Ligand:GLY4; B:GLN52—Ligand:LEU6; B:THR274—Ligand:LEU3; B:SER967—Ligand:VAL20; B:ASN969—Ligand:PRO15; B:ASN969—Ligand:HIS16; B:SER974—Ligand:LEU25; C:GLN755—Ligand:HIS16; B:CYS301—Ligand:GLY1; A:HIS519—Ligand:HIS12; A:ARG567—Ligand:ALA22; A:ARG567—Ligand:HIS12 | B:ASP228—Ligand:LYS11; B:ARG44—Ligand:GLU23; B:HIS49—Ligand:GLU23 | C:LEU754—Ligand:VAL5; C:LEU754—Ligand:PRO19; A:LEU518—Ligand:LYS11; A:LEU518—Ligand:VAL13; B:LYS964—Ligand:ILE21 |
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liscano, Y.; Oñate-Garzón, J.; Ocampo-Ibáñez, I.D. In Silico Discovery of Antimicrobial Peptides as an Alternative to Control SARS-CoV-2. Molecules 2020, 25, 5535. https://doi.org/10.3390/molecules25235535
Liscano Y, Oñate-Garzón J, Ocampo-Ibáñez ID. In Silico Discovery of Antimicrobial Peptides as an Alternative to Control SARS-CoV-2. Molecules. 2020; 25(23):5535. https://doi.org/10.3390/molecules25235535
Chicago/Turabian StyleLiscano, Yamil, Jose Oñate-Garzón, and Iván Darío Ocampo-Ibáñez. 2020. "In Silico Discovery of Antimicrobial Peptides as an Alternative to Control SARS-CoV-2" Molecules 25, no. 23: 5535. https://doi.org/10.3390/molecules25235535
APA StyleLiscano, Y., Oñate-Garzón, J., & Ocampo-Ibáñez, I. D. (2020). In Silico Discovery of Antimicrobial Peptides as an Alternative to Control SARS-CoV-2. Molecules, 25(23), 5535. https://doi.org/10.3390/molecules25235535